
Two dynamic N-terminal regions are required for function in ribosomal RNA adenine dimethylase family members
- Danielle A. McGaha,
- Alexandrea Collins,
- Luqman O. Ajisafe,
- Calvin C. Perdigao,
- Jordan L. Bondrowski,
- Karen Fetsch and
- Jack A. Dunkle
- Corresponding author: jadunkle{at}ua.edu
-
Handling editor: Marina Rodnina
Abstract
Prominent members of the ribosomal RNA adenine dimethylase (RRAD) family of enzymes facilitate ribosome maturation by dimethylating 2 nt of small subunit rRNA, including the human DIMT1 and bacterial KsgA enzymes. A subgroup of RRAD enzymes, named erythromycin resistance methyltransferases (Erm), dimethylate a specific nucleotide in large subunit rRNA to confer antibiotic resistance. How these enzymes regulate methylation so that it only occurs on the specific substrate is not fully understood. While performing random mutagenesis on the catalytic domain of ErmE, we discovered that mutants in an N-terminal region of the protein that is disordered in the ErmE crystal structure are associated with a loss of antibiotic resistance. By subjecting site-directed mutants of ErmE and KsgA to phenotypic and in vitro assays, we found that the N-terminal region is critical for activity in RRAD enzymes: The N-terminal basic region promotes rRNA binding, and the conserved motif likely assists in juxtaposing the adenosine substrate and the S-adenosylmethionine cofactor. Our results and emerging structural data suggest that this dynamic, N-terminal region of RRAD enzymes becomes ordered upon rRNA binding, forming a cap on the active site required for methylation.
Keywords
INTRODUCTION
In all organisms, RNA is posttranscriptionally modified in diverse ways, including methylation, thiolation, the conversion of uridines to pseudouridines, and many other chemical marks (Boccaletto et al. 2018). Methylation is among the most frequent modifications of RNA occurring on tRNA and rRNA in all organisms and on mRNA in eukaryotes (Grosjean 2009; Machnicka et al. 2014; Roundtree et al. 2017). Remarkably, a limited number of protein topologies are utilized to carry out methylation of diverse RNA substrates. The two most prominent protein topologies are the SPOUT (SpoU-TrmD) fold and the Rossmann-like fold (Czerwoniec et al. 2009). The SPOUT fold possesses five parallel β-strands with the topology ↑2-↑1-↑4-↑3-↑5 and the distinctive feature of a C-terminal α-helix, which forms a trefoil knot by threading between a loop linking β-strand 3 and β-strand 4 (Strassler et al. 2022). Methyltransferases with the Rossmann-like fold are also known as class I methyltransferases and possess seven β-strands with the topology ↑3-↑2-↑1-↑4-↑5-↓7-↑6 (Schubert et al. 2003). β-strands 1–3 of class I RNA methyltransferases serve as a platform for structural elements that bind the S-adenosylmethionine (SAM) cofactor, while β-strands 4–7 play the same role for RNA binding (Fig. 1A).
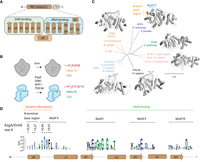
The structure and activity of RRAD family members. (A) RRAD enzymes consist of a Rossmann-like methyltransferase (RM) catalytic domain and a C-terminal domain. The secondary structure of the RM domain is shown indicating that α1–β3 are predominantly involved in SAM while β4–β7 are involved in rRNA binding. (B) RRAD family members dimethylate rRNA of either the large ribosomal subunit (LSU) or small ribosomal subunit (SSU). (C) Experimentally determined structures are available for each RRAD family member in some cases bound to rRNA (MTF1, PDB code 1i4w; ErmE, 6nvm; ErmC, 1qao; TFB1M, 6aax; DIM1, 7wtm; KsgA, 7o5h). An N-terminal basic region (orange) is disordered in these structures (except for MTF1), while the adjacent motif X (blue) is partially ordered. (D) The conservation of RRAD family members at key motifs throughout the α0–β3 region of the protein family is shown as a sequence logo. Residues in the N-terminal basic region and motif X investigated by site-directed mutagenesis in this study are marked with an asterisk (*).
Class I methyltransferases participate in critical functions in all organisms. For example, METTL1 in humans carries out m7G modification of tRNA (Li et al. 2023; Ruiz-Arroyo et al. 2023). In metazoans METTL3, along with its METTL14 partner, deposits m6A in mRNAs to regulate their stability, and the dynamic status of m6A marks in mRNA has been implicated in many disease states (Dominissini et al. 2012; Meyer et al. 2012; Murakami and Jaffrey 2022). In metazoans, METTL16 has been found to introduce m6A into the U6 snRNA and an intronic region of the MAT2A transcript regulating its stability and therefore the levels of its protein product, a SAM synthetase (Pendleton et al. 2017). In bacteria, deposition of m7G at position 1405 or m1A at position 1408 of 16S rRNA provides bacteria resistance to aminoglycoside antibiotics (Jeremia et al. 2023). The dimethylation, m62A2058, on 23S rRNA provides multidrug antibiotic resistance to macrolide, lincosamide, and streptogramin B antibiotics (MLSB phenotype) (Fyfe et al. 2016). Interestingly, while the overall architecture of these enzymes is diverse and the type of methylation varies, the class I methyltransferase catalytic domain is shared by all (Dunkle et al. 2014; Sledz and Jinek 2016; Wang et al. 2016; Doxtader et al. 2018; Srinivas et al. 2023).
The ribosomal RNA adenine dimethylase (RRAD) family comprise enzymes with sequence and structural homology that utilize a class I catalytic domain to dimethylate adenosine residues on small ribosomal subunit RNA or large ribosomal subunit RNA (Fig. 1B,C; Mistry et al. 2021). KsgA deposits m62A at positions 1518 and 1519 (Escherichia coli numbering) of 16 S rRNA in bacteria during ribosome biogenesis (Fig. 1B). The current model for KsgA function is that it binds a conformation of the 30S ribosome that is off-pathway for maturation, and this binding event promotes remodeling of the 30S to an on-pathway conformation (Connolly et al. 2008; Sun et al. 2023). Since ribosomes are able to assemble in ΔksgA cells, methylation is not required for ribosome biogenesis but rather promotes KsgA dissociation from the ribosome once the remodeling event has occurred (Connolly et al. 2008). DIMT1 (DIM1) performs the same methylations on the cytoplasmic ribosomes of eukaryotes (Lafontaine et al. 1994; Zorbas et al. 2015; Shen et al. 2020), whereas TFB1M in humans methylates the 12S rRNA of mitochondrial ribosomes (Metodiev et al. 2009; Liu et al. 2019). Erythromycin resistance methyltransferases (Erm), such as ErmE or ErmC, dimethylate a single position, A2058, in 23S rRNA of bacteria during large ribosomal subunit biogenesis to promote antibiotic resistance (Fig. 1B; Fyfe et al. 2016). ErmE is found in S. erythraea, a soil bacterium that biosynthesizes the protein synthesis inhibitor erythromycin and it protects the bacterium from this toxic molecule (Skinner et al. 1983). ErmC is found on mobile genetic elements that circulate widely among Gram-positive bacteria such as Bacillus subtilis and Staphylococcus aureus. ErmC is a significant contributor to multidrug antibiotic resistance in S. aureus.
In all cases, RRAD proteins are thought to bind SAM and the nucleotide targeted for methylation in a similar manner using conserved motifs (Fig. 1D). RRAD proteins contain conserved motifs observed in other 6-methyladenosine methyltransferases (Schubert et al. 2003). These include motif I, a Gly-rich region that sits adjacent to the kink between the ribose and methionyl moieties of SAM and has shape complementarity to it. Motif II possesses an acidic residue that hydrogen bonds to the ribose moiety of SAM. Motif IV possesses an aromatic residue that forms π-stacking interactions to the target Ade residue. The importance of these interactions has been validated by numerous functional studies (Farrow et al. 2002; Maravic et al. 2003; O'Farrell et al. 2012; Rowe et al. 2020; Shen et al. 2020; Goh et al. 2022; Sharkey et al. 2022).
We asked whether there may be unappreciated elements of the catalytic domain of RRAD proteins required for function. To answer this question, we performed random mutagenesis on the catalytic domain of ErmE and screened the resulting cells for an erythromycin-sensitive phenotype. Surprisingly, the screen indicated residues in an N-terminal region that is disordered in the crystal structure were associated with an erythromycin-sensitive phenotype (Stsiapanava and Selmer 2019). Site-directed mutants of ErmE were generated and characterized phenotypically and by in vitro biochemistry. These data indicated the existence of two unappreciated regions in ErmE that are critical for function: an N-terminal basic region and motif X (Fig. 1D). We next investigated whether these regions were functionally important in other Erm proteins using the ErmC model system and in RRAD proteins, broadly, using the small subunit rRNA methyltransferase KsgA as a model. We produced evidence that the N-terminal basic region and motif X are also required for function in these methyltransferases indicating they are a characteristic of RRAD family members generally. A synthesis of our biochemical data and available structural data suggests the N-terminal region undergoes a disordered to ordered transition when basic residues recognize the enzyme's specific rRNA substrate and the ordered conformation contributes key interactions to the active site.
RESULTS
We previously identified multiple residues within the catalytic domain of ErmE that are required for function (Rowe et al. 2020; Sharkey et al. 2022). Residues were targeted for site-directed mutagenesis based on sequence alignment to ErmC or because a Rosetta docking of ErmE to its rRNA substrate indicated they were likely to contact rRNA (Fig. 2A). We wondered whether there may be residues in ErmE critical for function that had not yet been identified by a model-driven approach. Therefore, we performed random mutagenesis of the catalytic domain of ErmE by error-prone PCR (Fig. 2A; Supplemental Fig. S1). The plasmid containing the resulting library was used to transform E. coli cells. Cells containing the ErmE plasmid were selected by their growth on an ampicillin plate, then were transferred to an erythromycin plate with the expectation that loss of catalytic activity in ErmE would result in an erythromycin-sensitive phenotype. From a pool of 182 transformants, we identified 146 colonies that were erythromycin sensitive. We sequenced these transformants, and all contained multiple mutations, as expected for our implementation of error-prone PCR. We plotted the frequency that each amino acid was altered in an erythromycin-sensitive colony (Fig. 2B). The mutations at S88 and A96 were frequently observed which could be rationalized based on their proximity to the SAM-binding site. Random mutagenesis of ErmB has previously been reported and also observed mutations in the SAM pocket associated with an erythromycin-sensitive phenotype (Farrow et al. 2002). Additionally, R163 is a position that closely approaches rRNA in our computational model and whose site-directed mutants are erythromycin sensitive (Sharkey et al. 2022). Unexpectedly, we also saw that P20 and R47 mutations often occurred among our erythromycin-sensitive colonies, which are both located in the N terminus that is not ordered in the crystal structure of ErmE (Stsiapanava and Selmer 2019).
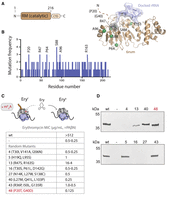
Random mutagenesis reveals that mutations in the dynamic, N-terminal region of ErmE are associated with an erythromycin-sensitive phenotype. (A) The crystal structure of the RRAD family member ErmE lacks the dynamic, N-terminal region. Docking of SAM onto the crystal structure and rRNA via Rosetta modeling gives the approximate positions of the substrates. The residues most frequently associated with an erythromycin-sensitive phenotype in random mutagenesis screens are shown as spheres. Residues P20 and G40 were not present in the coordinate model. (B) Random mutagenesis of the catalytic, Rossmann-like methyltransferase (RM) domain of ermE was performed. A histogram shows how often specific ermE residues were mutated among clones that were erythromycin sensitive. (C) Minimal inhibitory concentrations (MIC) for erythromycin were measured in E. coli cells expressing ermE mutants in the presence of the antibiotic adjuvant phenyl-arginyl-β-napthylamide (PAβΝ). Random mutagenesis produced variants with two to four mutations per clone. MIC values were measured from three replicates. The range of MIC values observed is reported. Where one value is listed, all replicates possessed this value. (D) Western blotting of cell lysates was used to detect whether ermE mutants produced a soluble and stable protein. Mutant 48 is highlighted in red to denote it is the sole mutant composed of only N-terminal changes.
Mutants of the basic region and motif X are associated with an erythromycin-sensitive phenotype
To scrutinize the observation that mutations in the dynamic N-terminal region may contribute to an erythromycin-sensitive phenotype, we performed a microdilution assay on a selection of these mutants. The assay confirmed that the selected colonies possessed very low minimal inhibitory concentrations (MIC) for erythromycin (Fig. 2C). We performed western blotting of these mutants to determine which random mutants may owe their phenotype to a destabilization of the ErmE structure and which may be explained by a loss of ErmE catalytic activity. Four out of the eight mutants assayed produced a signal comparable to wild type in a blot of the supernatant of lysed E. coli cells, suggesting the mutants did not destabilize ErmE (Fig. 2D). Interestingly, one mutant, the P20T G40D double mutant, was solely composed of mutations to the dynamic, N-terminal region of ErmE, suggesting it may play a previously unappreciated role in ErmE function.
Since the N-terminal region of ErmE is not ordered in the crystal structure, we inspected an AlphaFold2 model of ErmE to hypothesize how this region may contribute to function (Jumper et al. 2021). AlphaFold2 predicts the presence of an α-helix enriched in basic residues followed by a loop region with sequence conservation among many DNA and RNA methyltransferases that has been denoted as motif X (Figs. 1D and 3A; Malone et al. 1995). The N-terminal basic region and motif X are adjacent to the ErmE active site. We hypothesized that the basic region, while disordered in the apo structure of ErmE, may become ordered upon RNA binding. It may contribute to the binding affinity of ErmE for RNA or may influence the conformation of motif X in a manner that promotes SAM binding or positioning of the target A2058 nucleotide in the active site. To assess the importance of the basic region and motif X for ErmE function, we generated site-directed mutants of the two regions and scored the resulting phenotype of cells bearing these mutants in the presence of erythromycin (Fig. 3B). We altered basic Arg to the acidic Glu and polar or hydrophobic residues to Ala. Three site-directed mutants (R36P, R47S, and G40D) were constructed due to observations from random mutagenesis. Most, but not all, mutants displayed a dramatic reduction in MIC value. To dissect whether the erythromycin-sensitive phenotypes were derived from a loss of ErmE catalytic activity or destabilization of the protein, we again performed western blots. Within the basic region, site-directed mutants of R34, R36, and R37 were associated with erythromycin sensitivity yet produced levels of protein similar to wild type, suggesting these mutants are defective in catalysis (Fig. 3B,C). Random mutagenesis had indicated R47 may play an important functional role in ErmE, but the R47S mutant was not erythromycin sensitive (Fig. 3B). Mutants G40D, N42A, and F43A within motif X were associated with erythromycin sensitivity but had levels comparable to wild type by western blotting, suggesting a catalytic defect (Fig. 3B,C). Taken together, our phenotypic experiments identified multiple residues within both the basic region and motif X that appear to be crucial for catalysis.
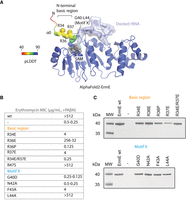
Site-directed mutants in the N-terminal basic region and motif X are associated with altered phenotypes in ErmE. (A) An AlphaFold2 model of ErmE suggests that some of the dynamic N-terminal residues form α-helix 0. Superposition of the AlphaFold2 ErmE model onto the model given in Figure 2A indicates α0 forms one wall of the active site. (B) MIC values for erythromycin were measured for E. coli cells expressing ermE site-directed mutants in the basic region and motif X. MIC values were measured from three replicates. The range of MIC values observed is reported. Where one value is listed, all replicates possessed this value. (C) Western blotting of cell lysates was used to detect whether ermE site-directed mutants produced a soluble and stable protein.
To further test the hypothesis that the basic region and motif X contribute to ErmE function, we performed in vitro methylation reactions. The reactions were carried out with wild-type and site-directed mutants of ErmE, a synthetic RNA that forms a hairpin mimicking helix 73 of 23S rRNA and 3H-SAM as methyl donor (Fig. 4A; Supplemental Fig. S2). The experiments were performed under single turnover conditions, that is, with excess enzyme and with SAM limiting. Mutants reporting on the contribution of the basic region, R34E/R37E and R36P, showed very low levels of methylation compared to wild type (Fig. 4A). However, R36E possessed an intermediate defect. Mutants reporting on motif X, G40D, and N42A had low levels of methylation, while the F43A mutant had an intermediate level of methylation (Fig. 4A). These results strongly suggest that both the basic region and motif X are critical for ErmE function and are consistent with the results from the phenotypic assays. The two site-directed mutants with intermediate levels of methylation, R36E and F43A, also had intermediate MIC values of 128 and 2 μg/mL erythromycin, respectively, in the phenotypic assays, demonstrating a qualitative correlation between methyltransferase activity and erythromycin resistance.
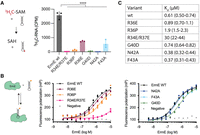
The N-terminal basic patch and motif X contribute to rRNA methylation, and the basic patch contributes to rRNA binding. (A) Methylation reactions under single-turnover conditions were conducted using site-directed mutants in the N-terminal basic region or motif X of ErmE. (B) Affinity binding of ErmE site-directed mutants to an analog of helix 73 of 23S rRNA was measured by fluorescence polarization. Three independent binding reactions were performed for each variant, and replicate measurements are shown as a scatter plot. A cartoon depicting the assays is given. The fluorescein label on the 48 nt rRNA analog is indicated by an orange circle. (C) Dissociation constant (Kd) values of ErmE variants for RNA. Parentheses indicate 95% confidence interval. (****) P ≤ 0.001.
Identifying the mechanistic contributions of the basic region and motif X to catalysis
To begin to dissect how the basic region and motif X contribute to rRNA methylation by ErmE, we performed a saturation binding assay to test how mutants affected affinity for rRNA. Purified site-directed mutants of ErmE were titrated against a helix 73 rRNA analog possessing a fluorescent label and binding was read by fluorescence polarization (Fig. 4B). ErmE wild type was used as a positive control for binding, and pepsin, a protein not known to interact with RNA, was used as a negative control. The basic region mutant R36E had a binding affinity similar to wild type. R36P possessed a modest defect in RNA binding, while an R34E/R37E double mutant displayed a major defect. The motif X mutants G40D, N42A, and F43A possessed binding curves similar to wild type. Taken together, these data suggest that motif X in ErmE may not make a major contribution to RNA binding. Alteration of a single residue in the basic region of ErmE does not lead to a major defect in RNA binding but alteration of multiple residues, as in R34E/R37E does.
With, the RNA affinity binding data in hand, we reasoned we could combine these data with structural information and a kinetic analysis of ErmE and its mutants to determine the most likely contribution of the basic region and motif X to ErmE catalysis. We sought to distinguish between contributions to rRNA binding, SAM binding or chemistry, such as positioning of the nucleophilic N6 of adenosine adjacent to the sulfonium of SAM. The structure of ErmC bound to SAM, the apo structure of ErmE, our Rosetta model of ErmE bound to a fragment of rRNA and the crystal structure of RRAD family member TFB1M bound to RNA informed our analysis (Schluckebier et al. 1999; Liu et al. 2019; Stsiapanava and Selmer 2019; Sharkey et al. 2022). While motif X is not fully ordered in these structures, the portion that is ordered, for example, N36, F37, and L38 in TFB1M, suggest it could contribute to SAM-binding affinity or positioning of the target Ade or SAM for chemistry. The RNA-binding affinity reported above shows it does not make a major contribution to rRNA binding. The possibilities for the basic region are more complex. While our data show that it makes a contribution to RNA binding, that interaction with RNA could influence the adjacent motif X residues to affect SAM binding or chemistry or both.
To distinguish between the possible ways the basic region and motif X could contribute to catalysis, we followed methylation over time, under conditions of limiting SAM and as a function of ErmE concentration (Fig. 5A). We extracted observed rate constants (kobs min−1) for these reactions and plotted them versus ErmE concentration fitting the data to a hyperbola to produce K1/2,SAM and kchem, which reflects the rate-limiting step prior to product formation. The K1/2,SAM parameter should approximate Kd,SAM since our reaction is under rapid equilibrium conditions (Fig. 5B; Table 1; Hou and Masuda 2015). We chose the mutants R36E and F43A for this analysis because they retained enough methylation activity to produce a signal that could be fit to our kinetic model.

Single turnover kinetics. (A) Methylated RNA versus time plots for increasing concentration of wild-type (wt) ErmE and site-directed mutants. (B) Methylation rates (kobs) are plotted versus ErmE concentration under limiting SAM and excess RNA conditions and fit to a model for single-turnover kinetics to extract kchem and K1/2,SAM values. An inset is shown of the F43 data with a y-axis range of 0–0.006 min−1.
Single turnover kinetics and selected RNA affinity binding parameters
R36E possesses a fourfold decrease in kchem compared to wild type (0.033 min−1 vs. 0.14 min−1) but possesses a K1/2,SAM = 0.83 μM, which suggests a slightly higher affinity than wild type and indicates there is no defect in SAM binding (Table 1). R36E does bind RNA with slightly lower affinity than wild type (0.89 μM vs. 0.61 μM), but since the kinetics assay is performed with [R36E] in excess of Kd, the binding defect does not explain the fourfold lower kchem. One possible explanation of the data is that the basic region binding to rRNA affects the structure in a way that enhances catalysis. F43A possesses a 23-fold decrease in kchem compared to wild type (0.0061 min−1 vs. 0.14 min−1) indicating a major defect in catalysis, yet possesses a K1/2,SAM = 2.6 μM, similar to wild type. Since we have shown above that motif X mutants do not possess a major binding defect for RNA, the synthesis of the available data suggests how the basic region and motif X affect the mechanism of ErmE. Binding of rRNA by the basic region enhances the affinity of ErmE for rRNA and subtly affects the structure of the neighboring motif X in a manner that promotes catalysis. Since the residues comprising motif X are not suspected of acid-base chemistry, it likely promotes catalysis by optimally positioning the N6 of the target Ade adjacent to the labile methyl group of SAM.
The basic region and motif X contribute to rRNA methylation by RRAD family members
Sequence alignments of ErmE with ErmC, other erythromycin resistance methyltransferases and other RRAD family members, suggests the presence of an N-terminal basic region and motif X is a general feature of these proteins (Fig. 1D; Supplemental Fig. S3). Additionally, comparison of the AlphaFold2 structures of ErmE and ErmC reinforces this (Figs. 3A and 6A). We generated site-directed mutants of ErmC within the basic region and motif X, both single and double mutants, and assayed cells containing them for erythromycin sensitivity. As a positive control, we assayed cells containing wild-type ErmC and as a negative control, cells containing an empty vector. The K4E single mutant possessed a reduced MIC value indicating erythromycin sensitivity and the K4E/K7E double mutant had a further reduced MIC value consistent with the basic region in ErmC playing an important functional role (Fig. 6B). Single mutants within motif X did not possess erythromycin sensitivity, but double mutants did (Fig. 6B). While the erythromycin sensitivity phenotype associated with these ErmC mutants was not as pronounced as in ErmE, the data suggest that both the basic region and motif X also play an important role in ErmC function.

Site-directed mutants in the N-terminal basic region and motif X are associated with altered phenotypes in ErmC. (A) An AlphaFold2 model of ErmC demonstrating the predicted location of basic region residues K4 and K7 and motif X residues S9-I13. (B) MIC values for erythromycin were measured for E. coli cells expressing ermC site-directed mutants in the basic region and motif X. MIC values were measured from three replicates. The range of MIC values observed is reported. Where one value is listed, all replicates possessed this value.
The Erm proteins are the only members of the RRAD family that methylate large ribosomal subunit rRNA; all other family members methylate small ribosomal subunit rRNA (Fig. 1B). KsgA (RsmA) in bacteria dimethylates two adenosine residues in helix 45 of 16S rRNA as part of its role in promoting 30S ribosome maturation. To determine whether the N-terminal basic region and motif X play a role in the function of the small ribosomal subunit rRNA methyltransferases, we used the bacterial KsgA protein as a model system.
We developed a phenotypic assay that reports on KsgA methylation of 16S rRNA by utilizing the aminoglycoside antibiotic kasugamycin. This molecule inhibits initiation of bacterial protein synthesis by binding to a pocket on the ribosome that overlaps with the path of mRNA between the E and P-sites of the 30S subunit (Fig. 7A; Poldermans et al. 1979; Schluenzen et al. 2006; Schuwirth et al. 2006; Zhang et al. 2022). This pocket is adjacent to the sites methylated by KsgA, m62A1518, and m62A1519. Loss of methylation on A1519, for example, in ΔksgA cells, leads to partial resistance to kasugamycin (Helser et al. 1972). It appears the loss of methylation affects the conformation of residues adjacent to A1519 that directly interact with the antibiotic (Fig. 7A). Notably, this phenomenon wherein methylation causes antibiotic sensitivity, works in the opposite manner of ErmE or ErmC where methylation causes antibiotic resistance.
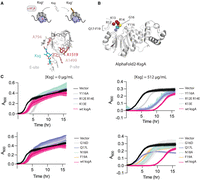
The N-terminal basic patch and motif X of RRAD family member, KsgA, contribute to function in vivo. (A) Kasugamycin (Ksg) inhibits protein synthesis by binding to ribosomes that possess the m62A1519 modification on 16S rRNA. Methylation is indicated by the red asterisk. The structure of Ksg bound to the E. coli ribosome is shown (PDB code 1vs5). A loss of methylation on 16S rRNA residue A1519 affects the local conformation leading to partial Ksg resistance. (B) An AlphaFold2 model of E. coli KsgA shows the predicted location of the basic region R12-R14 and motif X residues G16-F19. (C) Growth curves of E. coli ΔksgA in the absence or presence of kasugamycin. The cells are transformed with wild-type wt ksgA, a site-directed mutant of ksgA or empty vector (Vector). Y116A is included as a control because it is defective in methylation. Three independent cultures were monitored for each variant, and the mean and standard deviation of each time point is plotted.
Site-directed mutants of KsgA in the basic region and motif X were constructed and growth curves were measured for these cells in the presence or absence of kasugamycin (Fig. 7B,C). Three controls were employed to validate the assay worked in the intended manner, that is, growth of cells that efficiently form m62A1519 on 16S rRNA is inhibited by kasugamycin, but cells that do not efficiently methylate this residue are kasugamycin resistant. First, E. coli K12 BW25113 ΔksgA from the Keio collection, labeled as “Vector” in Figure 7C, were transformed with empty vector (Baba et al. 2006). As expected, these cells possessed a brief lag phase of 0.2 ± 0.1 h followed by exponential growth (Table 2). The lag phase of empty vector cells is similar in the presence or absence of kasugamycin confirming that loss of m62A1519 leads to resistance to the antibiotic. Second, we hypothesized that the Y116A mutant of KsgA would be deficient in methylation and found this was the case (Fig. 8A,B). Many class I RNA methyltransferases possess an aromatic residue in motif IV that π-stacks with the substrate nucleobase (Dunkle et al. 2014; Liu et al. 2019). This motif IV residue has been shown to be strictly required for rRNA methylation by Erm enzymes and a multiple sequence alignment shows that Y116A is the motif IV aromatic residue in KsgA (Supplemental Fig. S3; Maravic et al. 2003; Rowe et al. 2020). Cells bearing Y116A possessed a similar length of lag phase (2.8 ± 0.3 vs. 2.7 ± 0.4) in the presence or absence of antibiotic but a longer doubling time (0.48 ± 0.05 vs. 1.05 ± 0.05) (Table 2). The results are consistent with our expectations that the growth defect in the presence of kasugamycin is substantially dependent on rRNA methylation (Fig. 7C). Our final control was to measure the growth of the ΔksgA cells complemented with wild-type ksgA from a plasmid. When stationary phase bacterial cells are diluted into new, nutrient-rich media, they initiate complex gene expression changes to synthesize the proteins needed to reprogram their metabolism and initiate rapid cell division (Bertrand 2019). Cells expressing wild-type ksgA, in which initiation of protein synthesis is expected to be strongly inhibited by kasugamycin, possessed a pronounced phenotype: an exaggerated lag phase increasing to 7 ± 1 h (Fig. 7C). In sum, our controls showed that cells competent in m62A1519 formation demonstrated a pronounced lag phase in the presence of kasugamycin linking the lag phase of cells transformed with ksgA variants to m62A1519 formation in 16S rRNA.
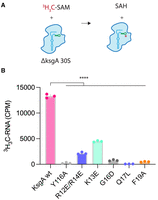
The N-terminal basic region and motif X of KsgA contribute to 16S rRNA methylation in vitro. (A) KsgA catalyzed methylation of 16S rRNA in vitro was measured using 30S ribosomes devoid of m62A1518 and m62A1519 (ΔksgA strain) and a radiolabeled SAM cofactor. (B) Wt KsgA robustly methylates 30S ribosomes in vitro. The Y116A active site mutant is methylation deficient and serves as a negative control. (****) P ≤ 0.001.
Growth parameters for E. coli expressing ksgA variants
Having determined that our phenotypic assay produces results (an exaggerated lag phase) that correlate with the methylation activity of KsgA, we subjected site-directed mutants in the basic region and motif X to the assay. The variants can be placed into two groups. The basic region mutants, K13E and R12E/R14E possessed lag phase lengths of 5.5 ± 0.4 and 5 ± 2 h, respectively (Fig. 7C; Table 2), which are shorter than wild type, but this difference is not statistically significant. These results suggest the basic region variants retain some methylation activity. Motif X mutants G16D, Q17L, N18A, and F19A possessed lag phases ranging from 0.8 to 3.6 h (Fig. 7C; Table 2). These lag phase lengths are significantly shorter than that of wild type consistent with the motif X mutants being defective in methylation. Taken together, these results are consistent with the hypothesis that motif X contributes to methylation activity in all RRAD family members and that the basic region may contribute to methylation in a more subtle way.
To further test the hypothesis that the basic region and motif X contribute to KsgA activity, we purified selected variants and performed in vitro methylation assays (Fig. 8A,B; Supplemental Fig. S4). The assays employed 30S ribosomal subunits purified from ΔksgA cells as substrate and 3H-SAM (Supplemental Fig. S5). Methylation of 30S subunits by wild-type KsgA was performed as a positive control and reactions containing the Y116A variant of KsgA were used as a negative control (Fig. 8B). Basic region mutants K13E and R12E/R14E had decreased, but detectable methylation activity (Fig. 8B). Motif X mutants G16D, Q17L, and F19A exhibited levels of methylation similar to our negative control (Fig. 8B). In sum, the in vitro methylation results are consistent with what we observed in the phenotypic assays and demonstrate that the basic region contributes to methylation and motif X is critical for rRNA methylation by KsgA.
DISCUSSION
We have provided functional evidence that two unappreciated regions of RRAD family enzymes are critical for their function. We demonstrated the importance of the N-terminal basic region and motif X using in vivo assays of site-directed mutants of ErmE, ErmC, and KsgA. We performed methylation assays with purified site-directed mutants of ErmE and KsgA to verify that mutants with a phenotype suggesting loss of function did in fact have perturbed methylation. Using ErmE as a model system, we performed RNA affinity binding assays and kinetics assays to determine the mechanistic contribution of these regions to rRNA methylation. The specific basic residues we mutated in the N-terminal basic region were not required for rRNA binding; however, the defects in RNA binding that emerged in the multimutant ErmE R34E/R37E suggest in aggregate they contribute to rRNA binding. The KsgA mutant R12E/R14E was not assayed for rRNA binding, but notably possessed a greater methylation defect than the single K13E mutant. Motif X mutants bind rRNA normally and our kinetics investigation of the motif X mutant, F43A, indicated it has a K1/2,SAM similar to wild type, indicating it binds SAM normally. This leads to the question: Why are motif X mutants defective in methylation? Given its structural location, adjacent to the sulfonium of SAM and the adenosine ring of the target nucleotide, and the fact that it does not possess ionizable residues typically associated with chemical steps in catalysis, motif X most likely is involved in optimally positioning the substrate to react with the sulfonium of SAM.
Do RRAD family enzymes use their N-terminal regions to drive methylation in a conserved manner? Considering our biochemical data alongside existing structural data, it seems possible that the N-terminal basic region and motif X are typically dynamic in the absence of rRNA, but the interaction of the basic region with its specific rRNA substrate, orders it and the neighboring motif X to promote catalysis (Fig. 9A). In support of this idea, most crystal structures of RRAD family enzymes in the absence of RNA have a disordered N-terminal basic region and a partially ordered motif X. Three examples of this are the apo structures of ErmC, ErmE, and E. coli KsgA (PDB codes 1qao, 6nvm, and 1qyr) (Schluckebier et al. 1999; O'Farrell et al. 2004; Stsiapanava and Selmer 2019). Interesting exceptions to this are the B. subtilis KsgA structures reported in PDB code 6ifs (Bhujbalrao and Anand 2019). Two copies of KsgA are present in the asymmetric unit with chain B possessing a partially ordered basic region and a gap of 10 disordered residues, containing part of motif X, before the chain becomes ordered again. Chain A in this structure has both regions fully ordered. Cryo-EM structures of B. subtilis KsgA bound to the 30S ribosome were recently solved capturing five distinct KsgA bound states (Singh et al. 2022). The state referred to as K5 by the authors has a disordered N-terminal basic region and motif X, as well as disordered helix 44 of 16S rRNA, but in the K1–K4 state both KsgA regions are ordered as is helix 44 (Fig. 9B,C). In recent structures of the human RRAD enzyme TFB1M bound to an oligo mimicking rRNA, the N-terminal basic region and part of motif X are disordered, but in cryo-EM structures of TFB1M bound to the small subunit of the mitochondrial ribosome these regions are ordered (PDB codes 6aax and 8csp) (Liu et al. 2019; Harper et al. 2023). However, in recent cryo-EM structures of E. coli KsgA bound to immature 30S ribosomes from the Ortega and Davis groups, a different mechanism from that in Figure 9A was proposed. It was argued that the N-terminal region of KsgA interacts with helix 24 (h24) of 16S rRNA and that this interaction keeps h24 from occluding the KsgA active site (Sun et al. 2023). While existing data leave room to debate the specific mechanistic role of the N-terminal region in RRAD enzymes, including the possibility of divergent mechanisms in the family, our biochemical data and the cryo-EM data agree that the N-terminal region features prominently in the function of the enzymes.
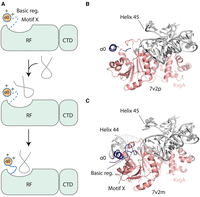
A model for the contribution of the basic region and motif X to rRNA methylation. (A) In the absence of rRNA, motif X is disordered. The electrostatic interaction of the basic region (Basic reg.) with rRNA promotes an initial encounter complex that then matures into a structure with an ordered motif X contributing to the active site of the RRAD methyltransferase. (B) In the KsgA-30S ribosome structure reported in PDB code 7v2p, helix 44 of 16S rRNA is disordered, as is the basic region and motif X of KsgA, but in C the structure reported in 7v2m helix 44 is ordered along with the basic region and motif X. The basic region is positioned to make electrostatic contacts to helix 44 of 16S rRNA.
The phenomenon of a dynamic N-terminal region that is critical for function has been reported recently for two other Rossmann-like, RNA methyltransferases indicating this may be a common mechanism among these enzymes. Human METTL1, with the assistance of partner protein WDR4, installs m7G46 on multiple tRNAs, a modification that regulates tRNA stability and is associated with disease states (Shaheen et al. 2015; Dai et al. 2021; Orellana et al. 2021). The N-terminal region possesses sequence conservation and is the location of a phosphorylation site, at residue S27, that regulates its activity, yet in structures of METTL1-WDR4 in the absence of its tRNA substrate this region is disordered (Li et al. 2023; Ruiz-Arroyo et al. 2023). In cryo-EM structures of METTL1-WDR4 bound to tRNA, the N-terminal region is ordered and forms interactions adjacent to the G46 target nucleotide thought to be important for positioning it for catalysis (Li et al. 2023; Ruiz-Arroyo et al. 2023). Functional assays by the same authors validate the importance of the N-terminal region: the site-directed mutant R24A and phosphomimetic mutants of S27 have dramatically reduced methylation. Rossmann-like methyltransferase TlyA methylates positions in both 16S and 23S rRNA and the presence of these methylations facilitates binding of the anti-tuberculosis antibiotics, capreomycin, and viomycin, to the ribosome similar to the situation with KsgA and kasugamycin (Johansen et al. 2006). Full-length TlyA is recalcitrant to crystallization, but TlyA-CTD readily crystallizes (Witek et al. 2017). Two crystal forms of truncated TlyA have different positions of the peptide that links the NTD and CTD, which extrapolate to different spatial arrangements of the full-length protein. The cryo-EM structure of TlyA bound to the 50S ribosome confirms a critical role for the NTD in rRNA recognition (Laughlin et al. 2022). Collectively, the available structural and functional data suggest that recognition of rRNA by the NTD of TlyA affects the RAWV linker peptide whose conformation affects SAM binding and methylation.
Two interesting future directions for understanding how the N-terminal region of RRAD proteins contributes to catalysis are: What are the specific interactions between motif X and the substrates that are driving catalysis, and is the N-terminal region a modular appendage that controls RNA substrate specificity? The former question arises because the KsgA and TFB1M ribosome-bound structures are determined by cryo-EM at moderate resolution and therefore do not produce certainty regarding side-chain positions. Additionally, the side chain-substrate interactions are likely dynamic across the reaction coordinate and therefore functional studies and structures of multiple reaction intermediates will be necessary to truly describe them. The latter question presupposes that since the fold of class I RNA methyltransferases is conserved, the variable appendages must be providing the RNA substrate specificity. Evidence supporting this model was recently reported by Bhujbalrao and colleagues, who showed that KsgA/ErmC chimeras that included the N terminus could alter substrate specificity (Bhujbalrao and Anand 2019). The many roles RNA methyltransferases play in health and disease warrants a thorough understanding of their structures and mechanisms.
MATERIALS AND METHODS
Random mutagenesis of ermE
Random mutants were created using pBAD/Myc-His-ermE as template DNA, a construct previously reported (Rowe et al. 2020). Briefly, ermE codon-optimized for E. coli was inserted into pBAD/Myc His A (Invitrogen), resulting in an open reading frame encoding the sequence given by Uniprot IDP07287, except that the 82 C-terminal residues, which encode a low complexity Gly-rich segment, are removed and the MYC and hexa-histidine tags are added to the C terminus. GeneMorph II Random Mutagenesis Kit (Agilent Technologies) was used according to manufacturer instructions, except that to achieve a low mutation frequency, 100 ng of target DNA was used as well as 20 PCR cycles for mutant megaprimer synthesis. Transformants were plated on LB-ampicillin (100 μg/mL) plates that were coated with 250 μL of 0.2% w/v L-arabinose. To determine the phenotype of each random mutant, an agar-based selection assay was performed. Transformants were transferred to a gridded 0.2% w/v L-arabinose coated LB-ampicillin (100 μg/mL) plate as well as a gridded 0.2% w/v L-arabinose coated LB plate containing 512 μg/mL of erythromycin. The plates were incubated at 37°C overnight. Colonies that did not grow on the LB-erythromycin plate were then collected from the LB-ampicillin plate for plasmid purification and sequenced via Sanger sequencing (Eurofins).
Erythromycin MIC assay
Microdilution MIC assays were performed similarly to a previous protocol with minor modifications (Sharkey et al. 2022). The strain under investigation was streaked on an LB-agar plate containing 100 μg/mL ampicillin and coated with 0.02% L-arabinose. The plate was incubated at 37°C overnight. Several colonies were lifted from the agar plate with a sterile toothpick, resuspended in 300 mM NaCl, and diluted to an A600 of 0.1. A twofold serial dilution was performed in a sterile, 96-well assay block of Mueller–Hinton broth containing erythromycin ranging from 0 μg/mL to 512 μg/mL, 0.02% w/v L-arabinose, and 20 μg/mL PAβN in a 200 μL final volume. A volume of 2 μL of the cell suspension was transferred to the assay block at each of the concentrations. This procedure was performed three times to generate three replicates. The assay block was incubated at 37°C for 16 h. A plate reader was used to assess growth, with an A600 value of 0.01 greater than the background signal indicating growth. The lowest concentration of erythromycin able to prevent cell growth was recorded.
Western blotting
Western blotting was performed similarly to a previous protocol with minor modifications (Rowe et al. 2020). Cells were harvested from the microdilution assay block from wells containing 0 μg/mL erythromycin media. The cells were harvested by centrifugation, resuspended in buffer E (50 mM Tris HCl pH 7.5, 250 mM NaCl, 1 mM DTT, 2% v/v glycerol) with lysozyme (0.1 mg/mL), and incubated on ice for 30 min. Cells were then lysed with two freeze/thaw cycles in liquid nitrogen followed by centrifugation to pellet cellular debris. The supernatant was subjected to SDS-PAGE on a 4%–20% gel, with wild type and pBAD (empty vector) as controls, then transferred to a nitrocellulose membrane. The membrane was blocked for 1 h in 3% w/v BSA in TBST, washed with TBST three times, and incubated for 1 h in 1:1250 anti-myc antibody at room temperature. The membrane was washed again with TBST and incubated in 1:2500 anti-mouse HRP conjugated antibody for 1 h. The membrane blots were developed with 1- Solution TMB and imaged.
ErmE and ermC site-directed mutagenesis
Site-directed mutagenesis of pBAD/Myc-His-ermE and pBAD/Myc-His-ermC was previously described (Rowe et al. 2020). Site-directed mutants were generated as indicated in Supplemental Table S1 along with the oligonucleotide sequences used.
ErmE purification
To purify the ErmE variants for in vitro assays, E. coli TOP10 cells containing either pBAD-ermE or site-directed mutants were cultured in LB media at 37°C until reaching an optical density of ∼0.6 at A600. Recombinant expression was induced by adding 0.02% w/v L-arabinose, and cell growth continued at 37°C for 4 h. Cells were harvested via centrifugation, and the pellet was resuspended in buffer E, along with 15 mM imidazole and 0.1% v/v Triton X-100. Sonication was employed to disrupt the cells, and after centrifugation at 4800g for 30 min, the clarified lysate was applied to HisPur immobilized Ni2+ resin. The resin was washed with 15 column volumes of buffer E supplemented with 15 mM imidazole. Protein elution was accomplished through a stepwise gradient of increasing imidazole concentrations (125, 250, and 500 mM) over 15 column volumes. Fractions were subjected to SDS-PAGE analysis to evaluate purity. Further purification of ErmE variants was conducted via size-exclusion chromatography using an S75 (Sephadex) column. Fractions containing monomeric ErmE were pooled and concentrated using a Pall centrifugal device with a 10,000 kDa MWCO membrane. Glycerol was then added to a final concentration of 10% v/v, and aliquots were flash-frozen for storage until required.
RNA affinity binding
RNA-binding affinity was assessed using fluorescence polarization as previously reported (Sharkey et al. 2022). Briefly a synthetic 48 nt RNA mimicking helix 73 of 23S rRNA (V48), purified via PAGE and labeled with fluorescein at the
5′ end (obtained from Horizon Discovery), served as the substrate (Vester et al. 1998). The RNA was present at 47 nM final concentration. Protein serial dilutions, ranging from 20 μM to 10 nM, were combined
with fluorescein labeled RNA and incubated for 2 h at room temperature prior to fluorescence detection on a Bio-Tek Synergy
H1 plate reader. The assay was performed in 0.5× buffer E. A titration of the protein pepsin served as a negative control.
Data from three replicates were fitted in GraphPad Prism version 9 to the equation: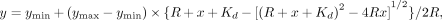 in which R indicates the concentration of V48 RNA. The parameters ymax and ymin were fit globally.
in which R indicates the concentration of V48 RNA. The parameters ymax and ymin were fit globally.
ErmE endpoint methylation assay
Methylation assays were conducted under single-turnover conditions following previously described methods with slight modifications (Rowe et al. 2020). All reactions were conducted in buffer E with ErmE at 10 μM, RNA at 10 μM, and 3H-SAM was present at 0.05 μM (55-85 Ci/mmol). The synthetic oligonucleotide V48 served as the RNA substrate (Vester et al. 1998; Rowe et al. 2020). After 60 min, 2.5 μL of the reaction mixture was withdrawn and quenched by diluting into 47.5 μL of 0.1 mg/mL salmon sperm DNA, followed by the addition of 200 μL of 10% TCA. The precipitated RNA was collected via vacuum filtration using a Millipore Multiscreen GF 96-well plate, washed with 10% TCA and ethanol, and subsequently dried. Scintillation counting was performed on a MicroBeta2 instrument using PerkinElmer Betaplate scintillation fluid. Three replicate assays were performed for each variant.
ErmE single turnover kinetics
Single turnover kinetic assays were performed similarly to limiting SAM methylation assays described above, except that each
ErmE variant was titrated from 6.0 to 0.2 μM and time points between 0 and 60 min were collected. Two replicate products versus
time curves were measured for each ErmE variant concentration. All wild-type product versus time data (Fig. 5A) were input to GraphPad Prism 9 and were fitted to the expression 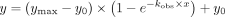 , with ymax and y0 fit globally. This produced values for kobs,wt, ymax, and y0. The ymax value reports on the maximum amount of SAM that can be converted to product SAH, which given the specific activity of our
3H-SAM substrate, was 2895 CPM. Product versus time curves for R36E and F43A variants were also fit to the single exponential
described above but with ymax set to 2895 CPM. The kobs values for each variant at each concentration were plotted versus ErmE (μM). These curves were fit, using GraphPad Prism
9, to the expression: kobs = kchem × x/(K1/2 + x), where x = [ErmE].
, with ymax and y0 fit globally. This produced values for kobs,wt, ymax, and y0. The ymax value reports on the maximum amount of SAM that can be converted to product SAH, which given the specific activity of our
3H-SAM substrate, was 2895 CPM. Product versus time curves for R36E and F43A variants were also fit to the single exponential
described above but with ymax set to 2895 CPM. The kobs values for each variant at each concentration were plotted versus ErmE (μM). These curves were fit, using GraphPad Prism
9, to the expression: kobs = kchem × x/(K1/2 + x), where x = [ErmE].
Construction of pBAD-ksgA and site-directed mutagenesis
Insertion of E. coli ksgA into pBAD/Myc-His was performed in the following manner. PCR was used to amplify the ksgA ORF from E. coli TOP10 genomic DNA. A second PCR reaction was used to add homology arms to pBAD/Myc-His. Finally, an NEB HiFi assembly reaction was performed with the product of the second PCR reaction and linearized vector. The primers used in the assembly are given in Supplemental Table S1.
Growth kinetics in the presence of kasugamycin
Keio Collection E. coli K-12 BW25113 ΔksgA cells were purchased from Horizon Discovery (Baba et al. 2006). E. coli K-12 BW25113 ΔksgA harboring pBAD-ksgA WT or site-directed mutants were streaked onto an LB agar plate containing 100 μg/mL ampicillin and coated with 0.02% w/v L-arabinose which was incubated at 37°C overnight. Several colonies were lifted from the agar plate with a sterile toothpick, resuspended in 300 mM NaCl and diluted to an A600 of 0.2. Mueller–Hinton broth containing 0.02% w/v L-arabinose, and either lacking kasugamycin or containing kasugamycin at 512 μg/mL was dispensed into a 96-well assay block with a 200 μL final volume. A volume of 10 μL of the cell suspension was transferred to the assay block. This procedure was performed three times to generate three replicates. The assay block was covered with a Breathe-Easy membrane, placed inside a plate reader set to 37°C, and A600 was measured every 10 min for 16 h. The Dashing Growth Curves webtool was used to extract values for the length of lag phase and the doubling time during log phase from the growth curves (Reiter and Vorholt 2024). The mean and standard deviation for these parameters is reported in Table 2. Growth curve fitting was performed with the Gompertz tight model in an automated manner with no smoothing except for two replicates that required interactive fitting.
30S ribosomal subunit purification
E. coli K-12 BW25113 ΔksgA was used to inoculate 10 mL of LB, which was grown overnight at 37°C. This culture was used to inoculate 1000 mL of LB, which was grown at 37°C until it reached A600 ≃ 0.6. The culture was placed on ice for 1 h and then the cells were collected by centrifugation. The cells were resuspended in buffer A, 20 mM HEPES pH 7.5, 100 mM NH4Cl, 10 mM Mg(OAc)2, and 6 mM 2-mercaptoethanol. The cells were lysed by sonication and DNase I (RNase-free) was added. Centrifugation was used to remove cell debris, and the supernatant was concentrated on a 100 kDa MWCO filtration device. The concentrated supernatant was layered onto a sucrose cushion made of buffer A with 30% w/v sucrose. The ribosomes were pelleted by ultracentrifugation at 32k RPM for 22 h. The pellet was dissolved in buffer B, 20 mM HEPES pH 7.5, 50 mM NH4Cl, 1 mM Mg(OAc)2, and 6 mM 2-mercaptoethanol and layered onto a sucrose gradient composed of 10%–40% sucrose w/v in buffer B. Ultracentrifugation of the gradient was performed in a SW32Ti rotor for 10 h at 32k RPM. Fractions were eluted from the gradient using a syringe pump loaded with heavy sucrose and collected on a Biocomp fraction collection system with a Triax flow cell.
KsgA methylation assay
Recombinant expression and purification of KsgA and its variants were performed in the same manner as the ErmE expression and purification described above. Methylation of 30S ribosomal subunits was performed in buffer E (described above) diluted 1:1 with ultrapure water. 30S subunits at 10 μM and 3H-SAM at 0.1 μM final concentration were incubated with 10 μM KsgA at 37°C for 60 min. Reaction mixture was aspirated at various time intervals, quenched with 10% TCA, and prepared for scintillation counting as described above for ErmE methylation assays.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R35GM142966.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080068.124.
- Received April 20, 2024.
- Accepted October 20, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Danielle McGaha is the first author of this paper, “Two dynamic N-terminal regions are required for function in ribosomal RNA adenine dimethylase family members.” Danielle is a senior PhD student, working in Dr. Jack Dunkle's lab at the University of Alabama. Her research has focused on better understanding the structure and function of Erm proteins and their broader impact in conferring antibiotic resistance.
What are the major results described in your paper and how do they impact this branch of the field?
We have provided functional evidence that two previously unrecognized regions of RRAD family enzymes are essential for their activity. Our research focused on the N-terminal basic region and motif X of ErmE, ErmC, and KsgA. Methylation assays confirmed that mutants exhibiting loss of function had impaired methylation. Using ErmE as a model, we conducted RNA affinity binding and kinetics assays, revealing that the N-terminal basic region aids in rRNA binding. Although motif X mutants bind rRNA normally and maintain SAM-binding affinity, they are still defective in methylation. This suggests that motif X may play a crucial role in positioning the substrate for reaction with SAM.
Integrating our biochemical findings with structural data indicates that both the N-terminal basic region and motif X are typically dynamic without rRNA. For example, crystal structures of RRAD enzymes show disordered N-terminal and motif X regions in the absence of RNA, while structures bound to ribosomes display ordered configurations. This is why we propose that binding rRNA stabilizes these regions, facilitating catalysis.
What led you to study RNA or this aspect of RNA science?
My work with RNA began with an interest in antibiotic resistance, a pressing issue that poses a significant threat to global public health. Many proteins modify rRNA, sometimes leading to antibiotic resistance, as is the case for some Erm proteins. Understanding the role these proteins play in modifying rRNA provides insight into potential targets for novel therapeutic drugs. Through this, we can provide innovative solutions that are critical in combating the growing worldwide concern that is antibiotic resistance.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
Having the opportunity to work in a biochemistry research lab during my undergraduate career opened my eyes to what it is truly like to be a scientist. It was an invaluable experience that transformed my understanding of scientific inquiry from abstract concepts in textbooks to hands-on experimentation and discovery. I found it extremely rewarding to follow a scientific question and find answers to that question through experimentation. My first time purifying DNA and running it on an agarose gel was a profound moment for me. It was tangible evidence of how far science has come and in turn how far it has advanced humanity as a whole. In these seemingly small moments, I knew that I also wanted to contribute to those advancements in knowledge by taking on the role of a scientist.
If you were able to give one piece of advice to your younger self, what would that be?
Being a scientist is a continuous endeavor for knowledge. You will never be able to know everything there is to know about a certain topic. The nature of science encourages constant questioning and exploration. Each discovery leads to new questions, and the more you learn, the more you realize how much remains unknown. To be a scientist means embracing this journey of lifelong learning, where each step forward reveals new paths to explore and new mysteries to uncover. It is a commitment to adaptability, open-mindedness, and the understanding that the pursuit of knowledge is never ending.








