
Natural human tRNAAla anticodon variants mistranslate the genetic code
- 1Department of Biochemistry, The University of Western Ontario, London, Ontario N6A 5C1, Canada
- 2Bioinformatics Solutions, Inc., Waterloo, Ontario N2L 3K8, Canada
- 3Department of Chemistry, The University of Western Ontario, London, Ontario N6A 5C1, Canada
- Corresponding author: patrick.odonoghue{at}uwo.ca
-
Handling editor: Eric Westhof
Abstract
Transfer RNAs (tRNAs) play an essential role in protein synthesis by linking the nucleic acid sequences of gene products to the amino acid sequences of proteins. There are >400 functional tRNA genes in humans, and adding to this diversity, there are many single-nucleotide polymorphisms in tRNAs across our population, including anticodon variants that mistranslate the genetic code. In human genomes, we identified three rare alanine tRNA (tRNAAla) variants with nonsynonymous anticodon mutations: tRNAAlaCGC G35T, tRNAAlaUGC G35A, and tRNAAlaAGC C36T. Since alanyl-tRNA synthetase (AlaRS) does not recognize the anticodon, we hypothesized that these tRNAAla variants will misincorporate Ala at glutamate (Glu), valine (Val), and threonine (Thr) codons, respectively. We found that expressing the naturally occurring tRNAAla variants in human cells led to defects in protein production without a substantial impact on cell growth. Using mass spectrometry, we confirmed and estimated Ala misincorporation levels at Glu (0.7%), Val (5%), and Thr (0.1%) codons. Although Ala misincorporation was higher at Val codons, cells misincorporating Ala at Glu codons had the most severe defect in protein production. The data demonstrate the ability of natural human tRNAAla variants to generate mistranslation, leading to defects in protein production that depend on the nature of the amino acid replacement.
Keywords
INTRODUCTION
Living cells depend on the accurate flow of genetic information encoded in the genome. Compared to genome replication and transcription of messenger RNAs (mRNAs), translation of mRNAs during protein synthesis is the most error-prone step (Steiner and Ibba 2019). Mistranslation can result from the incorrect incorporation of an amino acid into a growing polypeptide chain. Translational fidelity is normally high with varying estimates of basal translation error suggesting that 1 in 104 to 108 codons are misread (Manickam et al. 2014). Diverse cells can tolerate elevated mistranslation levels of 1%–10% per codon. Mistranslation can be caused by mutations to the ribosome (Hoffer et al. 2019) and ribosomal proteins (Agarwal et al. 2011) or by ribosome stalling (Collart and Weiss 2020). Mutations in aminoacyl-tRNA synthetases (aaRSs) (Wilhelm et al. 2023; Zhang and Ling 2025) and single-nucleotide substitutions in transfer RNA (tRNA) genes can lead to proteome-wide mistranslation (Lant et al. 2021).
The aaRSs are responsible for recognizing and accurately ligating each tRNA with its specific or cognate amino acid, producing aminoacyl-tRNA products that are the substrates for protein synthesis. Aminoacyl-tRNAs are adaptor molecules that translate the genetic code into protein sequences by delivering amino acids to the ribosome during translation (Schimmel and Söll 1979). The tRNA gene transcripts undergo posttranscriptional modifications and cleavages to become mature tRNAs that are 76–90 nt in length and have an L-shaped tertiary structure (El Yacoubi et al. 2012). The mature tRNA body is composed of several stem–loops. Base-pairing between the 5′ and 3′ end of tRNA generates the acceptor stem with the universally conserved 3′-CAA sequence that is necessary for the ligation of an amino acid to the 3′-terminal A76 (Alexander and Hendrickson 2020). On the ribosome, selection of the correct tRNA for each mRNA codon relies on complementary base-pairing with the anticodon nucleotides (34, 35, 36) in the tRNA (Agris et al. 2007). The 20 standard amino acids are encoded by 61 trinucleotide sequences that are referred to as sense codons in the genetic code table. Due to degeneracy of the genetic code, most of the amino acids are encoded by two or more codons. The tRNAs account for this apparent redundancy in the code with families of tRNA isoacceptors that accept the same amino acid and have different anticodons (Behura and Severson 2011), while tRNAs that share the same anticodon are called isodecoders.
Missense suppressor tRNAs cause mistranslation through different mechanisms. Identity elements are nucleotides in the tRNA body that allow recognition by the cognate aaRS (Giegé et al. 1998; Giegé and Eriani 2023). Each aaRS catalyzes a two-step ATP-dependent reaction that ligates a specific amino acid to its cognate tRNA (Schimmel and Söll 1979). Nucleotides in the anticodon often serve as identity elements in tRNAs (Giegé et al. 1998). The aaRSs for alanine (Ala), serine (Ser), leucine (Leu), and pyrrolysine (Pyl) do not recognize the anticodon sequence and rely on different nucleotides or structural elements for recognition of their cognate tRNA (O'Donoghue et al. 2012; Hoffman et al. 2017; Lant et al. 2018; Rozik et al. 2022; Hasan et al. 2023). For example, alanyl-tRNA synthetase (AlaRS) exclusively uses the G3:U70 base pair in the acceptor stem of alanine tRNA (tRNAAla) for recognition (Hou and Schimmel 1988), while the long variable arms in tRNASer and tRNALeu serve as major identity elements for SerRS and LeuRS (Giegé and Eriani 2023). Mutations to the anticodon sequences of tRNAAla, tRNASer, tRNALeu, and tRNAPyl do not affect or substantially reduce aminoacylation, thus these tRNAs can be programmed to incorporate their cognate amino acid at any other codon, resulting in missense suppression of those codons and mistranslation of the proteome.
Since tRNAs are capable of decoding multiple codons through G:U pairing and other pairings possible by modification of nucleotides in the anticodon, the theoretical minimum number of tRNA genes required for an organism to decode all 61 sense codons is 32 (Söll et al. 1966). There are examples of parasitic microbes (Jaffe et al. 2004) and mitochondria (Salinas-Giege et al. 2015) with fewer than 32 tRNA genes. In these species and organelles, expanded wobble decoding (Haag et al. 2016) and in some cases, tRNA import (Rubio et al. 2008) are needed to enable translation. Most eukaryotic genomes far exceed this theoretical minimum. Estimates suggest that of the 610 tRNA genes in the human genome, 416 are functional tRNAs and expressed differentially in some or constitutively in all cells and tissues (Chan and Lowe 2016). In comparison to the reference genome, individuals carry many single-nucleotide polymorphisms (SNPs) as well as variants with multiple SNPs in tRNA genes (Parisien et al. 2013; Berg et al. 2019; Lant et al. 2019). In a targeted and deep coverage tRNA gene sequencing study, an average of 60–70 SNPs and ∼10 rare variants were identified in the tRNA genes of each individual (Berg et al. 2019), including different kinds of mistranslating tRNAs that occur as rare or common variants in the population (Lant et al. 2019).
Amino acid misincorporation is not always deleterious to cells (Pezo et al. 2004), including mammalian cells (Gomes et al. 2016). We identified natural human tRNA variants that misincorporate Ala at Gly codons through either anticodon or identity element mutations (Hasan et al. 2023). Despite low fidelity of translation, cells expressing these tRNA variants had minor changes in protein production without cytotoxicity (Hasan et al. 2023). Moreover, misincorporation of methionine may serve as a protective mechanism in response to oxidative stress in cells (Stadtman et al. 2002; Wang and Pan 2016). Here, we characterized tRNAAla genes with anticodon variants that occur naturally in the human population (Table 1). We found that each of the tRNA variants caused defects in protein production compared to normal cells. Using mass spectrometry, we confirmed that naturally occurring variants cause mistranslation of Ala at glutamate (Glu), valine (Val), and threonine (Thr) codons, and we assessed the impact of each missense suppressor tRNA on cell growth and cell signaling in the unfolded protein response pathway.
Naturally occurring tRNAAla variants characterized in this study
RESULTS
Identifying mistranslating tRNAAla genes
By searching the genomic tRNA database (data release Sept 22, 2024, Homo sapiens GRCh38/hg38), we identified three human tRNAAla genes: tRNAAlaCGC (Ala-CGC-1-1), tRNAAlaUGC (Ala-TGC-4-1), and tRNAAlaAGC (Ala-AGC-8-1) that each have a naturally occurring variant in the population, resulting in a nonsynonymous anticodon change (Table 1; Fig. 1). Since AlaRS does not recognize the tRNAAla anticodon, mutations to the anticodon can lead to misincorporation of Ala at non-Ala codons. In comparison to a common tRNAAla variant that we characterized before (Hasan et al. 2023), which is found in 6.5% of the population (Table 1), each of the other three human tRNAAla anticodon mutants are rare variants. The most prevalent variant of the three, tRNAAlaUGC G35A (tRNAAlaUAC(Val)), occurs in 0.009% of the population and has the UAC anticodon that decodes Val codons. The tRNAAlaCGC G35T (tRNAAlaCUC(Glu)) and tRNAAlaAGC C36T (tRNAAlaAGU(Thr)) variants are even more rare. The tRNAAlaCGC G35T allele produces a CUC anticodon that has the potential to misread Glu codons and occurs at an allele frequency of 0.0004%. The tRNAAlaAGC C36T variant has a similar prevalence (0.0008% allele frequency) and has an AGU anticodon that may misread Thr codons with Ala. Our studies described below tested our hypothesis that expression of each of these tRNA variants in mammalian cells will lead to mistranslation.
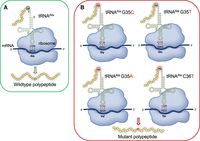
Schematic of tRNA-dependent Ala mistranslation. (A) Wild-type tRNAAla with anticodon NGC (3′-CGN-5′), (N = A/G/C/T) recognizes Ala codons (5′-GCN-3′). (B) Anticodon mutations to tRNAAla genes found in human genomes cause misreading of Gly (G35C) (Hasan et al. 2023), Glu (G35T), Val (G35A), and Thr (C36T) codons with Ala.
Quantifying the level of a tRNAAla anticodon variant in mammalian cells
We previously used mass spectrometry to confirm misincorporation of Ala at Gly codons in murine neuroblastoma (N2a) cells expressing the human tRNAAla G35C variant that misreads Gly codons (Fig. 1A; Table 1). According to spectral counts of the number of mistranslated relative to properly translated peptides that we identified, the tRNA generated ∼10% misincorporation of Ala at Gly codons in an mCherry reporter protein (Hasan et al. 2023). To determine the level of mutant tRNA that led to the observed misincorporation of Ala at Gly codons, here we conducted a tRNA sequencing experiment to quantify the levels of all tRNA transcripts in the indicated cell lines (Fig. 2). We compared endogenous tRNA expression levels in murine N2a cells transfected with wild-type human tRNAAlaAGC or with the Gly-decoding tRNAAlaACC variant (Fig. 2A). We identified only a single tRNA gene that was differentially regulated by more than twofold, which was a mitochondrial tRNAArg (mt-Arg-TCG) gene that was enriched in cells expressing wild-type relative to mutant tRNA. We observed two tRNA genes that were significantly increased by 1.5-fold in mistranslating cells, and several mitochondrial tRNA species (Ala-TGC, Asn-GTT, Asp-GTC, Glu-TCC) and one cytoplasmic tRNA (Lys-CTT-3-1) that were enriched more than 1.5 but less than twofold in cells expressing wild-type tRNAAla. Overall, expression of the Gly-decoding tRNAAla led to minimal changes in the tRNAome.
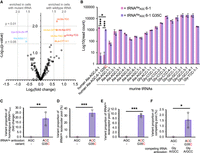
tRNA sequencing quantifies tRNAAla anticodon variant levels in cells. We used hydro-tRNA-seq (Gogakos et al. 2017) to sequence the complete tRNAome of murine N2a cells expressing wild-type tRNAAlaAGC or a Gly-decoding G35C variant (tRNAAlaACC(Gly)) that we showed previously causes misincorporation of Ala at Gly codons in N2a cells (Hasan et al. 2023). (A) The volcano plot shows the differential expression of endogenous tRNA genes as the significance of the difference (−log10 of the P-value) as a function of the log2 of fold change. Significant changes above 1.5-fold are annotated. Only one tRNA gene (mt-Arg-TCG) showed a significant change of more than twofold. (B) Levels of the indicated human tRNAAlaAGC (wild-type or G35C variant) and mouse tRNAAla and tRNAGly species expressed as log10 of the normalized read count. The level of the tRNAAla G35C variant is plotted as (C) a proportion of the level of human tRNAAlaAGC 6-1 G36C variant detected in cells relative to the level of the homologs mouse tRNAAla (Ala-AGC-1-1), as (D) a proportion of the tRNAAlaAGC isodecoder pool, as (E) a proportion of the total tRNAAla isoacceptor pool, and as (F) a proportion of the pool of competing tRNAGlyACC and tRNAGlyGCC isodecoders. Independent sample t-tests were computed (*** P < 0.001, * P < 0.05) based on at least n = 3 biological replicates.
The tRNA sequencing approach clearly identified expression of the wild-type human tRNAAlaAGC and the G35C variant only in cells transfected with the plasmid bearing the corresponding human tRNA allele, while no significant expression was observed of the mutant in cells transfected with wild-type human tRNA gene or vice versa (Fig. 2B). We then compared the level of the human wild-type and mutant tRNAs to the background of endogenous mouse tRNAAla and tRNAGly gene expression. We observed significantly more expression of the wild-type human tRNAAlaAGC compared to the mutant tRNA, since the G35C variant level was 64% that of wild-type. The most similar mouse tRNA to our human tRNAAla variants is expressed from the Ala-AGC-1-1 gene, which was expressed at a level of 5% relative to our transfected human tRNA alleles (Fig. 2C). In the murine N2a cells, several other tRNAAlaAGC genes are more abundantly expressed. In comparison to the total level of all tRNAAlaAGC isodecoders, the human tRNAAlaACC(Gly) mutant comprises ∼30% of the tRNAAlaAGC pool (Fig. 2D) and 9% of the total pool of tRNAAla isoacceptors (Fig. 2E). Thus, the plasmid-based expression of the tRNAAla mutant leads to expression that contributes to but does not dominate the native tRNA pool.
In translation on the ribosome, the mutant tRNAAla competes with a large pool of tRNAGly for the decoding of Gly codons. We found that due to the high level of tRNAGly expression, tRNAAlaACC(Gly) makes up just 1.5% of the Gly-decoders in the cell (Fig. 2F). Even at this apparently low level, the Gly-decoding tRNAAla mutant is effective in competing with tRNAGly for decoding (Hasan et al. 2023). In future studies, we used advanced tRNA sequencing approaches to examine tRNA abundance and modification states (Davey-Young et al. 2024) of other tRNAAla anticodon variants.
Naturally occurring tRNAAla variants cause defects in protein production
A hallmark response to tRNA-dependent mistranslation is the downregulation of protein synthesis (Geslain et al. 2010). Our recent studies, however, showed that the common tRNAAlaACC(Gly) variant generated significant mistranslation of Ala at Gly codons yet caused only a minor impact on protein production (Hasan et al. 2023). To determine the impact of the rare human tRNAAla variants on protein production in a human cell line known for efficient transfection and transgene expression, we transfected human embryonic kidney (HEK) 293T cells with plasmids coexpressing the wild-type or mutant tRNAAla genes and mCherry as a marker for protein production. Using fluorescence microscopy, we measured red fluorescence (ex. 542 ± 20 nm, em. 593 ± 40 nm) of individual transfected HEK 293T cells expressing the indicated tRNA variants (Fig. 3).
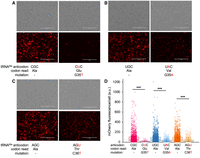
mCherry fluorescence in normal and mistranslating cells. (A–C) Brightfield (above) and fluorescent images (below) showing mCherry production in cells coexpressing wild-type or the indicated (A) Glu-decoding, (B) Val-decoding, or (C) Thr-decoding tRNAAla variants. Scale bars, 400 μm. (D) Box and whisker plot and quantitation of mean mCherry fluorescence in individual cells. Independent sample t-tests were computed (*** P < 0.001) based on at least n = 3 biological replicates. We imaged 1500–1700 individual cells in each biological replicate.
A substantial and significant reduction of mCherry fluorescence per cell was seen in HEK 293T cells carrying each tRNAAla anticodon variant compared to cells expressing the wild-type tRNA allele (Fig. 3). Compared to the cells expressing wild-type tRNA, cells expressing tRNAAlaCUC(Glu) had a ∼4.8-fold reduction in mCherry fluorescence per cell, while cells with tRNAAlaUAC(Val) and tRNAAlaAGU(Thr) had more modest twofold reductions in mCherry production. Although fluorescence microscopy of mCherry provided a quantitative approach to measure protein levels over a broad dynamic range, we also used western blotting to confirm the defects in mCherry protein production relative to a GAPDH loading control (Fig. 4). According to densitometry of the western blots, an 84-fold decrease in protein production was observed in cells expressing tRNAAlaCUC(Glu), while higher levels of mCherry were detected in cells expressing tRNAAlaUAC(Val) and tRNAAlaAGU(Thr). In comparison to their wild-type counterparts, however, protein production was still significantly reduced in cells expressing tRNAAlaAGU(Thr) or tRNAAlaUAC(Val) that showed fourfold reductions in mCherry production. No significant difference in protein levels was observed between cell lines expressing wild-type tRNAAla.

mCherry protein levels in normal and mistranslating cells. (A) Western blots of mCherry and a GAPDH loading control from cells transfected with plasmids bearing mCherry and the indicated wild-type or mutant tRNAAla. (B) Quantitation of mCherry protein levels normalized by GAPDH. Independent sample t-tests were computed (** P ≤ 0.001, *** P ≤ 0.001) based on at least n = 3 biological replicates.
Proliferation in cells expressing naturally occurring tRNAAla anticodon variants
Some mistranslating tRNAs have been shown to elicit cytotoxicity in mammalian cells, such as the tRNASerAGA G35A mutant (Lant et al. 2023). Conversely, other kinds of mistranslation have little or no toxicity and are well tolerated in human cells (Hasan et al. 2023; Hou et al. 2024). To determine whether the tRNAAla anticodon variants cause defects in cell proliferation, we used a quantitative cell counting assay to measure cell growth over a 72 h time course. The assay employs a water-soluble tetrazolium salt that produces an orange formazan dye upon bio-reduction by dehydrogenases. Absorbance (A450) is proportional to cellular dehydrogenase activity, which is a proxy for cell growth. We observed significantly lower cell viability at the 24 h time point in cells expressing tRNAAlaCUG(Glu) relative to cells expressing no additional tRNA (vector only) and cells expressing wild-type tRNAAlaCGC (Fig. 5A). At 48 and 72 h time points, we observed no significant difference between the vector only control and the Glu-decoding tRNAAla, but the wild-type tRNAAlaCGC significantly stimulated cell growth. The Val-decoding tRNAAla shows a defect in viability at the 24 h time point relative to the wild-type tRNA and vector only controls (Fig. 5B). Cells expressing the Thr-decoding tRNAAla showed proliferation levels indistinguishable from cells transfected with the vector only or with the wild-type tRNAAlaAGC.
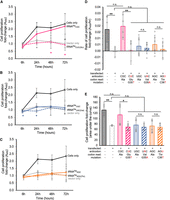
Proliferation of cells expressing wild-type or mutant tRNA. Cell growth was monitored using Cell Counting Kit-8 for HEK 293T cells only or cells transfected with a plasmid expressing no additional tRNA or (A) wild-type tRNAAlaCGC or the Glu-decoding G35T mutant, (B) wild-type tRNAAlaUGC or the Val-decoding G35A mutant, and (C) wild-type tRNAAlaAGC or the Thr-decoding C36T mutant. (D) The rate of proliferation was calculated as the slope of each growth curve over 72 h, and (E) the level of cell proliferation was estimated from the area under the curve. Independent sample t-tests were (n.s., not significant, * P < 0.05) based on at least n = 3 biological replicates. In A–C, significant differences between the mutant tRNA and the wild-type tRNA (colored *) or vector only control (gray *) are indicated.
We then analyzed the relative growth rates (Fig. 5D) and the total level of cell proliferation (Fig. 5E) observed during the time course. Transfecting the cells with the vector only shows a significant reduction in growth relative to the untransfected controls, as expected. Interestingly, transfection with the wild-type tRNAAlaCGC stimulated growth rate and level to that observed in untransfected cells. We found that the growth rate and levels of the mutant tRNAs were not significantly different form the vector only control. The data suggest that although we recorded defects in cell viability for the Glu and Val-decoding tRNAs are discrete time points (Fig. 5A,B), the overall growth was not substantially impacted by the mistranslating tRNAs. Wild-type tRNAAlaCGC stimulated cell proliferation, while the wild-type tRNAAlaUGC and tRNAAlaAGC showed no impact on growth.
Cells expressing tRNAAla variants cause misincorporation of Ala
To identify and estimate the misincorporation levels of Ala in our mCherry reporter caused by each tRNAAla variant, we used liquid chromatography tandem mass spectrometry (LC-MS/MS). The mCherry protein was isolated from HEK 293T cells coexpressing either a mutant tRNAAla or the wild-type tRNAAlaCGC as a control. MS/MS analysis of immunoprecipitated mCherry purified from cells expressing potentially mistranslating tRNAs identified multiple peptides corresponding to Ala misincorporation (>2% ion intensity) at Glu, Val, and Thr codons (Fig. 6).
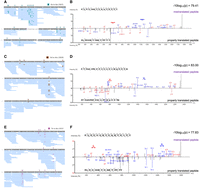
tRNA-dependent misincorporation of Ala detected by mass spectrometry. Tandem mass spectrometry analysis revealed Ala misincorporation in mCherry isolated from HEK 293T cells expressing (A, B) tRNAAlaCUC(Glu), (C, D) tRNAAlaUAC(Val), and (E, F) tRNAAlaAGU(Thr) at (A, B) Glu, (C, D) Val, and (E, F) Thr codons, respectively. (A, C, E) Misincorporation events in mCherry are depicted in peptide coverage maps alongside (B, D, F) example mirror spectra comparing mistranslated peptides (top) to the properly translated matching peptide (bottom). Colored boxed symbols above the peptide sequence annotate the indicated mistranslated residues identified with minimal ion intensities of ≥2%. The data are based on n = 3 biological replicates.
The Glu codon GAG appears 24 times in the mCherry sequence, which tRNAAlaCGC G35T has the potential to decode. There are no occurrences of the Glu codon GAA in mCherry. We confirmed Ala misincorporation at eight of the 24 GAG codons, where the probability of random hit score (−10logP) was ≥60 (Table 2; Fig. 6A,B). These high scoring hits have multiple ions with high signal-to-noise ratio confirming misincorporation. The higher ion intensity and higher quality scores of peptides from cells expressing the mutant tRNAs indicate confident identification of the mistranslated peptides. Seven more potential peptide hits with −10logP scores of ∼40–60 were also identified, which contain more unidentified peaks and lack multiple y and b ions that cover the site of interest, though the hits do match the full peptide mass. Some areas of the mCherry protein (e.g., residues near 100–120) had relatively lower coverage than other regions of the protein, which may explain why misincorporated peptides were not found at these sites.
Summary of Ala misincorporation identified by MS/MS
The tRNAAlaUGC G35A variant has the Val anticodon UAC, and due to G:U wobble base-pairing, it can decode both GUA and the more commonly used GUG codons. In the mCherry sequence, there are 15 Val residues, and we detected misincorporation of Ala at six of these sites (Fig. 6C,D). Three of the four highest scoring hits were at GUG codons. The highest scoring mistranslated peptide was associated with Ala misincorporation at the V140 GUA codon (−10logP = 83).
The tRNAAlaAGC C36T variant and its wild-type counterpart have a known posttranscriptional modification of the A34 base to inosine (I) by tRNA-adenosine deaminases (Rafels-Ybern et al. 2019). Functionally, I34 allows the tRNA to form wobble pairs with C, U, and A nucleotides in the third codon position (Söll et al. 1966). In the mCherry sequence, Thr appears 12 times with the codon ACC used at each occurrence. Without the modification, tRNAAlaAGC C36T would not be able to decode ACC efficiently, as its AGU anticodon should only be able to efficiently read the ACU Thr codon. However, the I34 modification allows normal base-pairing with the ACC Thr codon as well. Indeed, we confirmed misincorporation of Ala at eight Thr codons (Fig. 6E,F). Double misincorporation events were observed in individual peptides at Thr residues 184 and 185 as well as in cells expressing Glu-decoding tRNAAla at mCherry Glu33 and Glu39 (Supplemental Fig. S1).
Ala misincorporation was not detected in cells expressing wild-type tRNAAlaCGC, but several other non-Ala mistranslation events were observed that may represent background or basal misincorporation (Table 2; Supplemental Fig. S2). Compared to peptides identified in cells expressing mutant tRNAs (Table 2; Fig. 6), quality scores of mistranslated peptides detected in the wild-type cell line were generally lower (−10logP = 26–48) and ion intensities were <2%, indicating greater uncertainty in the match of peptide to spectrum and reduced abundance. Therefore, these spectra may represent natural mistranslation, an unknown modification at a nearby site, or a misidentification of the peptide, which may explain the low peptide quality scores of potential mistranslated peptides in normal cells.
Estimating the level of Ala misincorporation
We used the MS/MS data to provide a quantitative estimate of the level of misincorporation generated by each tRNAAla variant. Using the area under the isotopic peak to estimate peptide abundance, we compared the proportion of mistranslated to matching properly translated peptides observed in protein extracted from each cell line. Based on the quantitation, we found that cells expressing tRNAAlaAGU(Thr) misincorporated Ala at Thr codons at an average level of 0.1%. There was significantly more mistranslation in cells expressing tRNAAlaCUC(Glu) and tRNAAlaUAC(Val) (Fig. 7). The Val-decoding tRNAAla generated the highest level of misincorporation with Val codons being read as Ala 5% of the time on average, while the Glu-decoding tRNAAla displayed an intermediate level of misincorporation with Glu codons read as Ala at an average level of 0.7%. We observed no significant correlations (r2 = 0.75, P = 0.2) between growth rate and mistranslation level (Supplemental Fig. S3A). We also found no significant correlation (r2 = 0.02, P = 0.9) between the observed defects in mCherry production and mistranslation level (Supplemental Fig. S3B). Reminiscent of our findings with Leu misincorporation (Davey-Young et al. 2024) and related studies on Ala misincorporation in yeast (Cozma et al. 2023), the data suggest the nature of the amino acid substitution rather than the level of misincorporation may be a dominant factor related to the phenotypic impacts of mistranslation.

Tandem mass spectrometry-based estimation of mistranslation levels in cells with tRNAAla variants. The plots show the proportion of mistranslated peptides in mCherry from HEK 293T cells expressing the indicated wild-type or mistranslating tRNAAla variants. (A, B) The percentage of Ala misincorporation was calculated by comparing the area under the isotopic peak observed for matching mistranslated relative to properly translated peptides and presented in (A) standard and (B) logarithmic plots. Independent sample t-tests were (* P < 0.05, ** P < 0.01, *** P < 0.001) based on at least n = 3 biological replicates.
Probing the integrated stress response in mistranslating cells
Previous studies have shown that mistranslation can lead to activation of the integrated stress response, which is a branch of the unfolded protein response (Mohler et al. 2017). As a result of the upstream activation of stress-responsive kinases, phosphorylation of eukaryotic translation initiation factor 2-alpha (eIF2α) at Ser51 leads to inhibition of translation initiation and a reduction in protein production (Geslain et al. 2010). Thus, we probed eIF2α and phospho-eIF2α (p-eIF2α) relative to a vinculin loading control in cells transfected with no additional tRNA or with the Glu-, Val-, or Thr-decoding tRNAAla variants. Although we found a small but significant decrease in eIF2α levels in cells expressing the Thr-decoding variant (Supplemental Fig. S4A,B), none of the mutant tRNAs induced a change in p-eIF2α levels (Supplemental Fig. S4C,D) or the stoichiometry of eIF2α phosphorylation (Supplemental Fig. S4E). The data indicate that Glu-, Val, and Thr-decoding tRNAAla variants do not induce the integrated stress response. In future studies, we will probe other branches of the unfolded protein response pathway in mistranslating cells.
DISCUSSION
Mistranslation from natural tRNAAla variants in human cells
The human genome contains a diverse family of tRNAs (Parisien et al. 2013). While some the ∼600 tRNA genes may be pseudogenes, recent predictive methods that consider both experimental data and sequence characteristics of tRNA genomic loci, suggest that 300–400 tRNAs are both functional and expressed (Chan and Lowe 2016; Thornlow et al. 2020). Human tRNA isoacceptors and isodecoders which bear the same amino acid but differ in parts of the tRNA body are found in multiple identical or similar copies. For example, human tRNAAla can be expressed from 34 high-confidence genes (Chan and Lowe 2016) that use one of three anticodon sequences (AGC, UGC, or CGC) to decode the four Ala (GCN) codons. Variations in human tRNA genes go beyond isoacceptors and isodecoders within a genome, and many common and rare variants are found throughout the body of all tRNA genes across the human population (Parisien et al. 2013; Chan and Lowe 2016; Lant et al. 2019).
Some of these variants produce tRNAs with nonsynonymous anticodon mutations. Expression of these tRNAs can cause misreading of the genetic code, and mistranslation that affects the entire proteome. Anticodon mutations to tRNASer, tRNAAla, and tRNALeu in particular are more likely to cause amino acid misincorporation because their cognate aaRSs do not use the anticodon as an identity element (Giegé and Eriani 2023). Several anticodon variants are surprisingly common in the human population, including variants that decode Gly codons with Ala (6%–8%) (Hasan et al. 2023) and a variant that misincorporates Ser at Phe codons (2%–3%) (Lant et al. 2021). We found that Ser misincorporation at Phe codons caused a significant decrease in protein production and cytotoxicity in the context of proteosome inhibition (Lant et al. 2021), while Ala misincorporation at Gly codons showed minor impacts on protein production and no cytotoxicity in mammalian cells despite low fidelity translation (Hasan et al. 2023). The tRNAAla variants characterized here occur much less frequently in the human population. Nonetheless, the most common variant of the three, tRNAAlaUAC(Val), occurs at a frequency of 0.009% (Table 1), which translates to nearly two million people globally. The allele frequency data are from the TOPMED project, which has a large sample size of 150K genomes (Taliun et al. 2021).
Each tRNA variant studied here causes misincorporation of Ala through a single-nucleotide anticodon mutation. These tRNA variants retain Ala accepting activity as a result of their G3:U70 base pair, which is the sole recognition element for AlaRS (Hou and Schimmel 1988). In mammalian cells, we confirmed that the tRNAAlaCGC G35T variant was able to decode Glu codon GAG, while the tRNAAlaCGC G35A variant decoded Val codons GUA and GUG due to wobble base-pairing. Previously, a study focused on a naturally occurring tRNA variant with mutations that generate a G4:U69 base pair in tRNAThr, and these tRNAs can be mischarged by AlaRS to generate Ala-tRNAThr. The mischarged species, however, is deacylated by an editing activity in threonyl-tRNA synthetase (ThrRS) (Chen et al. 2020). The ThrRS editing activity apparently prevented mistranslation of Ala at Thr codons in mammalian cells and murine tissues. Here, we show a different route that indeed generates mistranslation of Thr codons with Ala in proteins as confirmed by mass spectrometry, wherein a C36T anticodon mutation to tRNAAlaAGC allowed misincorporation of Ala at Thr codons. Since our Thr-decoding tRNAAla has a tRNAAla and not a tRNAThr body, it is not likely to be recognized by the ThrRS editing domain for deacylation. If, however, the Thr anticodon conferred some level recognition by the ThrRS editing domain, such an activity could help explain the low levels of Ala misincorporation at Thr codons that we observed. In future studies beyond the scope of this work, we will examine aminoacyl-tRNA editing and other factors, including further studies on tRNA expression levels using advanced tRNA sequencing approaches (Davey-Young et al. 2024) to complement and expand on our characterization of the Gly-decoding tRNAAla (Fig. 2). Overall, these findings demonstrate that even very rare tRNA variants may cause mistranslation when expressed ectopically in mammalian cells. Our future studies will focus on the characterization of genome-encoded tRNA mutants.
tRNA anticodon variants cause defects in protein production in mammalian cells
Taking advantage of the fact that SerRS does not recognize the anticodon, human tRNASer was mutated to read 10 non-serine codons in human cells (Geslain et al. 2010). Most of the anticodon variants caused a reduction of green fluorescent protein (GFP) production, and only some (e.g., Ser misincorporation at isoleucine codons) resulted in stimulation of the integrated stress and unfolded protein responses (Geslain et al. 2010). Here, we compared a series of tRNAAla variants that misread Glu, Val, or Thr codons with Ala. The effect of each of these tRNA variants on cells is influenced by both the degree of mistranslation and perhaps more so by the nature of the substitution. This fact was also evident in studies of tRNAAla anticodon variants in yeast (Cozma et al. 2023), and tRNALeu anticodon variants in yeast and mammalian cells (Davey-Young et al. 2024).
With the tRNAAla variants characterized here, we found the greatest defect in protein production was in cells misreading Glu codons with Ala. According to the Blosum 62 substitution matrix, Ala substitutions at Gly, Val, and Thr residues (Blosum score = 0) are neutral mutations in protein evolution, while Ala to Glu is a nonconservative substitution that is underrepresented (Blosum score = −1) in alignments of homologous proteins (Henikoff and Henikoff 1992). In terms of their chemical properties and in the context of a folded protein, Ala and Glu are also the most dissimilar out of the three substitutions investigated here. Thus, Ala substitutions at Glu residues are most likely to have a negative impact on protein folding, structure, and function. Indeed, when we previously investigated Ala misincorporation at codons for the more chemically similar Gly, only minor changes in protein synthesis were observed (Hasan et al. 2023). Conversely, the Val-decoding tRNAAla caused relatively greater defects in protein synthesis, despite Ala and Val having more similar physiochemical properties than Ala and Gly.
Of the three tRNAAla variants investigated here, Ala misincorporation was most abundant at Val sites, while misincorporation at Glu codons was most impactful on mCherry protein production. The differential impact of protein synthesis could also be attributed to the number of mistranslation sites in the mCherry protein. We confirmed Ala misincorporation at eight of the 24 Glu codons (GAG), four out of 15 Val codons (GUA and GUG), and six of the 12 Thr codons (ACC). In the wild-type cells, there was no Ala mistranslation reported at any of the Glu, Val, or Thr codons in mCherry. While the level of observed misincorporation is certainly impacted by the pool of competing tRNAs, it is also possible that rare or more abundant codons (i.e., codon usage) could also impact mistranslation level. We recently demonstrated that codon usage does not correlate with impact on growth from tRNALeu missense suppressors by surveying a complete library of tRNALeu anticodon mutants (Davey-Young et al. 2024).
At this stage, we conclude that the impacts on mCherry production are the result of amino acid misincorporation in proteins, including those involved in translation and in regulating translation. The compound impact of these misincorporation events likely then leads to defects in protein production. Certainly, other factors could contribute to the protein production defects we observed. In future studies, we will examine mutant tRNA aminoacylation levels (Lant et al. 2018; Hernandez-Alias et al. 2023) and determine if the tRNA mutants impact the broader transcriptome and mRNA levels or if they lead to the generation of tRNA fragments (tRFs) that can in turn regulate protein production (Guzzi et al. 2022).
Translation error and cell growth
In considering each of the tRNAAla mutants that misread codons for Val, Thr, and Glu, we found minor or no defects in cell proliferation. These findings agree with our earlier observations that the Gly-decoding tRNAAla does not cause cytotoxicity in mammalian cells (Hasan et al. 2023). Studies in yeast that surveyed all tRNAAla anticodon variants found that the Thr-decoding tRNAAla induced almost no change in relative growth, while the Val and Glu tRNAAla missense suppressors caused modest but significant decreases in cell growth rate (Cozma et al. 2023). This finding was similar to our results in human cells, where minor defects in viability were recorded at some time points for the Glu- and Val-decoding tRNAs, while the Thr-decoding tRNAAla showed cell proliferation levels indistinguishable from cells with the vector control or with the wild-type tRNA. The impact and level of tRNA-dependent mistranslation in cells may be influenced by several factors; however, the anticodon sequence and the nature of the amino acid substitution are the dominant factors that impact cell growth in the context of tRNA-dependent mistranslation (Cozma et al. 2023; Davey-Young et al. 2024). For example, tRNALeu anticodon mutants with GC-rich anticodons show a more potent impact on growth in yeast, while tRNAs that generate less conservative substitutions according to the Blosum62 amino acid similarity matrix led to generally greater growth defects (Davey-Young et al. 2024).
Applications of missense suppressor tRNAs in medicine
Normal levels of translation error in human cells suggest 0.001%–0.0001% of codons are misread (Drummond and Wilke 2009). The ectopically expressed tRNA variants studied here far exceed this basal frequency and generated mistranslation without substantially compromising cell viability. Although mistranslation is normally considered a mistake in protein synthesis, an emerging field is repurposing tRNA anticodon variants to act as therapeutics by suppressing missense mutations that account for 50% of the alleles that cause genetic disease in humans (Hou et al. 2024; Ward et al. 2024; Tennakoon et al. 2025). For example, the Ala628Thr mutation in the FGFR2 gene causes lacrimo-auriculo-dento-digital syndrome (LADD) as a result of reduced tyrosine kinase activity in cells (Lew et al. 2007). The tRNAAlaAGU(Thr) variant characterized here represents a starting point to a tRNA therapeutic that can effectively reverse this mutation to produce some wild-type protein from a disease-causing allele without impacting cell growth. In addition, missense suppressor tRNA variants may be able to alleviate gain-of-function phenotypes by slowing translation and reducing protein aggregation linked to Huntington's disease (Tennakoon et al. 2025). Engineering missense suppressor tRNAs, inspired by the natural variants we characterized here, will provide opportunities to enable a balanced level of missense suppression to provide just enough of the wild-type protein from a defective allele to restore function without generating too many mistakes and overwhelming protein quality control pathways that maintain cell fitness and protein homeostasis in the context of reduced translation fidelity.
Conclusion
We found three naturally occurring human tRNAAla variants with anticodon mutations that cause mistranslation in cells through misincorporation of Ala at Glu, Val, and Thr codons. Each of the tRNAAla missense suppressors directed mistranslation that caused defects in protein synthesis without causing substantial defects in cell growth. Our data show that natural human tRNAAla variants are able to misincorporate Ala at different codons with phenotypic impacts on protein production that depend on the nature of the replaced amino acid.
MATERIALS AND METHODS
Cloning and plasmid purification
tRNA genes of interest were amplified from genomic DNA of HEK 293T cells via polymerase chain reaction (PCR) following the PfuUltra II polymerase protocol (Agilent). Primers were designed to amplify 500 bp upstream of and downstream from the tRNA genes of interest (Supplemental Table S1). PCR products were separated on a 1.5% agarose gel and visualized with ethidium bromide (EtBr) using a ChemiDoc MP (Bio-Rad). Bands corresponding to the appropriately sized product were excised and purified using the Geneaid Gel Extraction Kit (Geneaid). Purified products were used as templates for subsequent PCR reactions. For wild-type tRNAs, a second round of PCR was performed using primers flanking the tRNA gene 300 bp upstream and downstream. Mutant tRNAs were synthesized by overlap extension PCR, using the first-round products as a template. Mutant tRNA genes were amplified as half molecules, where the mutation was incorporated in overlapping 5′ ends of the primers (Supplemental Table S1). The two overlapping tRNA halves were then used as templates in a second round of PCR, amplifying the full-length mutant tRNA and 300 bp upstream of and downstream from the flanking sequence. PciI or NcoI restriction sites were added to the 5′ end of each primer to allow for PciI or NcoI digestion and subsequent ligation into the PciI site on pPANCherry (Lant et al. 2023) using T4 DNA ligase (NEB). pPANCherry was assembled by removing the EGFP gene (using SpeI and NheI restriction sites and re-ligation) from the EGFP-mCherry construct in WT-PAN (Addgene #99638). mCherry serves as a marker for transfection and protein production levels in individual cells expressing each tRNA variant.
Cell lines and fluorescence microscopy
HEK 293T (ATCC no. CRL-3212) were used for all cell culture experiments, except tRNA sequencing studies (see below) which were conducted in murine N2a neuroblastoma cell (ATCC no. CCL-131) to enable better differentiation of the human tRNA variants from the mouse background. Cells were cultured in Dulbecco's modified Eagle medium (4.5 g/L; Gibco) with 10% fetal bovine serum (Gibco by Life Technologies) and 1% penicillin-streptomycin (P: 100 IU mL−1; S: 100 µg mL−1; Wisent Bioproducts). Cells were maintained and grown at 37°C with 5% CO2 and humidity. Cells were transfected with plasmids using Lipofectamine 3000 (Invitrogen, Thermo Fisher Scientific), according to the manufacturer's instructions for the appropriate plate size. Fluorescent images were taken using the EVOS FL Auto Imagining System (Thermo Fisher Scientific). mCherry fluorescence was visualized using brightfield and also the EVOS LED RFP light cube (excitation 542 ± 20 nm; emission 593 ± 40 nm) to capture mCherry fluorescence. Cell images were captured 24 h after transfection and the level of mCherry fluorescence per cell was determined using a custom semi-automated ImageJ macro (Lant et al. 2023).
Cell harvesting and lysis
HEK 293T cells were transfected with plasmids containing mCherry and the indicated tRNA variant alleles in 6-well plates with Lipofectamine 3000 (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's protocol. After 24 h, cells were harvested by aspirating the existing media and incubating the cells with 1× phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) supplemented with 10 mM ethylenediaminetetraacetic acid (EDTA) for 10 min. Cells were then resuspended and transferred to a sterile 1.5 mL microcentrifuge tube and centrifuged at 500g for 10 min. The supernatant was aspirated, and cell pellets were stored at −80°C until use. A 50 µL volume of radio immunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, and 1 tablet/10 mL complete mini EDTA-free Protease Inhibitor Cocktail) was added to cell pellets and centrifuged for at 21.1g for 20 min at 4°C to lyse the cells. Supernatants were transferred to sterile 1.5 mL microcentrifuge tubes. To determine lysate protein concentrations and appropriate loading volumes, the Pierce bicinchoninic acid (BCA) assay (Thermo Fisher Scientific) was used.
Western blotting
Samples of 20 µg of protein from cell lysates prepared as above were separated using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 15% acrylamide) at 70 V for 30 min followed by 120 V for 2 h. Proteins were transferred from gel to methanol-activated polyvinylidene difluoride (PVDF) membranes using a Trans-Blot Turbo transfer system (25 V, 1.3 A, 15 min; Bio-Rad). All blocking and wash solutions were prepared using tris-buffered saline (TBS; 50 mM Tris-HCl, pH 7.5, 150 nM NaCl). Following transfer, membranes were incubated in blocking solution (5% bovine serum albumin [BSA], 0.1% Tween-20, 1× TBS) for 1 h. Membranes were then incubated overnight at 4°C in primary antibody at dilutions of 1:5000 for anti-GAPDH (Sigma-Aldrich MAB374) and 1:1000 for anti-mCherry (AbCam ab213511). Membranes were washed the following day with 1% BSA in 1× TBS and 0.1% Tween-20 3 × 10 min, then incubated at room temperature for 2 h in in IRDye 680RD anti-mouse (GAPDH; Li-Cor Inc., 926–68070) or IRDye 800CW donkey anti-rabbit (mCherry; LiCor Inc., 926–32213) fluorescent secondary antibodies at dilutions of 1:5000. Membranes were then washed 3 × 15 min each in 1× TBS and 0.1% Tween-20 and once in 1× TBS for 10 min. Membranes were then imaged using fluorescence on a ChemiDoc MP imager (Bio-Rad). Specific antibodies for eIF2α (Invitrogen, anti-EIF2-alpha, #AHO0802), p-eIF2α (Abcam ab32157 Anti-EIF2S5, phospho S51) were used to determine p-eIF2α levels. The IRDye 800CW donkey anti-rabbit secondary 600 antibody was used to detect the eIF2α antibodies and the vinculin (anti-vinculin produced in mouse, V9131, Sigma) loading control was visualized using chemiluminescence as described before (Tennakoon et al. 2025). Briefly, secondary ECL anti-mouse IgG horseradish peroxidase linked whole antibody (from sheep, GPR NXA931V, GE Healthcare UK) was used for detection of anti-vinculin and chemiluminescence was conducted using the Clarity Western Blotting ECL Substrate (Bio-Rad) per manufacturer's instructions and imaged using a ChemiDoc MP imager (Bio-Rad) as above.
Cell proliferation assay
Cell proliferation was measured using the Cell Counting Kit (CCK)-8 (Sigma-Aldrich 96992) assay, which is based on the conversion of water-soluble tetrazolium salt, WST-8 [2-(2-methoxy-4-nitrophenyl)−3-(4-nitrophenyl)-5-(2,4-disulfophenyl)−2H-tetrazolium, monosodium salt] to a water-soluble formazan dye upon reduction in the presence of an electron carrier by dehydrogenases. HEK 293T cells were transfected in 96-well plates with Lipofectamine 3000 (Invitrogen, Thermo Fisher Scientific) according to the manufacturer's protocol. At 6, 24, 48, and 72 h after transfection, cells were treated with 10 µL of the CCK-8 reagent, then incubated for 4 h at 37°C with 5% CO2 and humidity. Absorbance was measured at 450 nm using Cytation C10 imaging plate reader (BioTek).
Mass spectrometry
mCherry protein was immunoprecipitated from HEK 293T cells expressing wild-type tRNAAlaCGC, tRNAAlaCGC G35T, tRNAAlaUGC G35A, and tRNAAlaAGC C36T at 24 h after transfection. For each transfected plasmid, biological triplicates of cells grown in 10 cm plates were harvested and lysed with ice-cold lysis buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.5% Nonidet P40 Substitute, pH adjusted at 4). mCherry was purified through immunoprecipitation using the RFP-Trap Agarose kit (ChromoTek), according to the manufacturer's instructions. An acid elution was performed three times per sample in the final step to elute mCherry. The protein samples were digested with trypsin and a separated by nanoflow liquid chromatography using an UltiMate 3000 chromatography system (Thermo Fisher Scientific), then injected into the Thermo Orbitrap Fusion Lumos (Thermo Fisher Scientific) to identify Ala misincorporation in mCherry proteins. Detailed mass spectrometry methods can be found in the Supplemental Material.
tRNA sequencing
Samples were prepared for tRNA sequencing exactly as before (Lant et al. 2023). N2a cells were transfected with plasmids co-expressing tRNAAlaAGC (Ala-AGC-6-1) or the variant tRNA-Ala-ACC (Ala-AGC-6-1 G35C) and mCherry as a transfection marker. After 48 h, cells were harvested and resuspended in TRIzol reagent (Thermo Fisher Scientific) and then stored in liquid nitrogen before sending to Arraystar, Inc. for tRNA sequencing using the hydro-tRNA-seq method (Gogakos et al. 2017). Normalized hydro-tRNA-seq read counts are listed in Supplemental Data File 1. The file lists read counts that uniquely map to a specific tRNA as well as multimapped reads that align to transcripts from multiple identical or nearly identical tRNA genes. The levels of our expressed human tRNA genes can be confidently identified among read counts that uniquely map to the human wild-type or mutant tRNAAla sequence. Since most of the mouse background tRNAs are expressed from two or more identical gene copies, the multimapped read counts provide an accurate estimate of the level of genomically encoded mouse tRNAs in N2a cells.
DATA DEPOSITION
Mass spectra were deposited in the PRDIE database. Project accession: PXD053463. Project Webpage: https://www.ebi.ac.uk/pride/archive/projects/PXD053463. FTP Download: https://ftp.pride.ebi.ac.uk/pride/data/archive/2025/03/PXD053463.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to Ilka Heinemann for insightful discussion and suggestions. This work was supported by the Natural Sciences and Engineering Research Council of Canada (04282 and 580241 to P.O.), Canada Research Chairs (232341 to P.O.), and the Canadian Institutes of Health Research (165985 to P.O.). P.O. is also supported by the Huntington Society of Canada Research Chair at Western University.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080450.125.
- Received March 5, 2025.
- Accepted March 6, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Rasangi Tennakoon is the first author of this paper, “Natural human tRNAAla anticodon variants mistranslate the genetic code.” Rasangi holds an MSc in biochemistry from Western University in London, where she explored the effect of human tRNA missense suppressors in nature and Huntington's disease. She is continuing her research in the field at the University of Toronto as a PhD student in chemistry, where she is focusing on the role of aminoacyl-tRNA synthetase mutations in white matter brain disorders.
What are the major results described in your paper and how do they impact this branch of the field?
Our study reveals that rare human tRNA variants can introduce mistakes during protein production, leading to defects in how cells make proteins. We found that some of these tRNAs cause alanine to be inserted where it shouldn't be, and while certain mistakes happened more often than others, the severity of their impact depended on which amino acid was replaced. This suggests that even small changes in tRNA can affect how proteins are made in cells. Our findings provide new insight into how naturally occurring tRNA variants can influence translational fidelity in human cells. This work expands our understanding of the potential consequences of tRNA mutations in the human population and underscores the need to further explore their role in disease and cellular function. By identifying specific mistranslation events that impair protein synthesis, our study also lays the groundwork for future research into how cells cope with or exploit tRNA-driven mistranslation.
What led you to study RNA or this aspect of RNA science?
My interest in RNA science began during my undergraduate studies, where I became fascinated by how RNA acts as both an information carrier and a functional molecule. This curiosity deepened during my MSc, where I worked on tRNA-based therapeutics for neurodegenerative diseases, as well as tRNA variations in nature. Being part of both of these projects showed me firsthand how RNA can be harnessed to correct genetic mutations in ways we're only beginning to understand. That experience solidified my interest in RNA's therapeutic potential and led me to explore how disruptions in RNA processes contribute to disease. In my current research, I'm particularly interested in RNA–protein interactions and their role in mitochondrial function, which has direct implications for rare neurological disorders. Studying RNA offers a unique intersection of fundamental biology and translational potential, and that's what keeps me excited about this field.
If you were able to give one piece of advice to your younger self, what would that be?
I would tell my younger self to embrace the unexpected turns in research. Science rarely follows a straight path, and some of the most exciting discoveries come from experiments that don't go as planned. Instead of seeing setbacks as failures, I've learned to treat them as opportunities to ask better questions and refine my approach. Also, don't be afraid to reach out to others, as collaborations and conversations often lead to insights you wouldn't have found on your own. Most importantly, enjoy the process! The excitement of discovery is what makes the challenges worthwhile.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
During this work, I was incredibly fortunate to be mentored by my Principal Investigator, Dr. Patrick O'Donoghue, whose passion for science is truly infectious. His enthusiasm for tRNA biology made every discussion exciting, and his curiosity-driven approach showed me how much fun research can be. He encouraged me to explore new ideas, and approach challenges with creativity rather than frustration. His excitement for discovery was a constant reminder of why I love doing science.
Beyond that, my labmates played a huge role in shaping my approach to research. Science is rarely a solo endeavor, and I was lucky to be surrounded by colleagues who were just as eager to discuss ideas, troubleshoot experiments, and celebrate the small wins along the way. Their support made the challenges of research more rewarding, and I'm grateful for the collaborative environment that helped bring this project to life.
What are your subsequent near- or long-term career plans?
In the near term, I'm continuing my research as a PhD student at the University of Toronto, where I'm investigating the role of aminoacyl-tRNA synthetases in white matter brain disorders, and establishing tRNA sequencing methods in the lab to better understand how RNA processing is disrupted in disease.
Long term, I hope to contribute to the growing field of RNA therapeutics, whether in academia or industry. The potential of RNA-based treatments is still unfolding, and I want to be part of efforts to translate fundamental RNA biology into real-world therapies. My goal is to work at the interface of discovery and application, advancing our understanding of RNA function while developing innovative strategies to treat disease.








