
Probing the epitranscriptome and RNA damage with nanopore direct RNA sequencing
- Corresponding authors: afleming{at}chem.utah.edu; burrows{at}chem.utah.edu
Abstract
Nanopore direct RNA sequencing (DRS) is revolutionizing our ability to analyze the epitranscriptome to evaluate nucleoside modifications in both cellular and synthetic RNA. The process involves minimal handling of fragile RNA strands, one round of reverse transcription to provide a DNA:RNA duplex, and library preparation to directly read nucleotides with their modifications as they pass through a protein nanopore embedded in a membrane. Simultaneous sequencing of hundreds of strands on a chip provides unprecedented access to whole transcriptome information. A key advantage is the long-read length that permits, for example, operon-specific epitranscriptomics of ribosomal RNA modifications as a function of cellular stress. By analyzing the entire transcriptome, the interplay of different modifications on the same RNA, or the correlation of changes in different RNAs in the same cell type, can be monitored. This review presents several recent examples of the types of experiments that are suitable for nanopore DRS as well as some of the current challenges and future expectations.
Keywords
INTRODUCTION
From a sketch in David Deamer's notebook to a nanopore sequencing device on the market took a quarter of a century (Deamer et al. 2016). The next decade saw truly remarkable advancements in the chemistry of nanopore sequencing, the software for analysis of canonical nucleotides and their common modifications, and the creative application of nanopore sequencing to answer questions in nucleic acid chemistry and biology (Deamer et al. 2016; Begik et al. 2022; White and Hesselberth 2022). The present-day nanopore sequencer employs a transmembrane protein and helicase. The helicase slowly delivers the nucleic acid into the nanopore protein at the rate of ATP hydrolysis, while the ionic current levels for the nucleotides are recorded as they pass the central constriction of the pore (Fig. 1). In 2022, Oxford Nanopore Technology's (ONT's) MinION sequencer was key to finishing the complete human genome sequence in the “Telomere-to-Telomere” project because the long reads possible with the ONT system permitted sequencing and alignment of repeat sequences that make up nearly 10% of human DNA (Nurk et al. 2022). For example, human genomes have hundreds of copies of ribosomal DNA, and each sequence is nearly, but not necessarily completely, identical (Welfer et al. 2025). Human telomeres can be >10,000 bp long with a consistent hexamer repeat. Alignment of short fragments from such sequences by standard next-generation sequencing (NGS) methodology would not be possible, but read lengths using nanopore sequencing can now reach 4 Mb for DNA and thousands of nucleotides for RNA.
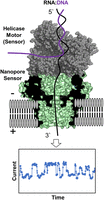
Schematic of the nanopore sequencer illustrating the protein nanopore and helicase positioning to provide ionic current versus time traces as the RNA moves through the sensor. The figure for the nanopore sequencer was constructed from PDB 4UV3 and 2P6R (Büttner et al. 2007; Goyal et al. 2014).
Direct whole transcriptome sequencing from cellular extracts without PCR amplification is now made possible by nanopore sequencing. Consequently, recent nanopore research has focused on understanding the signatures of base and ribose modifications because the native RNA strands, not DNA amplicons, are being analyzed in the nanopore in what is called nanopore “direct RNA sequencing” (DRS). As an example, modifications in ribosomal RNA can be analyzed using ∼105 cells from which ∼100 ng of RNA can be extracted; tRNA is even more abundant on a per-strand basis, although its relatively short lengths require special adaptors to be ligated because of the first 3 nt at the 3′ end (Thomas et al. 2021). Challenges persist for sequencing the last 15–20 nt at the 5′ end due to high error rates and poor coverage, which is most significant for small RNAs, such as tRNA, requiring an additional 5′ end adaptor (Thomas et al. 2021; Lucas et al. 2023). The low abundance of mRNA requires analysis of ∼107 cells, which remains a challenge for many biological studies. Even with these limitations, new questions are readily addressable by nanopore sequencing of cellular RNAs.
Nanopore sequencing suffered in the early days from a reputation for high error rates compared to sequencing-by-synthesis methods. However, steady improvements in the nanopore protein design, the helicase “brake” used to control translocation speed, and the machine-learning tools that base call electrical signals as either a canonical nucleotide or a modification have vastly improved (Zhang et al. 2024). Many biological questions are now efficiently addressed by nanopore sequencing rather than the more expensive and time-consuming NGS methods or mass spectrometry, of which the latter remains the gold standard for accurate identification and quantification of modified nucleotides (Kang et al. 2025). In this short review, we will highlight several examples where nanopore direct RNA sequencing has provided ready access to data that would have been cumbersome to obtain by more traditional means. These examples are meant to showcase the types of experiments that are appropriate for nanopore sequencing as opposed to NGS sequencing-by-synthesis methods. We hope it is useful to readers assessing prior literature and to researchers considering nanopore experiments. For a more detailed treatment of base-calling tools that use nanopore-generated data, see a recent review (Cruciani and Novoa 2026).
NANOPORE SYSTEM AND TECHNOLOGICAL ADVANCES
The basic design of a nanopore sequencing device includes a transmembrane pore-forming protein (α-hemolysin, MspA, CsgG, etc.) with an internal pore diameter just larger than 1 nm to allow translocation of single-stranded DNA or RNA through the pore (Fig. 1; Branton and Deamer 2019). The polyanionic nucleic acid strand is driven electrophoretically by an electrical potential applied across the membrane. As nucleobases with different sizes and solvation properties pass through the narrowest point of the pore, they constrict electrolyte flow to varying extents, thereby reducing the ionic current level in the picoampere range (Wolna et al. 2013). To control the otherwise rapid translocation, a molecular “brake,” typically a helicase enzyme, unravels a DNA:RNA duplex at a rate dependent on the ATP concentration in solution (Derrington et al. 2015). When the helicase loses grip of the duplex ∼15–20 nt from the 5′ end of the RNA, the strand traverses through the nanopore too fast for sequencing to be achieved, resulting in loss of the data for this region of RNA. To achieve sequencing of the 5′ end of the RNA, an additional adaptor must be added to this end (Ibrahim et al. 2021). In the MinION sequencer, >1000 individually addressable membrane-embedded nanopores are present on a chip for parallel sequencing. Although the data at each pore are acquired in a single-molecule fashion, each current–time trace shows rather low signal-to-noise, such that only by superposition of data from hundreds or thousands of identical RNA strands can one obtain confident base calling for sequencing.
Until 2024, it was most common to use ONT's version 9.4.1 flow cell chemistry for collecting current versus time data. These data are then “resquiggled” in the time domain to assign a standard event period or dwell time for each nucleotide (Loman et al. 2015; Furlan et al. 2021). This is necessary because the helicase does not consistently pull apart base pairs, and some base pairs are more stable than others, which can be used as a point for identifying modifications in certain cases (vide infra). After 2024, updates to the system include a more processive helicase introduced for greater data collection and a flow cell with a more accurate protein nanopore. These advancements came alongside updated software to collect and analyze the data with greater accuracy. These updates are anticipated to continue in the future, which can impact long-term projects that require continuity in the technology.
Next, base-calling algorithms (formerly “Guppy,” now “Dorado”) are used to evaluate current signatures to assign a nucleotide as A, C, G, or U. It is important to note that the current signature for a given nucleotide is influenced by its surrounding sequence context, and for the ONT RNA system, it is a ∼5 nt window that is being read at any given time (Esfahani et al. 2025). Thus, the presence of an adenosine influences the ion current when 5′…ANNNN…3′ is first in the reading frame, and next with 5′…NANNN…3′, and so on, until finally 5′…NNNNA…3′ is read, recalling that the nanopore threads RNA strands from the 3′ end. For this nanopore, the 5 nt sequence surrounding the nucleotide of interest, that is, 5′…NNANN…3′, is called a “k-mer.” For comparison, the k-mer for sequencing DNA with ONT's version 10.4.1 nanopore is a ∼9 nt window (Straver et al. 2025). Consequently, for each RNA nucleotide, there are 44 = 256 different sequence contexts of a centrally located nucleotide, assuming no modifications are present. The version 9.4.1 flow cells using RNA002 chemistry led to error rates in base calling that were typically 87% accurate, with exceptions being homonucleotide sequences ≥4 nt that display much higher errors (Esfahani et al. 2025). In contrast, the recent RNA flow cells released in late 2024 using RNA004 chemistry employ a much faster helicase and are therefore higher throughput to achieve greater coverage. The new base caller Dorado, with current chemistry, provides accuracies near 98% (Esfahani et al. 2025), which is clearly good enough for most applications.
Additional advancements include data storage from fast5 to pod5 formats that create smaller data files, which are highly desirable for file transfers, manipulation, and storage. The Dorado base call software operates using three different levels of accuracy: fast, hac, and sup. As the accuracy increases, the demand for high-performance computational resources becomes essential, which may be an issue for some researchers. The most recent version of Dorado (v.5.2.0) includes modification-aware base-calling for m6A, m5C, I, Ψ, and all four Nm nucleotides. These newest modification-aware models use the highest accuracy sup models, which are computationally demanding to employ. The choice in accuracy of the base calls represents a choice researchers must make when analyzing their data. It is anticipated that many of the future key advancements in the technology will occur in the data analysis component of the technology.
The detection of base and ribose modifications in RNA using nanopore technology has seen a dramatic increase in the past 5 years, and it is primarily based on errors in base calling. For example, Figure 2 shows averaged base calls (v.9.4) for pseudouridine in synthetic RNA in which Ψ replaces a U at 100% occupancy in 16 different sequence contexts (Fig. 2A,B; Fleming et al. 2021). The signals range from nearly 100% U base calling to nearly 100% C, even though the actual nucleotide is 100% Ψ in each of these sites (Fig. 2C). This example underscores the complexity of the data analysis that must occur during base calling of a modification in every possible sequence context. Furthermore, without any other information, the mixture of C + U base calls is indistinguishable from the presence of C/U sequence variants, which are common in rRNA and probably elsewhere (Fleming et al. 2023b). A number of scientists took on this challenge and developed algorithms for the detection of Ψ (Begik et al. 2021; Huang et al. 2021; Leger et al. 2021; Hassan et al. 2022; Alagna et al. 2025). To be convinced of the presence of a modification, one can knock out or knock down the writer enzyme, assuming it is known, in order to provide the unmodified signal (Liu et al. 2019; Leger et al. 2021; Fleming et al. 2023a). Conversely, synthesis of long RNAs containing the modification in the correct sequence context is valuable as a positive control (Begik et al. 2021; Fleming et al. 2023a). For extreme accuracy, synthesis of an mRNA-length strand with a synthetic insert containing one modification leads to a “ground truth” signal in that sequence (Gamper et al. 2023; Tavakoli et al. 2023). Verification of modifications via mass spectrometric sequencing provides excellent confirmation of structure and stoichiometry, but it can be time-consuming (Kang et al. 2025). For modifications like Ψ, an isomer of U, MS analysis requires isotopic labeling (Taoka et al. 2018), and it may be more convenient to obtain verification of a site by chemical methods—several have now been perfected for this modification (Khoddami et al. 2019; Dai et al. 2023; Zhang et al. 2023; Xu et al. 2024).
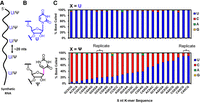
Range of base miscalls for Ψ embedded in synthetic RNA at 100% frequency. (A) Layout of the synthetic RNA to study base calling for U versus Ψ present at 100% occupancy and positioned >20 nt apart to ensure only one modification at a time influences the signal. (B) The structures for U and Ψ isomers. (C, top) Bar chart of base calls for U in 22 different 5 nt k-mers and (bottom) base calls for Ψ in the same k-mers. Two replicates are provided to demonstrate reproducibility in the base call data. These data were obtained using Guppy v.3 and aligning to the reference with minimap2. The figure and data are reconstructed from our prior report (Fleming et al. 2021).
In our own studies, we also sought an orthogonal method of detecting modifications using a different feature of the ONT system, namely the other protein—the helicase (Fleming et al. 2021, 2023a). Before entering the constriction zone of the nanopore protein, a modified nucleotide must first be processed by the helicase motor. Many modifications are destabilizing to the DNA:RNA duplex and are therefore unwound more rapidly than average by the helicase; this is reflected in a shorter dwell time. In contrast, a few modifications, including pseudouridine, likely due to stronger base stacking interactions, and 2′-OMe, possibly due to steric interactions between the methyl protrusion and the helicase cavity wall, have longer dwell times. Suitable analysis of dwell times using RNA002 chemistry helped establish pseudouridine assignments in the SARS-CoV-2 viral RNA and verify sites in the Escherichia coli rRNA strands (Fleming et al. 2021, 2023a). However, the dwell-time signal needs to be analyzed at a position ∼10–11 nt toward the 3′ direction of the sequence, because the modification causing a dwell-time change in the helicase will not arrive at the electrical sensing zone until 10 nt later. The present use of 2024 RNA004 chemistry with a much faster helicase now means that only Ψ and Nm modifications can be analyzed in this way; modifications that decrease the dwell time are now simply too fast to measure reliably.
Since late 2024, the Dorado program for base calling includes modification-aware base callers for Ψ, m6A, I, and m5C. These new modification-aware models have significant potential for DRS of known RNA modifications; however, they do display a high false-positive rate and cross-reactivity with other RNA modification sites (Copoulos et al. 2025; Diensthuber et al. 2025). In 2025, Dorado modification-aware models for the four Nm nucleotides have been added. In our experience, m6A and m5C were previously very difficult to distinguish from their unmodified parent nucleotides in rRNA (Fleming et al. 2023a). These models have changed this observation for known sites of m6A and m5C in rRNA. However, in mRNA, m6A is written into DRACH sequences, and when a purine “R” resides on the 5′ side of the m6A site (underlined), the current level differences are more apparent for analysis (Zou et al. 2025). The advancement of modification-aware base call models is a significant step for this technology to be a routine analytical tool used in the biological sciences.
To summarize, the ONT MinION sequencer is currently the only instrument available for direct RNA sequencing that provides information about the presence or absence of multiple modifications in the whole transcriptome. Other methods require reverse transcription to DNA, with or without additional chemical or enzyme treatment, in which case the RT signature of a modification might be readable after amplification (Khoddami et al. 2019; Dai et al. 2023; Zhang et al. 2023; Guo et al. 2024; Xu et al. 2024). Many chemical methods of sequencing take advantage of this process, and they have been reviewed (Behm-Ansmant et al. 2011; Bartee et al. 2021).
We and others have proposed that nanopore sequencing of modification-specific chemistries would be an ideal way to enhance the electrical signal, which includes the study of bisulfite adducts to pseudouridine or acrylonitrile adducts to inosine (Schibel et al. 2010; Ramasamy et al. 2022; Fleming et al. 2023b). These “chemical modifications of chemical modifications” seem like good ideas on the surface; however, such adducts have three barriers to overcome in the nanopore sequencing process. (1) The first step is one round of reverse transcription to feed a DNA:RNA duplex to the helicase. Reverse transcriptases (RTs), though processive, have limits to their promiscuity. Currently in favor is the Induro RT, which offers exceptionally long DNA copies with good processivity through RNA modifications (Unlu et al. 2024; Nakano et al. 2025). In this case, it does not matter how the RT copies opposite the modification, because the DNA strand will be discarded after helicase unwinding. (2) Next, the modified nucleotide must be a suitable substrate for the helicase, and here again, bulky adducts such as CMC-Ψ or large 2′-O-acyl groups appended during traditional SHAPE-seq do not work (Stephenson et al. 2022). In fact, they are likely stopped at the first step, RT, in any case. (3) Finally, the adducts must pass through the constriction zone of the nanopore protein, if they have made it this far.
Despite these challenges, some success has been seen with modified RNA modifications. As examples, the SHAPE-seq experiment works when the 2′-OH group is acetyl (Stephenson et al. 2022), the bisulfite adduct to Ψ provides some data but with a high rate of pore failure (Fleming et al. 2023b), and acrylonitrile adducts of inosine and Ψ have both been used to enhance nanopore error signals (Ramasamy et al. 2022). We suspect that other adducts could be studied if parameters in the MinION were adjustable. For example, in an experiment involving the series N1-methyl- versus ethyl- versus propyl-pseudouridine, we observed a continuous drop-off in the success of the nanopore that we attributed to long dwell times in helicase processing (Fleming et al. 2024b). After 100 µsec, the strand is presumably “stuck” due to the hydrophobicity of the sidechain and better base stacking of modified Ψ on its 5′ neighboring base; a current reversal then automatically ejects the strand from the nanopore.
In a final note about advancements in the ONT sequencer, comparing v.9.4.1 flow cells and RNA002 chemistry to the newer RNA flow cells with RNA004 version chemistry, the measurement of quantitation or occupancy of a modification at a given site has greatly improved. Nanopore data in the early 2020s could give a percent occupancy that was generally quite different from the actual value, and base calls of unmodified nucleotides were ∼90% accurate. With the new chemistry and modification-aware models, quantitation has generally improved for target RNA modifications, and unmodified base calling is >97% accurate for RNA. Importantly, the occupancy values for modifications are reproducible, so that if one is monitoring changes in a modification, for instance, due to application of cellular stress, the percent change is accurate and reproducible, even if the initial values are not (Fleming et al. 2023a; Milenkovic et al. 2025). Further advancements in the nanopore structure and computational models will see this technology achieve near-quantitative sequencing for target RNA modifications; however, many controls will be needed to minimize the false positives that remain in the data.
SEQUENCING ENZYMATICALLY INSTALLED RNA MODIFICATIONS
Although there are more than 150 known modifications of RNA nucleotides that are enzymatically written into the transcriptome, only about 20 of them are commonly studied (Wang and He 2014); many others are particular to tRNAs and have intermediates in the pathways to more elaborate final products (Table 1; Bommisetti and Bandarian 2023). The scope of ribosomal modifications in bacteria and eukaryotes is largely known (Natchiar et al. 2017; Watson et al. 2020; Holm et al. 2023), although the occupancies of these modifications under different stress and disease conditions remain to be fully characterized (Fleming et al. 2023a; Milenkovic et al. 2025). Similarly, tRNAs have abundant modifications and have been mapped in many organisms (Huber et al. 2019). Mapping mRNA modifications presents the greatest challenges because of the low density of modifications and the low abundance of mRNA strands. On the one hand, rRNA strands are abundant, and all strands have nearly the same sequence and modifications; in contrast, alternative splicing of mRNA sequences and their low abundance create challenges for identifying the distribution of modifications. In this section, we highlight some of the studies performed using nanopore DRS and show their advantages and limitations.
Values for copies and modification density in tRNA, rRNA, and mRNA in human and E. coli cells (Milo et al. 2009)
Ribosomal RNA
The long-read capability of nanopore sequencing, along with the fact that most ribosomal modifications are written at high occupancy at known (or suspected) positions, means that rRNA modification mapping by nanopore DRS provides a showcase of the technology. For E. coli and closely related ribosomal structures, the modifications are relatively well characterized from structural investigations (Polikanov et al. 2015; Watson et al. 2020), and the enzyme writers are known (Sergeeva et al. 2015). Using RNA002 chemistry, our laboratory combined base-calling error, current levels, and dwell-time analysis to monitor the 17 types of modifications occurring in 36 positions of the 16S and 23S subunits of the E. coli ribosome (Fig. 3; Fleming et al. 2023a). In general, nanopore analysis that relies heavily on base-calling error does a modest job of determining the modification occupancy at most sites, particularly if occupancy is <50%, although it does an excellent job of recording changes in the occupancies.
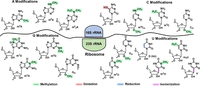
The 17 different structures that occupy 36 different positions in E. coli 16S and 23S rRNA strands. The figure is reconfigured from our prior report (Fleming et al. 2023a).
For E. coli rRNA, most modifications are written at a high level, ≥90% (Popova and Williamson 2014); therefore, we studied the impact of heat shock, cold shock, and nutrient stress on rRNA modifications, and a recent article also examined heat shock in greater detail for the entire E. coli transcriptome (Fleming et al. 2023a; Riquelme-Barrios et al. 2025). Many modifications did not change in occupancy under stress, although a few changed dramatically and others posed new questions. For example, the 16S m4Cm1402 modification decreased in occupancy under all stressors, but it was not immediately clear which methyl group was lost—from the base or from the ribose, or possibly both (Fig. 4A; Fleming et al. 2023a). Dwell-time analysis was particularly useful for making this assignment because “two” dwell-time changes between the E. coli RNA and synthetic RNA without modifications were observed in the m4Cm1402 data relative to unmodified C1402, at positions 1412 (i.e., +10 nt) and at 1414 (+12 nt) (Fig. 4B,C). During stress, the change at 1412 disappeared and the peak at 1414 remained; we suspected that the 1412 dwell-time change was due to the m4C modification by analogy to other base modifications changing in the active site of the helicase located 10 nt in distance from the nanopore reader (Stephenson et al. 2022; Fleming et al. 2023a). This observation demonstrated that the helicase can serve as an RNA modification sensor, in addition to the motor protein that slowly delivers RNA into the nanopore for sequencing.
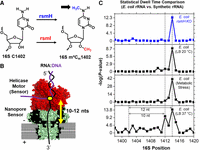
The helicase motor protein in the ONT sequencer is a sensor of base pair integrity for RNA modification sequencing. (A) Structure for m4Cm1402 in the E. coli 16S rRNA. (B) Schematic to illustrate the RNA sequence distance between the nanopore constriction zone and the helicase active site. (C) Pairwise dwell-time population analyses between E. coli rRNA and a synthetic 16S rRNA sequence without modifications to locate sites that differ with a statistical difference. The figure was reconstructed from our prior report (Fleming et al. 2023a).
The dwell-time perturbation at 1414 must be due to an interaction between the 2′-O-methyl group and the helicase that occurred 2 nt earlier, that is, in the 3′ direction, since this end threads first. We speculate that this interaction could be with a protruding amino acid such as Lys289, based on a crystal structure of a DNA duplex being unwound in the helicase active site (Fig. 5). In the end, our assignments for the two methyl signals of 16S m4Cm1402 were verified by analyzing in separate experiments the writer knockouts for m4C (RsmH) and Cm (RsmI) (Fleming et al. 2023a). Additionally, at the request of reviewers, we performed the more tedious experiment of analyzing this segment of rRNA by LC-MS that confirmed the conclusion that stress greatly lowers the base methylation, but ribose methylation was little affected. Interestingly, this hypermodification site is conserved in bacteria and located at the P-site of the ribosome, where it makes contact via N4 with the second and third bases of the P-site codon (Polikanov et al. 2015). Loss of the N4-methyl group under stress increases AUG readthrough but with a loss of ribosome fidelity, which may be one of the ways that cells adapt to stress (Kimura and Suzuki 2010). In another study, the tandem modifications in the small subunit, 16S m6,6A1518 and 1519, both increased during heat stress as well as modifications at nearby sites linked to the decoding center (Riquelme-Barrios et al. 2025). Clearly, there is much to learn about ribosomal stress responses.
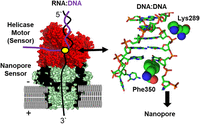
Structural illustration to rationalize the base and sugar points of contact in the helicase for detecting the difference in methylation status of E. coli 16S m4Cm1402 under thermal or metabolic stress. Note that in this model, Phe350 and Lys 289 contact nucleotides near the active site that are 2 nt apart. The helicase structure is for Hel308 (PDB 2P6R; [Büttner et al. 2007]), based on the patent literature from ONT, as the one that has been modified for sequencing (Bruce et al. 2015). Because we do not know the exact identity of the helicase or the modifications made by ONT, this structure is our best guess based on the available information.
Another unique advantage of nanopore sequencing is the ability to investigate operon-specific epitranscriptomic modifications, and here again, the E. coli ribosome provided such an opportunity for the first time. The E. coli genome has seven operons encoding rRNA, and each one contains sequence variants (Kurylo et al. 2018). Taking advantage of the long reads provided by the nanopore method, one can assign epitranscriptomic modifications to each of the seven different rRNA sequences (Fleming et al. 2023a). This was applied to the analysis of Ψ sites, of which there are 11 in the E. coli ribosome, one of which is a hypermodified m3Ψ. Under normal benchtop growth conditions (aerobic, 37°C, LB media), all seven rDNA operons are about equally expressed, but during metabolic stress, the operon usage levels of rrnA, rrnB, and rrnE decrease substantially, while others increase, notably rrnH >> rrnD > rrnG, compared to rrnC chosen as a standard because it is the closest to the origin of replication. Notably, those that increased in expression levels were more distal from the Ori (rrnH, D, and G), confirming an earlier report based on cDNA sequencing (Kurylo et al. 2018).
Next, the Ψ levels were analyzed in stressed conditions as a function of the operon (Fleming et al. 2023a). Many did not change, but three sites were noteworthy. First, 23S Ψ2604 increased in all transcribed operons except rrnC, where it was already high. Secondly, 23S Ψ2457 decreased the most, but only in operon rrnD. Finally, 23S Ψ746 does not appear to change until one examines operon specificity. This modification increases in most cases but decreases in rrnH, which is more than twofold more highly expressed, so that on average, one does not see a change without examining individual operons. Ultimately, the mechanism by which metabolic stress fine-tunes both operon expression and operon-specific epitranscriptomic modifications in the ribosome will require more study, but nanopore DRS is the enabling technology for assessing these changes.
Exposure to antibiotics presents a different type of stress imposed upon bacteria, and in many cases, the bacterial ribosome is a major target (Peng et al. 2025; Sherry et al. 2025). The Novoa laboratory studied the impact of streptomycin versus kasugomysin antibiotics on rRNA modifications in E. coli and found that modifications surrounding the A and P-sites decreased the most, but it depended upon the antibiotic, and in this case, no operon-specific changes were noted (Delgado-Tejedor et al. 2024).
Resistance to ribosome-targeted antibiotics occurs when either the genes encoding rRNA undergo mutation or the RNA modification levels or types change, or both (Darby et al. 2023). A recent example showcases nanopore sequencing as a method to uncover operon-specific epitranscriptomic changes in generating a Staphylococcus aureus mutant strain that is resistant to naphthyridone antibiotics (Copoulos et al. 2025). In this case, it was found by long-read nanopore DNA sequencing that a single operon, operon 2, of the six in S. aureus, was mutated at a single site, 23S T1732C (Fig. 6A). This result could not be determined by using short-read sequencing as a consequence of the long repeat nature of rDNA sequences (Fig. 6B). This T:A to C:G mutation likely arises under stress from deamination of A, generating I, an isocoder of G. Replication would then lock in the mutation, and this operon would now produce a ribosome with C at position 1732 of the large subunit, a position that may impact naphthyridone binding. It is interesting that operon 2 also contains multiple tRNA genes and is likely to be undergoing active expression during antibiotic exposure, making this gene more susceptible to cellular stressors such as NO-derived deaminating agents. S. aureus has a nitric oxide synthase that responds to antibiotic stress (van Sorge et al. 2013), suggesting a potential chemical source of mutagenesis.

Antibiotic stress in S. aureus results in a resistant strain with a 23S T1732C mutation in the second rDNA operon. (A) Long-read nanopore DNA sequencing to reveal the mutated operon. (B) Short-read DNA sequencing results in ambiguity in which operon is mutated. The C miscall frequency in each of the six operons based on the short-read sequencing data is 17% or 1/6th. The figure was adapted from our prior work (Copoulos et al. 2025).
Next, in the nanopore studies, sequencing rRNA from the six operons in the mutant S. aureus compared to wild type identified changes in modification levels to uncover several positions that responded to antibiotic stress, including 23S Ψ791, 23S D2476, and 23S ho5C2528 in the operon 2 variant of the mutant strain. In the other operons of the mutant strain, there were even greater changes in the epitranscriptomic modification levels, most of them mapping to key sites in the peptidyl transferase center and the decoding center, indicating that the modifications are probably sculpting ribosome active sites for more efficient protein synthesis under stress. Overall, these insights into operon-specific epitranscriptomics provide new clues to possible mechanisms of antimicrobial resistance and immediately suggest further studies.
Recent work has shown that the >220 modifications in human and mouse rRNAs can be mapped by nanopore DRS (Milenkovic et al. 2025). Interestingly, the modification levels show dependencies on cell type and disease type, and such studies pave the way for the rRNA epitranscriptome to provide a fingerprint indicating healthy and disease states. We expect this to be a very active area of investigation in the near term; ribosomal RNA is comparably long-lived in mammalian cells and can therefore serve as a reporter of cellular health.
Transfer RNA
tRNA strands are abundant in cells and heavily modified; however, they are short, only ∼80 nt on average (Dedon and Begley 2022). Because nanopore DRS does not perform well on the ends, the problem of sequencing tRNAs has been solved by adding adaptors to both the 5′ and 3′ ends to lengthen the strand by 54 nt before single-pass reverse transcription and ligation of Y-shaped adaptors compatible with the ONT system (Thomas et al. 2021; Lucas et al. 2023).
In a recent example, the Novoa laboratory was able to overcome limitations of bias toward longer tRNAs and the problem of some significant amounts of data being discarded by using redesigned adaptors to lengthen the RNA and by developing a new computational platform to capture more data (Lucas et al. 2023). Their protocol, Nano-tRNA-seq, enabled reliable mapping of modifications, as well as noting an oxidative stress–induced deadenylation of the 3′-CCA terminal sequence that had been previously discovered (Czech et al. 2013; Lucas et al. 2023). Nano-tRNA-seq allows rapid monitoring of this reversible 3′ adenylation/deadenylation process that apparently acts as a rapid response to cellular stress.
Nanopore DRS analysis of tRNA modifications before and after heat stress in E. coli was particularly adept at monitoring closely spaced Ψ sites. For example, Ψs at positions 35, 38, 39, and 40 could be individually measured by DRS but proved difficult to analyze by mass spectrometric sequencing (Riquelme-Barrios et al. 2025). Lastly, intact aminoacylated tRNAs were sequenced by DRS to identify the amino acids attached to all tRNAs in a cellular sample (White et al. 2025). This method has significant potential to study the tRNA aminoacylation in cells and how this process contributes to cellular stress. Nevertheless, some modifications are easier to analyze than others; m6A, Gm, and Cm show only a small change in base-calling error analysis and so changes are difficult to quantify, whereas Ψ, m6,6A, m1G, m2G, and Um are easier to measure.
mRNA
Chemical modifications on mRNA regulate several aspects of their function including stability, splicing, nuclear export, and translation, and therefore mapping mRNA modifications is an important endeavor (Gilbert and Nachtergaele 2023). N6-methyladenosine (m6A) and Ψ are the most abundant, but important roles for m5C and the four Nm methylated nucleosides have also been reported. Among these, only m6A is reversible (Shi et al. 2019), although m5C can be further oxidized by TET2 to hm5C (Fu et al. 2014). As a consequence of these important roles in gene expression, mRNA modifications were the focus of many early studies by nanopore DRS (Liu et al. 2019; Jenjaroenpun et al. 2020; Parker et al. 2020; Leger et al. 2021; Ramasamy et al. 2022; Tavakoli et al. 2023; Huang et al. 2024). A recent study of 2′-O-methylation at internal sites in mRNA has pointed to the importance of this modification to stability and highlighted a role in cancer (Li et al. 2024).
An experiment that takes advantage of the ability of the nanopore to read multiple modifications simultaneously examined the cross talk between N6-methylation of A and Ψ in mRNA. These modifications are typically measured separately in chemical sequencing protocols, but the use of nanopore DRS by the Pan laboratory led to a proposal that m6A and Ψ are inversely correlated in mRNA transcripts; that is, those strands that contained m6A were less likely to contain Ψ, and vice versa (Huang et al. 2024). They also found that the two modifications had a synergistic effect on translation efficiency in polysome-associated mRNA with Ψ exerting a larger impact than m6A.
Viral RNA and vaccines
Several laboratories have been interested in the presence of modified nucleosides in viral RNA, with a particular focus on Ψ because it was reported to be present at overall high levels for several viruses, according to LC-MS/MS analysis (McIntyre et al. 2018), and because of its known properties in diminishing immune responses (Netzband and Pager 2020). The latter point was validated by the success of the mRNA vaccines that replaced all uridines with N1-methylpseudouridine (m1Ψ) (Nance and Meier 2021). Indeed, our first examination of ONT data in 2020 asked the question of deposition of Ψ in the SARS-CoV-2 genome (Fleming et al. 2021). Because the cytosol of an infected cell contains abundant copies of subgenomes to express more copies of essential proteins (Kim et al. 2020), we focused on the 3′ portion representing ∼30%, or 10,000 nt, of the gRNA. The modifications were initially identified by base-calling error analysis using ELIGOS2 (Jenjaroenpun et al. 2020), and these were then further justified by the observation that many of the eight sites identified in TRS-S, which includes the spike protein gene through the 3′ terminus, were also found in other subgenomes. Ionic current level changes and helicase dwell-time increase at about +10 nt, characteristic of Ψ, were found for these sites using Nanocompore (Leger et al. 2021), providing a consensus in the signals for stronger confidence in their modification in the viral RNA (Fig. 7; Fleming et al. 2021). Three other abundant subgenomes, TRS-3a, -E and -M, contained five sites in common, and four of these were in common with those of TRS-S. Synthetic RNAs containing some of these sequence contexts were verified as valid substrates for the pseudouridine writer enzymes PUS1 and/or PUS7 (Fleming et al. 2021). At a later stage (unpublished), we reanalyzed the data using Nano-Psu (Huang et al. 2021) and were able to quantify the Ψ depositions as shown in Table 2. We are therefore highly confident that pseudouridinylation occurs in the 3′ subgenomes of SARS-CoV-2.
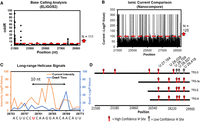
Consensus analysis of DRS data for discovery of possible pseudouridinylation sites in SARS-CoV-2 gRNA. (A) ELIGOS2 base call analysis comparing viral RNA against a synthetic control without RNA modifications. (B) Nanocompore pairwise analysis of the ionic currents between the viral and synthetic RNAs. (C) Helicase dwell-time differences between the viral and synthetic RNA. (D) Sites identified to be pseudouridinylated based on the consensus analysis. The viral and synthetic data were obtained from publicly available sources (Kim et al. 2020; Miladi et al. 2020), and the figure is adapted from our prior work (Fleming et al. 2021).
The percent pseudouridinylation at SARS-CoV-2 sites measured using DRS and Nano-Psu analysis
In concert with the above findings, Giambruno and coworkers found in 2023 that these sites and many more were pseudouridinylated in the SARS-CoV-2 sgRNA (Giambruno et al. 2023). They focused on the highly structured regions of the 5′ and 3′ termini where the most modifications were found. Additionally, they discovered through a mass spectrometric analysis that PUS7 was bound to sites predominantly in the 5′ and 3′ terminal regions. PUS7 is one of the RNA-modifying enzymes that is known to relocate to the cytosol during viral stress, accounting for its activity on viral RNA (Blanco-Melo et al. 2020). Other RNA viruses (e.g., the flaviviruses Zika and HCV) have Ψ at >1% of the U nucleotides when analyzed by LC-MS/MS, suggesting this may be a common feature of viral RNA genomes (McIntyre et al. 2018). Together, these results argue for substantial pseudouridinylation of the SARS-CoV-2 genomic RNA. However, contrasting with these results is a chemical sequencing protocol for Ψ in which no pseudouridine at all was found in the viral RNA from infected cells (Xu et al. 2024). This raises questions about which method, chemical sequencing versus nanopore sequencing, is best able to capture information about modifications in highly structured viral RNA. This feature has also hampered studies of the HIV genome, in which tightly structured ends resist unfolding and analysis (Gener and Kimata 2019).
We and others have employed nanopore DRS to monitor the installation of modified nucleotides into synthetic RNA prepared by in vitro transcription (IVT) (Liu et al. 2019; Fleming et al. 2021, 2023a; Leger et al. 2021; Ramasamy et al. 2022). In the present mRNA vaccine technology, synthesis is performed using a DNA template and an NTP mixture in which UTP is replaced by m1ΨTP, resulting in the replacement of all Us by m1Ψs (Nance and Meier 2021). The RNA strand produced is more heavily modified than any naturally occurring RNA, but it works! mRNA vaccines saved millions of lives per year in the height of the COVID-19 pandemic. Nevertheless, one wonders if a lower level of modification, for example, replacement of only 1%, 5%, or 10% of uridines with m1Ψ, would work equally well or better. To achieve this would require either highly efficient phosphoramidite chemistry or related programmable methods, or perfect enzymatic control of the installation of modifications at only certain sites in the full-length vaccine.
Alternatively, we asked the question, could one dope in modifications by using a prescribed mixture of UTP and m1ΨTP (Fleming and Burrows 2023)? Would an RNA polymerase discriminate between the two, or would it perform an unbiased insertion opposite template adenosines? We used nanopore DRS to examine the distribution of modifications in the RNA product, mimicking vaccine synthesis via IVT, in which the NTP cocktail contained a 1:1 mixture of either UTP + ΨTP or UTP + m1ΨTP (Fig. 8A). Although Ψ and m1Ψ were not readily distinguishable in nanopore reads, both were different from U, and therefore, we were able to examine sequence context effects of the RNA polymerase inserting a modified versus a canonical nucleotide. Using the most common enzyme for IVT, namely T7 RNA polymerase, we designed a DNA template that would interrogate >100 sequence contexts, 5′-VXV-3′ and 5′-VXXV-3′ (V = A, C, G; X = U, Ψ, m1Ψ), all of which are found in the original COVID-19 vaccines of Moderna and Pfizer/BioNTech (Nance and Meier 2021; Fleming and Burrows 2023).
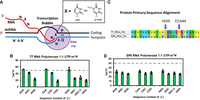
Monitoring RNA polymerization with competing nucleotide triphosphates using nanopore DRS. (A) Schematic of in vitro transcription for monitoring insertion yields of competing nucleotide triphosphates. (B) The insertion yields of m1ΨTP when competed against UTP for the active site of T7 RNA polymerase. (C) Sequence comparison of T7 and SP6 RNA polymerases to display differences in active site residues. (D) The insertion yields of m1ΨTP when competed against UTP for the active site of SP6 RNA polymerase. The figure was adapted from our prior work (Fleming and Burrows 2023).
The analysis of the IVT-synthesized sequences then needed quantification at each U/Ψ site (Fleming and Burrows 2023). Current levels provided by the Tombo tool were not reliable in this case because they often do not occur exactly at the modification, but rather 1 nt on either side. The ELIGOS2 tool was selected because it was specifically developed to compare a sequence of unknown modification level to one that is void or depleted in the modification (Jenjaroenpun et al. 2020). In the first analysis, we found that ΨTP always outcompeted UTP, by varying amounts, and we attribute this to the ability of the Ψ to enter the active site of the polymerase in either the syn or anti conformations with respect to the “glycosidic” bond, either of which could productively base pair with a template A. In contrast, UTP always outcompeted m1ΨTP, likely because the latter favors the nonproductive syn conformation (Chang et al. 2008), and then perhaps it is lost from the active site before bond rotation. However, neither of these observations addresses sequence context. It was found that sequence context was particularly impactful to the selection of UTP versus m1ΨTP, in which those sequences with a 3′ A in the RNA strand, that is, a T:A base pair in the template DNA, led to twice as much m1Ψ insertion compared to those sequences with a 3′ flanking G (Fig. 8B; Fleming and Burrows 2023).
At first glance, it is a complete mystery as to why the identity of a not-yet-present nucleotide, A or G, should impact the selection at the prior site of synthesis. The answer seems to lie with the fact that T7 RNA polymerase uses a duplex DNA template, and the n + 1 base pair, T:A versus C:G in this case, is still intact during selection at the n site (Temiakov et al. 2004; Yin and Steitz 2004). Phe644 in the O′ helix of the polymerase is π-stacked on this base pair, and perhaps the weaker H-bonding of the T:A base pair allows more time for sampling the anti conformation of the incoming m1ΨTP. We tested this hypothesis by replacing the C:G base pair in the template DNA with a C:I (hypoxanthine) base pair with nearly identical π-stacking ability but H-bonding strength similar to T:A. In this case, the synthetic RNA showed 2–3× higher insertion of m1ΨTP, mimicking the result with T:A (Fleming and Burrows 2023).
To further examine the role of Phe644, we examined the sequence context effects of another RNA polymerase, SP6—one that shares many structural features with T7 but has a leucine at position 644 (Fig. 8C; Czech et al. 2013). The insertion of UTP versus m1ΨTP always slightly favored UTP and was sequence context agnostic, within error, leading to the conclusion that if systematic doping of vaccine sequences with low levels of m1Ψ were the goal, then SP6 could provide this capability (Fig. 8D). As a side note, we submitted a patent application based on this concept, and it generated zero interest in the biopharma community, probably because the thousands of different entities produced by such a synthesis would present a nightmare for characterization and validation. Nevertheless, we found this to be a scientifically fascinating challenge and one that the nanopore DRS technique was ideally suited to address. After we reported on m1Ψ nanopore sequencing, VAX-seq was introduced for mRNA vaccine quality control to verify modification status, poly(A) tail length, overall length, and purity (Gunter et al. 2023).
SEQUENCING RNA DAMAGE MODIFICATIONS
Most RNA chemical modifications—from methylations to isomerizations—are performed by enzymes at selected sites during maturation of the biopolymers or when a change in function, for example, RNA editing or alternative splicing, is desired (Wang and He 2014; Gilbert and Nachtergaele 2023). Such modifications are written into the polymer sequence at relatively high stoichiometries, typically >20% and often >90%. This level of occupancy is ideal for analysis by nanopore DRS.
Modifications in RNA nucleotides that result from cellular chemistry rather than enzymology, such as oxidation and deamination of bases that occur during inflammation or oxidative stress conditions, pose an intriguing challenge to nanopore analysis. For example, oxidative stress leads to the formation of 8-oxo-7,8-dihydroguanosine (OG) in RNA, and levels are estimated at 1 in 104, or about 50-fold higher than in DNA (Hofer et al. 2005; Mangerich et al. 2012). If there is no sequence or secondary structure preference for the occurrence of OG, this would mean an occupancy level on the order of 0.01% at any given G. This is far, far below the threshold for reliable detection and quantification in a de novo sequencing experiment. Nevertheless, we present two examples of the detection of RNA damage sites based on the concept that while nanopore data are not accurate enough for mapping low-level modifications, it is at least precise in quantifying changes, even small ones.
In the first example, the question being asked was whether oxidative stress in cells led to oxidized bases at all four nucleotides, A, C, G, and U, or whether oxidation was base-specific (Fleming et al. 2024a). Of further interest was whether oxidation chemistry in the cytosol versus the mitochondria was the same or different. In this case, we were monitoring the Fenton reaction: FeII + H2O2 as a function of bicarbonate added to the media (Fleming and Burrows 2020). In the nanopore experiment, we analyzed both cytosolic and mitochondrial ribosomes in the same experiment and separated the data accordingly. Next, error analysis for all reference G sites were pooled, and A, C, and U were treated similarly. In each case, nucleotides residing in the same k-mer as an enzymatically written modification were discarded from the analysis because enzymatically written modifications can be impacted by stress. An increase in base-calling error was expected if any of the bases were susceptible to oxidation. For example, oxidation reactions are known to convert G to OG, and correspondingly A to OA, C to ho5C, and U to ho5U, and other base oxidation products are also possible. If the cellular Fenton reaction were to generate either hydroxyl radical (HO•) or ferryl species (Fe = O2+) (Meyerstein 2021), oxidation of all four bases would be expected, and this was what we observed when no bicarbonate was added to the media (Fleming et al. 2024a).
Mammalian cells typically have 25 mM 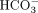 in the cytosol, and gut bacteria such as E. coli are bathed in 20 mM bicarbonate in the mammalian intestine. Consequently, before stressing cells with 100 µM H2O2 for 15 min, up to 20 mM NaHCO3 was added and allowed to equilibrate with isolated cells (Fleming et al. 2024a). After treatment with peroxide and nanopore analysis, only G sites showed increased base-calling error, indicative of chemical
changes. This observation is characteristic of a switch in the Fenton reaction to generate carbonate radical,
in the cytosol, and gut bacteria such as E. coli are bathed in 20 mM bicarbonate in the mammalian intestine. Consequently, before stressing cells with 100 µM H2O2 for 15 min, up to 20 mM NaHCO3 was added and allowed to equilibrate with isolated cells (Fleming et al. 2024a). After treatment with peroxide and nanopore analysis, only G sites showed increased base-calling error, indicative of chemical
changes. This observation is characteristic of a switch in the Fenton reaction to generate carbonate radical, 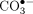 , rather than HO• (Fleming and Burrows 2020). Carbonate radical has a lower redox potential and reacts by a one-electron mechanism with DNA and RNA, leading to principally
G oxidation in nucleic acids (Fleming and Burrows 2017). In mitochondrial rRNA, the overall level of RNA oxidation was less compared to cytosolic rRNA, but it was again entirely
focused on G sites, consistent with the fact that mitochondria, at pH 7.8, are especially rich in
, rather than HO• (Fleming and Burrows 2020). Carbonate radical has a lower redox potential and reacts by a one-electron mechanism with DNA and RNA, leading to principally
G oxidation in nucleic acids (Fleming and Burrows 2017). In mitochondrial rRNA, the overall level of RNA oxidation was less compared to cytosolic rRNA, but it was again entirely
focused on G sites, consistent with the fact that mitochondria, at pH 7.8, are especially rich in 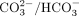 (Radi 2022). Thus, even though the modifications could not be accurately mapped, nanopore DRS provided convincing and reproducible evidence
that a change in reactive oxygen species (ROS) corresponding to a less harmful oxidant,
(Radi 2022). Thus, even though the modifications could not be accurately mapped, nanopore DRS provided convincing and reproducible evidence
that a change in reactive oxygen species (ROS) corresponding to a less harmful oxidant, 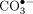 rather than HO•, was being generated under the conditions of cellular oxidative stress. In doing so, a half-century of misunderstanding about
cellular ROS was overturned.
rather than HO•, was being generated under the conditions of cellular oxidative stress. In doing so, a half-century of misunderstanding about
cellular ROS was overturned.
A second example of nanopore sequencing for RNA damage took the above analysis one step further (Fleming et al. 2025). Human cell culture (HEK293T cells) was exposed to tumor necrosis factor-α (TNF-α), which induces nitrosative stress in
addition to oxidative stress. The reactive nitrogen species formed include nitric oxide (NO•), peroxynitrite (ONOO−), and nitrogen dioxide 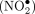 , and via radical combination, N2O3, an excellent nitrosating agent that leads to deamination of exocyclic amines, preferably those of C and A (Dedon and Tannenbaum 2004). Deamination of A yields inosine, I, for which there is now modification-aware base-calling in Dorado. Meanwhile, deamination
of C provides U, a canonical nucleoside in RNA. Therefore, it became possible to monitor the formation of I and U when sequencing
rRNA under conditions of inflammatory stress; G oxidation could be analyzed in the same experiment.
, and via radical combination, N2O3, an excellent nitrosating agent that leads to deamination of exocyclic amines, preferably those of C and A (Dedon and Tannenbaum 2004). Deamination of A yields inosine, I, for which there is now modification-aware base-calling in Dorado. Meanwhile, deamination
of C provides U, a canonical nucleoside in RNA. Therefore, it became possible to monitor the formation of I and U when sequencing
rRNA under conditions of inflammatory stress; G oxidation could be analyzed in the same experiment.
In cytosolic human ribosomes exposed to TNF-α, the major sites of A→I, C→U, and G→Gox were all at solvent-exposed sites (Fleming et al. 2025). Importantly, this study highlights the GC-rich expansion sequences, or “tentacles,” as being particularly reactive with RONS. Looking at the top 30 C→U and top 30 G→Gox sites, more than half of them were in tentacles. These expansion sequences in mammalian rRNA are typically hundreds of nucleotides in length and >80% G + C with very little A content; their structures do not appear in X-ray crystallographic or cryo-EM analyses because they are too dynamic, but they are likely to be long hairpins, often with branching (Bowman et al. 2020). These structural motifs are enigmatic in terms of function; might they serve to sweep up and respond to chemical stressors in the cytosol while protecting the core of the ribosome? Nanopore DRS will be one of the tools to help elucidate this biochemistry in the future.
SUMMARY AND FUTURE DIRECTIONS
Nanopore direct RNA sequencing is currently the only method available for long-read analysis of RNA modifications without chemical intervention and PCR amplification. The present ONT technology now delivers high-accuracy base calling (∼98%) and includes modification-aware software for Ψ, m6A, m5C, I, and all four Nm, all of which can be analyzed in a single experiment. Furthermore, the long-read technology means that operon-specific epitranscriptomic analysis is now available with relative ease, and one can begin to evaluate cross talks between modifications during different cellular conditions.
Challenges remain, particularly with the discovery of new modifications or in discriminating between modifications that behave very similarly in the nanopore such as nucleobase methylations at different sites. In particular, modifications at C5 of pyrimidines often give very similar signals to unmodified bases. The new modification-aware base callers need to be further benchmarked by the user community. Therefore, current nanopore studies need to be validated by positive and negative controls, either by synthesizing authentically modified RNA, the use of enzyme writer knockouts, complementary chemical sequencing methods, or by mass spectrometric analysis, the gold standard.
Accurate levels of modification occupancy at a given site will remain a challenge, although already the data show excellent reproducibility, meaning that experiments that look at changes in modifications will provide better data than those that aim for quantitative mapping. In the future, we can expect steady improvements in accuracy and additional modification-aware base callers. A feature that would help us understand many different sequencing problems in RNA would address homopolymer runs; for example, these are common in human rRNA tentacles and present a problem for nanopore sequencing of Nn, with n ≥ 4. Lastly, the introduction of barcoding kits to the commercial RNA library preparation kits and associated software to demultiplex the data will streamline the collection of more data for profiling epitranscriptomic changes in biological studies. Competing technologies may offer advantages to some of these challenges, but we will have to wait for them to be commercially available.
In this review, we have attempted to illustrate the types of studies in which nanopore DRS can be employed to learn new biological chemistry of cellular RNA. The timing is right to jump into this field because of the rapid progress made recently in base-calling accuracy and the ability to call common modifications. We add some cautionary notes: (1) more than once we have needed to do an additional sequencing experiment, for example, after manuscript review, and we then found that in the intervening few months, there has been a software update. This can necessitate reprocessing of your entire set of data with new software in order to make valid comparisons between new and old experiments. Plan accordingly, so that you do all the experiments with the same kits and software. (2) The computing needs are considerable! We found that data analysis during final examination week at a large university was unexpectedly slow, even though we are blessed with exceptional computing capacity at our institution. (3) Finally, whenever nanopore DRS delivers an unexpected finding, consider alternative methods of validation. Is it a new pseudouridine site or a C/U variant? An unexpected methylation? How good is that new software, anyway? Nevertheless, nanopore DRS offers creative applications to epitranscriptomics on a scale not imagined a decade ago.
ACKNOWLEDGMENTS
For nanopore research done in our laboratory, we are grateful to the National Institute of General Medical Sciences for past and ongoing support via grants R01 GM093099 and R35 GM145237. We also thank our coauthors and collaborators, especially Professor H.S. White (University of Utah), who introduced us to this fascinating field of nanopore technology. We also wish to thank the University of Utah Center for High Performance Computing for making computing time available for data processing.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080908.125.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








