
DIS3L, cytoplasmic exosome catalytic subunit, is essential for development but not cell viability in mice
- Michał Brouze1,2,
- Marcin Szpila3,
- Areta Czerwińska1,4,
- Wiktor Antczak1,
- Seweryn Mroczek1,5,
- Tomasz M. Kuliński1,2,
- Anna Hojka-Osińska6,
- Dominik Cysewski2,8,
- Olga Gewartowska3,
- Dorota Adamska2,7,
- Jakub Gruchota1,2,
- Ewa Borsuk1,9 and
- Andrzej Dziembowski1,2,9
- 1Laboratory of RNA Biology, International Institute of Molecular and Cell Biology in Warsaw, Warsaw 02-109, Poland
- 2Laboratory of RNA Biology and Functional Genomics, Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw 02-106, Poland
- 3Genome Engineering Facility, International Institute of Molecular and Cell Biology in Warsaw, Warsaw 02-109, Poland
- 4Department of Cytology, Institute of Developmental Biology and Biomedical Sciences, Faculty of Biology, University of Warsaw, Warsaw 02-096, Poland
- 5Institute of Genetics and Biotechnology, Faculty of Biology, University of Warsaw, Warsaw 02-096, Poland
- 6Bioinformatic Facility, International Institute of Molecular and Cell Biology in Warsaw, Warsaw 02-109, Poland
- 7Genomics Core Facility, Centre of New Technologies, University of Warsaw, Warsaw 02-097, Poland
- 8Clinical Research Centre, Medical University of Bialystok, Białystok 15-276, Poland
- 9Department of Embryology, Institute of Developmental Biology and Biomedical Sciences, Faculty of Biology, University of Warsaw, Warsaw 02-096, Poland
- Corresponding author: adziembowski{at}iimcb.gov.pl
-
Handling editor: Fatima Gebauer
Abstract
Among numerous enzymes involved in RNA decay, processive exoribonucleases are the most prominent group responsible for the degradation of entire RNA molecules. The role of mammalian cytoplasmic 3′-5′ exonuclease DIS3L at the organismal level remained unknown. Herein, we established knock-in and knockout (KO) mouse models to study DIS3L functions in mice. DIS3L in mice is indeed a subunit of the cytoplasmic exosome complex, the disruption of which leads to severe embryo degeneration and death in mice soon after implantation. These changes could not be prevented by supplementing extraembryonic tissue with functional DIS3L through the construction of chimeric embryos. Preimplantation Dis3l−/− embryos were unaffected in their morphology and ability to produce functional embryonic stem (ES) cells, showing that DIS3L is not essential for cell viability. There were also no major changes at the transcriptome level for both ES cells and blastocysts, as revealed by RNA-seq experiments. Notably, however, Dis3l KO led to inhibition of global protein synthesis. These results point to the essential role of DIS3L in mRNA metabolism, which is crucial for proper protein synthesis during embryo development.
Keywords
INTRODUCTION
In recent years, RNA decay pathways emerged as the RNA control mechanisms of importance comparable to transcription regulation or posttranscriptional modifications. While RNA-degrading enzymes consist of different types and families, processive exoribonucleases are the most prominent group, as they are responsible for the degradation of entire transcripts during mRNA turnover or in quality control pathways. The mammalian genomes encode six processive exoribonucleases, divided into three families. The first family of 5′ to 3′ exonucleases consists of XRN1 and XRN2, expressed, respectively, in the cytoplasm (Bashkirov et al. 1997) and nucleus (including nucleolus) (Wang and Pestov 2011). The second family consists of three hydrolytic 3′ to 5′ exoribonucleases. Two of them, nuclear DIS3 (which is also endowed with endoribonucleolytic activity) and cytoplasmic DIS3L, are parts of the ring-shaped exosome complex (Staals et al. 2010; Tomecki et al. 2010), which by itself remains inactive in eukaryotes. DIS3L2 exoribonuclease is also expressed in the cytoplasm, albeit in a free, monomeric form (Lubas et al. 2013). Finally, phosphorolytic 3′ to 5′ ribonuclease PNPase is responsible for RNA decay in mitochondria (Piwowarski et al. 2003). While the enzymes mentioned above are responsible for the bulk of RNA decay, plenty of less potent enzymes contribute to RNA metabolism, including nucleolar enriched, exosome-associated distributive exoribonuclease EXOSC10/RRP6 (Liu et al. 2006; Januszyk et al. 2011). The substrates and mechanisms of recognition appearing in concert with activatory complexes are quite well established for those enzymes thanks to years of biochemistry, structural biology, and functional studies using cell lines. However, the knowledge about their role in vivo is more fragmentary.
The DIS3 family of proteins, specifically, was shown to be associated with cancer. Mutation of DIS3L2, a nuclease responsible for the degradation of ARE-containing transcripts (Lubas et al. 2013) and oligouridylated ncRNAs in their quality control pathway (Pirouz et al. 2016), is the leading cause of Perlman syndrome and, consequently, Wilms tumor (Astuti et al. 2012; Chang et al. 2013; Hunter et al. 2018). Exosome-associated DIS3, which in the nucleus targets pervasive transcription products (promoter upstream transcripts and enhancer RNAs), snoRNAs, and premature termination products (Szczepinska et al. 2015), is among the most frequently mutated genes in multiple myeloma patients (Chapman et al. 2011; Lohr et al. 2014; Weissbach et al. 2015). Unlike DIS3L2, however, DIS3 is an essential gene in mice (Kuliński et al. 2023) and flies (Hou et al. 2012). This lethality can be attributed to the exoribonucleolytic activity of DIS3, as mutations inactivating or inhibiting this enzyme lead to early embryonic lethality in mice (Kuliński et al. 2023, 2024). Interestingly, the second nuclear exosome-bound exoribonuclease, RRP6, was recently identified to be essential for mouse embryo development past the morula stage (Petit et al. 2022).
Regarding DIS3L, our knowledge is minimal and comes mainly from stable cell lines. The Mus musculus Dis3l gene (Chromosome 9: 64,214,038–64,248,570) has eight known splice variants but notably encodes only one canonical isoform with the RNB catalytic domain localized in position 465–816 of the amino acid sequence. In the studied context of human cells, DIS3L was shown to act solely as an exoribonuclease associated specifically with the cytoplasmic exosome complex, where it can be involved in the degradation of 28S rRNA (Staals et al. 2010; Tomecki et al. 2010). Notably, essentially nothing is known about the role of DIS3L at the organismal level, with the only reporting showing that conditional knockout (KO) of Dis3l recently does not influence the fertility of male mice (Wang et al. 2024).
Here, we present the results of our study of mammalian DIS3L in vivo through the analysis of the KO mutation phenotype of the Dis3l gene in mice. We demonstrate that DIS3L is essential for mouse embryo development beyond embryonic day E6.5. The lethal phenotype in Dis3l KO embryos could not be rescued by supplementation of wild-type (WT) cells to extraembryonic tissue. Although preimplantation embryos lacking functional DIS3L were able to produce embryonic stem (ES) cells, this occurred with reduced efficiency. Dis3L KO ES cells accumulated a large number of mRNA, as shown by RNA-seq. Curiously, the accumulation of some of those transcripts was not reflected by higher protein levels. Further examination showed overall impaired protein production in Dis3L KO preimplantation embryos.
RESULTS
DIS3L is a part of the cytoplasmic exosome in mice
The composition and functionality of the mammalian cytoplasmic exosome complex and its catalytic subunit, DIS3L, were previously analyzed only in highly modified cell lines (HEK293 and HeLa) (Tomecki et al. 2010). Thus, we have generated a knock-in mouse line with DIS3L C-terminally tagged with GFP in the endogenous locus using the CRISPR/Cas9 system to study it in a more physiologically relevant system. sgRNA was designed to cut the DNA exactly before the stop codon of exon 17, and the GFP sequence was inserted by providing a GFP-encoding DNA repair template with 60 amino acid-long overhangs facilitating homology-directed repair (Supplemental Fig. S1). These mice in the homozygous state were healthy, with no apparent developmental, morphological, or physiological abnormalities. Then, we used protein extracts from three DIS3LGFP/GFP mice livers to confirm DIS3Ls association with cytoplasmic exosome complex in mice cells by performing immunoprecipitation on anti-GFP resin. Extracts from one WT mouse liver that did not contain a GFP tag and could not specifically bind to anti-GFP resin served as a background control of unspecific resin binding. Out of all 173 identified proteins (Fig. 1A; Supplemental Table S1), quantitative mass spectrometry analysis showed 98 proteins significantly enriched in the averaged results of three Dis3lGFP/GFP samples (identifiable as a cluster of proteins in Fig. 1A with peptide abundance and peptide specificity statistics above 2.5). From this set, we discarded all keratins, immunoglobulins, hemoglobin, and unidentified proteins as obvious nonspecifically bound peptides, narrowing the enriched set down to 82 proteins (Fig. 1B; Supplemental Table S1). Within this set, DIS3L co-precipitated with all nine core exosome proteins with the highest abundance, as well as one of the cytoplasmic exosome's cofactors—HBS1L.
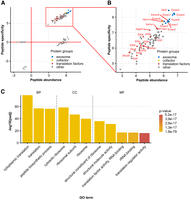
Analysis of proteins interacting with DIS3L in mice. (A) Distribution of proteins identified by mass spectrometry analysis of DIS3L-GFP co-precipitated peptides from Dis3lGFP/GFP protein liver extracts presented as peptide abundance [log10(mean LFQ intensity in GFP samples/molecular weight of identified protein)] relative to peptide specificity [log10(mean LFQ intensity in GFP samples/LFQ intensity in WT sample)]. (B) Blow out of specifically enriched proteins section of A marked by red rectangle. All nine mammalian core exosome proteins co-precipitated with DIS3L-GFP as the most specific and abundant proteins detected in the experiment. Also marked are all identified factors involved in translation regulation. (C) Functional enrichment analysis of proteins co-precipitated with DIS3L-GFP. Only several terms with the highest P-value are presented. Identified terms are grouped by data source subgroups: biological process (BP), molecular function (MF), and cellular component (CC). A full list of identified terms is available in Supplemental Fig. S2.
The majority of other proteins co-precipitating with the DIS3L are associated with the protein synthesis apparatus. This includes 32 ribosomal proteins and 15 proteins acting as translational factors (including nine proteins of the EIF family of elongation initiation factors). This is also supported by the functional annotation analysis of identified proteins—molecular function, biological process (BP), and cellular component gene ontology terms identified with the highest P-value are all associated with the translation process and its regulation (Fig. 1C; Supplemental Fig. S2).
Thus, we confirmed that DIS3L is indeed a subunit of the exosome complex, which in the cytoplasm of murine cells is associated with the translation machinery, mainly the ribosome and EIF2 family of proteins.
Dis3l−/− embryos die at embryonic day E6.5
Having established that DIS3L in mice is indeed a part of the cytoplasmic exosome complex, we aimed to determine its role in shaping mice transcriptome and its potential influence on mice physiology. Using the CRISPR/Cas9 system, we induced cleavage and random indel mutation in exon 10 of the Dis3l gene (Supplemental Fig. S3A), generating a mice line bearing c.1258_1259ins169 frameshift mutation resulting in a premature stop codon after 59 amino acids (His421fs59*) (Supplemental Fig. S3B) in a sequence encoding the exoribonucleolytic RNB domain, leading to disruption of the protein functionality. In our efforts to breed the Dis3l−/− mouse line, we were unable to obtain mice bearing homozygotic mutation. Therefore, we attributed it to the embryo lethality of homozygotic Dis3l mutation. Genotyping embryos at different developmental stages obtained from breeding Dis3l+/− males and females further confirmed the lethality.
Preimplantation development was unaffected in the progeny of those matings. The genotype ratio of obtained blastocysts did not differ significantly from the expected Mendelian ratio, with 28.95% Dis3l−/− and 31.58% Dis3l+/+ among them (Fig. 2A). Lack of the functional catalytic subunit of the cytoplasmic exosome also did not disturb the overall development of a preimplantation embryo. Staining all nuclei and immunostaining of specific markers of different cell lineages in developing blastocysts (CDX2 in trophectoderm, GATA4 in primitive endoderm, epiblast as the rest of unstained cells) also allowed us to assess the overall development of embryos lacking functional DIS3L. Both WT and KO blastocysts had a similar mean total cell count (84.8 ± 6.27 and 77.8 ± 8.13, respectively, Fig. 2B), and the ratio of all three cell lineages (i.e., trophectoderm, epiblast, and primitive endoderm) present at that stage of development (Fig. 2C,D).
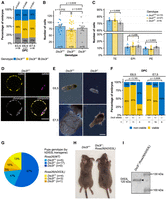
DIS3L KO phenotype in mice embryo. (A) Genotype distribution in embryos of different developmental stages obtained from Dis3l+/− × Dis3l+/− matings. (B) Total cell count of blastocysts in relation to the genotype. (C) Percentage ratio of trophectoderm (TE), epiblast (EPI), and primitive endoderm (PE) in blastocysts in relation to the genotype. (D) Example of Dis3l+/+ embryo staining used to determine the number of differentiated cells in the blastocyst. CDX2 marker of trophectoderm in yellow, GATA4 marker of primitive endoderm in magenta, and chromatin in gray. The number of epiblast cells was determined by subtracting the number of CDX2 and GATA4 positive cells from the total cell number. Scale bar, 50 µm. (E) Example of incorrectly developing Dis3l−/− embryos 6.5 and 7.5 dpc (days post coitum) compared to valid Dis3l+/+ embryos. Scale bar, 200 μm. (F) The ratio of viable to nonviable embryos of different genotypes at 6.5 and 7.5 dpc. (G) Distribution of genotypes of pups born from Dis3l+/−; Rosa26(hDIS3L) × Dis3l+/− matings. In blue scale pups without a transgene, in orange pups with a transgene. N denotes the number of pups of a given genotype born. (H) The only Dis3l−/−; Rosa26(hDIS3L) mice born (right) compared to WT mice (left) of the same sex and age (5 weeks old). (I) Western blot analysis of expression of human Dis3l transgene in Dis3l−/−; Rosa26(hDIS3L) mice tissue. Black bars with kilodalton values on the right mark protein ladder band sizes, protein names with predicted size values on the left mark the target of the used antibody, and black arrowhead marks specific bands detected. In all panels, individual data points represent single blastocysts analyzed, bars represent mean value, and error bars represent SEM.
We observed the first signs of impaired development early after implantation. On 6.5 and 7.5 dpc of embryo development, we identified only 14.29% and 16.67% Dis3l−/− embryos, respectively, although the entire genotype distribution on both stages remained within a range similar to the Mendelian ratio (Fig. 2A). However, the lack of functional DIS3L has already affected embryo morphology and developmental potential. While on embryonic day 6.5, >85% of Dis3l+/− and Dis3l+/+ embryos were properly developed, two out of three identified KO embryos were smaller and deformed compared to their WT counterparts. This effect became stronger at embryonic day 7.5 when all six identified KO embryos were highly degenerated and nonviable for further development, without any change in the viability of the embryos of other genotypes (Fig. 2E,F).
To confirm that the lack of DIS3L causes the embryo lethality effect, we decided to perform a rescue experiment targeting human WT DIS3L into the safe harbor ubiquitously expressed Rosa26. For this, embryos from Dis3l+/− × Dis3l+/− matings were microinjected with Cas9 mRNA, gRNA targeting the Rosa26 locus, and dsDNA coding sequence of human hDIS3L (Supplemental Fig. S4). Out of 37 pups born, one had the correct insertion of hDIS3L. The founder had a heterozygotic mutation of mouse Dis3l and was then crossed with Dis3l+/− females to investigate whether Dis3l−/−; Rosa26(hDIS3L) pups would be born. Out of 30 pups born from these matings, 22 did not carry the hDIS3L transgene, which suggests the potential toxicity effect of hDIS3L cDNA expression from the Rosa26 locus. Nevertheless, out of eight pups carrying the transgene, one indeed had homozygotic Dis3l mutation (Fig. 2G). This pup did not suffer apparent developmental disorders and was morphologically indistinguishable from its littermates, both with and without hDIS3L (Fig. 2H). Western blot analysis of DIS3L expression in Dis3l−/−; Rosa26(hDIS3L) tissue also showed full-length DIS3L protein product (Fig. 2I), proving the successful rescue experiment and specificity of our methodology.
It is concluded that the Dis3l KO mutation leads to embryo lethality at the implantation stage, but its cause remains to be established.
Embryo lethality of Dis3l KO cannot be rescued by supplementing WT extraembryonic tissues
As Dis3l KO early embryos fail to develop beyond early implantation stages, we decided to test the indispensability of DIS3L for the development of extraembryonic tissues responsible, among other functions, for proper implantation. To this end, we constructed chimeric embryos. Individual 4- to 8-cell embryos (of unknown genotype) from Dis3l+/− × Dis3l+/− mating were joined with two 2-cell tetraploid WT embryos (Fig. 3A). In such a model, tetraploid cells contribute only to developing extraembryonic tissue—compensating for the lack of DIS3L in KO embryos and potentially rescuing the phenotype. Chimeric embryos were then transferred to recipient females previously mated with vasectomized males. All females were euthanized 18 days after an embryo transfer to account for all implanted embryos and resorption sites in the uterus.

Development of chimeric embryos. (A) Schematic representation of chimeric embryo construction. Diploid 4-cell embryos from Dis3l+/− × Dis3l+/− mating (white) were covered with two previously prepared tetraploid 2-cell WT embryos (magenta). (B) Chimeric embryos implantation rate after transfer to recipient females and percentage of those implanted embryos that fully developed to term. (C) Genotype distribution of chimeric pups developed to term. No Dis3l−/− embryos developed fully.
Out of the total of 98 chimeric embryos transferred to recipient females, 63% of embryos were successfully implanted in the uterus. However, only 44% of those embryos developed to term, and the other 56% (35 out of 62) died and were subsequently resorbed, leaving only implantation sites (Fig. 3B). Genotyping of developed embryos revealed that our attempt did not produce any Dis3l−/− embryos, resulting in 67% of Dis3l+/− and 33% of Dis3l+/+ pups developed to term (Fig. 3C), all of them without any morphological signs of disturbed development. These results exclude the notion that the implantation process and, more broadly, the development of the trophectoderm is the only cause of embryo lethality.
Dis3l KO does not affect cell viability
Having established that Dis3l−/− embryos cannot develop properly after implantation and degenerate soon after, even with WT DIS3L supplemented in extraembryonic tissues, we assumed lethality may be related to changes at the transcriptome level at this developmental stage. Thus, we next focused on analyzing the effect of Dis3l KO mutation on the developmental potential of the preimplantation embryo.
To test the potential of the inner cell mass (ICM) of the preimplantation embryo to develop the embryo body, we first derived ES cell lines from blastocysts obtained from Dis3l+/− × Dis3+/− matings. We observed that Dis3l−/− ES lines are obtained with a slightly lower ratio compared to Dis3l+/− and Dis3l+/+ embryos. Of 28 blastocysts from which we derived ES cells, 14.29% were Dis3l−/− (Fig. 4A). Notably, both KO and WT ES cells were also able to form embryoid bodies (EBs). However, out of three tested KO cell lines, two produced EBs that, on days 2 and 5 of culture, displayed a decrease in expression of Foxa2 and Tbxt transcripts coding proteins responsible for differentiation of endodermal and mesodermal organs, respectively. In addition, all three lines also displayed an atypical expression pattern of Pax6, involved in ectoderm development, with a spike of transcript accumulation on day 2 of culture and a drop below the starting level on day 5 (Fig. 4B; Gersdorff et al. 2005).
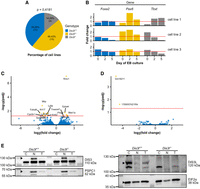
Phenotype and transcriptome analysis of Dis3l KO ES cells. (A) Genotype distribution of ES cell lines derived from blastocysts from Dis3l+/− × Dis3l+/− matings. (B) Transcript level fold change of three germ layer-specific differentiation factors: Foxa2, Pax6, and Tbxt, in EBs through 5 days of culture in three obtained Dis3l−/− ES cell lines related to mean result of three WT cell lines, obtained by RT-qPCR experiment. The red horizontal line marks a fold change of 1, meaning no change compared to WT. (C) Differential expression analysis of protein-coding genes identified in RNA-seq results of three Dis3l−/− and three Dis3l+/+ ES cell lines. Positive and negative log2(fold change) value means upregulation and downregulation, respectively, of transcript in KO cells. Red horizontal line marks adjusted P-value of 0.05 with transcripts above it considered significantly changed in Dis3l−/− ES cell lines compared to Dis3l+/+ ones. (D) Differential expression analysis of long noncoding RNAs genes identified in RNA-seq results of three Dis3l−/− and three Dis3l+/+ ES cell lines. Positive and negative log2(fold change) value means upregulation and downregulation, respectively, of transcript in KO cells. Red horizontal line marks adjusted P-value of 0.05 with transcripts above it considered significantly changed in Dis3l−/− ES cell lines compared to Dis3l+/+ ones. (E) Western blot analysis of the presence and localization of DIS3 and DIS3L proteins in the nucleoplasm (N), cytoplasm (C), and total protein extract (T) fractions of Dis3l+/+ and Dis3l−/− ES cells. Black bars with molecular weight values (in kilodaltons) on the left mark protein ladder band sizes, protein names with predicted size values on the right mark the targets of used antibodies, black arrowheads mark specific bands detected, PSPC1 protein served as a nucleoplasm fraction control, and EIF2α served as cytoplasm fraction control.
Having established the cellular model, we investigated the molecular basis of embryonic lethality under the DIS3L absence phenotype, using the RNA-seq after Ribo Zero rRNA depletion. We sought to identify genes specifically regulated by DIS3L in the derived ES cell lines since DIS3L, as a part of the cytoplasmic exosome ribonuclease subunit, can take part in the degradation of a wide range of RNA molecules. Looking globally, the transcriptome of Dis3l−/− ES cell lines showed no dramatic changes compared to the transcriptome of Dis3l+/+ ES cell lines. To our surprise, the differential gene expression analysis revealed both genes with increased and decreased expression levels when comparing Dis3l−/− with Dis3l+/+ ES cell lines. With in-depth analysis of the different RNA type transcriptome profiles, among protein-coding genes we identified only six genes with increased and nine with decreased expression levels in Dis3l−/− ES cell lines (Fig. 4C); whereas among the lncRNA coding genes, there were only two downregulated upon Dis3l KO (Fig. 4D). Overall, we could not observe any meaningful defect of global RNA degradation in the Dis3l−/− ES cell lines. However, it should be noted that due to very high variability between samples, some of the genes showed inconsistent changes between the replicates. This is also in line with the described individual gene expression levels analyzed by RT-qPCR for the same cell line model. Such variability may be due to the process of differentiation being initiated at the time of cell collection. As the size of the effect of transcriptome deregulation stood in contrast with the expected effect of Dis3l mutation, we decided to check separate cellular compartments of KO ES cells for traces of the full-length DIS3L protein and possible localization of nuclear DIS3 to the cytoplasm, which could produce a compensation effect. In western blot assay on nucleoplasm and cytoplasm protein extracts, we detected DIS3 only in the nucleus of both WT and KO cells, while DIS3L was present exclusively in the cytoplasm of WT cells, with no signs of expression in Dis3l KO cells (Fig. 4E).
It is concluded that although it is possible to derive and maintain a culture of Dis3l KO ES cells, and those cells have very subtle changes on the transcriptome level, they display some abnormalities affecting differentiation factors, which are difficult to explain mechanistically.
Dis3l KO mutation negatively affects global protein production in blastocysts
Since the analysis of ES cells did not reveal the cause of the lethality induced by the Dis3l KO mutation, and RNA-seq from those cells yielded results contrary to the expected effects of DIS3L absence, we returned to studying embryos at the blastocyst stage as more physiologically relevant models. Using a modified single-cell RNA-seq protocol based on oligo(dT)20 priming, we sequenced the RNA of three Dis3l−/− and three Dis3l+/+ blastocysts. Similarly to ES cell sequencing, we identified only a small number of differentially expressed protein-coding transcripts (15 out of 17,368 all identified). This time, however, only two of them were downregulated, and 13 were upregulated (differential expression analysis, Padj < 0.05). All these transcripts were also changed at least twofold (|log2foldchange| > 1) (Fig. 5A), and the upregulated ones may represent direct substrates of the cytoplasmic exosome. We also investigated potential changes in polyadenylated rRNA species assumed to undergo RNA surveillance. However, we observed no significant changes (Fig. 5B). Consequently, we focused only on protein-coding genes. Out of four BP terms that the gene ontology analysis annotated as the identified deregulated transcripts, three were regarded as cell death (the same seven transcripts were annotated to each). Additionally, three transcripts were successfully annotated to five different human phenotype (HP) terms (one transcript annotated to five terms, one to four, and one to three), four of which are related to developmental disorders (Fig. 5C).
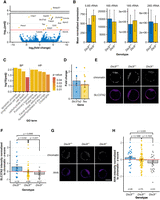
Transcriptome and translation analysis in Dis3l KO blastocysts. (A) Differential expression analysis of RNA-seq results of three Dis3l−/− and three Dis3l+/+ blastocysts. Positive and negative log2(fold change) value means upregulation and downregulation, respectively, of transcript in KO embryos. Red horizontal line marks adjusted P-value of 0.05 with transcripts above it considered significantly changed in KO embryos compared to WT ones. (B) Mean normalized expression of 4 rRNA species with sufficient coverage in RNA-seq of three Dis3l−/− and three Dis3l+/+ blastocysts. No significant changes were identified. Bars represent mean value, and error bars represent SD. (C) Functional enrichment analysis of significantly deregulated transcripts identified in blastocyst RNA-seq. Identified terms are grouped by data source subgroups: BP and HP. (D) Fold change of two of significantly downregulated transcripts identified in blastocyst's RNA-seq of Dis3l−/− embryos, obtained by RT-qPCR. Each data point represents fold change for one KO blastocyst. The horizontal red line marks a fold change of 1, meaning no change. (E) Example of SLC37A2 protein immunofluorescence staining used to measure protein level in blastocysts. (F) Normalized z-score value of SLC37A2 protein level in blastocysts depending on the genotype. Each data point represents one blastocyst. (G) Example of protein synthesis labeling with methionine analog AHA used to determine the amount of newly produced peptides in blastocysts. (H) Normalized z-score value of detected AHA-containing proteins’ levels in blastocysts depending on the genotype. In E and G, each data point represents one blastocyst. Scale bars, 50 µm. Bars represent mean value, and error bars represent SEM.
Notably, two genes upregulated in Dis3l−/− blastocysts are directly involved in preimplantation and early postimplantation development. Testin (Tes) is known for interacting with the cytoskeleton and affects actin dynamics to facilitate cell movements and migration (Coutts et al. 2003; Garvalov et al. 2003). In blastocysts, it is highly expressed in ICM and polar trophectoderm and after implantation at embryonic day E6.5 in the epiblast, anterior visceral endoderm precursors, and extraembryonic ectoderm (Crompton et al. 2007). SLC37A2 is a glucose-6-phosphate antiporter responsible for its transport from cytoplasm to ER lumen (Pan et al. 2011)—one of the steps in the metabolism of the primary blastocyst energy source—glucose (Gardner and Leese 1986; Martin and Leese 1995). RT-qPCR analysis of those two transcripts’ levels from seven Dis3l−/− and ten Dis3l+/+ freshly isolated blastocysts, revealed that while the Slc37a2 transcript was accumulated in most of the KO samples, with a mean fold change of 1.31 ± 0.22, the mean fold change of Testin-coding transcripts was only 1.05 ± 0.2 (Fig. 5D). Thus, we focused on Slc37a2. Surprisingly, immunofluorescence staining of SLC37A2 protein in blastocysts revealed that an elevated amount of transcript did not produce higher protein levels. On the contrary, the standardized z-score of the SLC37A2 protein amount in Dis3l KO (and even Dis3l+/−) blastocysts significantly decreased compared to WT embryos (mean z-scores −0.35 ± 0.10, −0.05 ± 0.14, and 0.57 ± 0.28, respectively) (Fig. 5E,F). All this suggests that the lethality of Dis3l KO is not related to changes in the transcript levels.
Decreased levels of SLC37A2 protein, despite increased mRNA expression and the fact that the cytoplasmic exosome is connected with the cytoplasmic quality control pathways, may suggest global problems with protein synthesis resulting from stress responses. To measure the global protein synthesis levels, we performed metabolic labeling with clickable methionine analog L-Azidohomoalanine (AHA). Quantification of AHA incorporation in all newly synthesized proteins in embryos revealed the negative impact of Dis3l KO mutation on global protein synthesis in the preimplantation embryo (mean z-scores −0.41 ± 0.22 and 0.23 ± 0.18 for WT embryos) (Fig. 5G,H).
It is concluded that Dis3l KO leads to the inhibition of protein synthesis in early embryos without major changes at the transcriptome level.
DISCUSSION
In this paper, we studied the role of mammalian DIS3L processive exoribonuclease at the organismal level. DIS3L, which co-precipitates with all nine core exosome proteins (Liu et al. 2006), is essential for mouse embryo development. Dis3l KO mutation causes severe embryo degeneration and death between days 6 and 7 of its development. This is due not only to the implantation process mediated by extraembryonic tissue but mostly to the changes in subcellular processes that occur before implantation. While KO preimplantation embryos do not display any changes in morphology or differentiation of the first embryonic cell lineages and ability to produce ES cells from ICM, the translation rate was reduced, despite small changes at the transcriptome level. This presents a picture of the deregulated state of the early embryo at the level of subcellular processes.
Despite the disruption of global protein production, Dis3l−/− embryos are still able to implant in the uterus and function there for a brief period, but their fate is most probably determined at the preimplantation stage. The state of both relies heavily on the dynamic regulation of the transcriptome in which DIS3L is actively involved as a key component of mRNA turnover and quality control pathways. Thus, its absence would most probably lead to the accumulation of transcripts in the cytoplasm. Those transcripts could be both naturally deadenylated mRNAs that should enter turnover pathways, as well as aberrant mRNAs that should be degraded in quality control pathways (most notably no-go [NGD], nonsense-mediated [NMD], and nonstop [NSD] decay pathways of RNA degradation [van Hoof et al. 2002; Mitchell and Tollervey 2003; Takahashi et al. 2003]), both reliant on cytoplasmic exosome degradation. Considering that transcript deadenylation is followed by decapping, which allows the mRNA molecule to be degraded in a 5′ to 3′ direction by Xrn1 exoribonuclease (Bashkirov et al. 1997; Lejeune et al. 2003) alongside 3′ to 5′ degradation by DIS3L, correct but naturally deadenylated mRNAs would be less impacted by the absence of functional DIS3L than the aberrant, polyadenylated and actively translated transcripts, the detection and degradation of which rely on Ski and exosome complexes, respectively, before they can be actively deadenylated.
This mechanism of aberrant transcript degradation was described to be triggered by ribosome stalling and ribosome collision events on abnormal mRNA particles. SKIV2L, part of the Ski complex, responsible for unwinding and feeding RNA molecules into the exosome channel (Brown et al. 2000; Zhu et al. 2005; Halbach et al. 2013), was shown to be required for the recruitment of the exosome in mRNA quality control pathways through its interaction with stalled ribosomes (Zinoviev et al. 2020). This allows for their extraction through the degradation of stalling-causing mRNA molecules, making those ribosomes available for dissociation and recycling by Pelota/Hbs1/ABCE1 complex (Pisareva et al. 2011). Hence, the debilitating effect of the cell's inability to degrade those problematic mRNAs would be twofold: occurring ribosome collision events could not be properly resolved, and the number of stalling-causing mRNA particles would grow in time, causing new collision events.
While local mechanisms recruited to colliding ribosomes counteract negative consequences of these events, like EDF1 and GIGYF2 working together to inhibit translation from mRNA particles on which collision occurred (Juszkiewicz et al. 2020; Sinha et al. 2020), they can be overloaded by the global scale of the occurring problems. In such a case, more general stress response mechanisms are triggered, leading to global translation initiation block by the action of GCN2 and eIF2α phosphorylation or, in the case of stress being unresolved by the cell, apoptosis activated by MAPKKK ZAKα (Wu et al. 2020). As we present, this might be the case for Dis3l−/− preimplantation embryos, where the global protein amount is lowered despite the accumulation of multiple transcripts. Additionally, recently SKI2VL was also shown to be essential for mouse development, with whole-body deletion leading to embryo lethality before embryonic day E13.5 (Yang et al. 2022). One questionable aspect of this conclusion is the size of the effect observed by us in the blastocysts. Both changes in transcriptome and protein synthesis are clearly identifiable, but rather small. This might be due to a gradual process of detrimental changes’ accumulation—developmentally unaffected blastocysts show the first sign of transcriptome deregulation. These changes can quickly accumulate after implantation, leading to observed rapid deterioration of the embryo state. More in-depth analysis of the correlation between mRNA transcriptome and translation in multiple peri-implantation stages of development would be required to better understand the dynamics of DIS3L activity and its importance. Also, more in-depth analysis of other RNA types could also be explored as potential DIS3L targets, but concerning the challenges of studying a model of early embryo lethality, the feasibility of such experiments would be questionable.
In conclusion, we propose that DIS3L, while dispensable for cell viability, is essential for embryo development as a part of RNA quality control response for translational stress, with its mutation possibly causing translation inhibition across the preimplantation embryo, and, further on, due to accumulation of the stress-inducing events, wide apoptosis after the implantation, leading to embryo death after embryonic day E6.5.
MATERIALS AND METHODS
Animals
Mice lines were generated using the CRISPR/Cas9 method in C57BL/6/Tar × CBA/Tar mixed background mice, following the procedure described previously (Gewartowska et al. 2021). Knock-in Dis3lGFP/GFP mutation was introduced in the ORF of the Dis3l gene at the 3′-end through homologous recombination by insertion of a GFP coding single-stranded DNA donor (mutagenesis strategy and full donor sequence in Supplemental Fig. S1) with 60 bp homology arms on both ends (gRNA sequence: GACAAAGGTCTTTAATGACA). The loss-of-function Dis3l−/− mutation was generated by random insertion of a 169 bp fragment (mutagenesis strategy and insertion sequence in Supplemental Fig. S3) through a nonhomologous end joining repair mechanism in exon 10 of the cDNA sequence of the Dis3l gene (gRNA: TCCAGGTTGCCGTTATTCA).
The Rosa26(hDIS3L) mouse line was generated in Dis3l+/− background. The coding sequence of hDIS3L preceded by an adenoviral splice acceptor (ADSA) with 60 bp homology arms (mutagenesis strategy and full donor sequence in Supplemental Fig. S4) was inserted into an intron of Rosa26 (gRNA sequence: CGCCCATCTTCTAGAAAGAC).
All animal experiments were approved by the Local Ethical Committee in Warsaw affiliated with the University of Warsaw, Faculty of Biology (approval nos. WAW/527/2013, WAW/176/2016, and WAW2/074/2024) and were performed according to Polish law (Act no. 266/15.01.2015).
Embryo isolation and culture
Superovulation was artificially stimulated in females by intraperitoneal injection of 10 units of pregnant mare's serum gonadotropin (PMSG, BioVendor) and human chorionic gonadotropin (hCG; MSD Animal Health), respectively, in 48 h intervals. Stimulated females were mated with males in the evening and terminated by cervical dislocation and dissected in the following days to obtain either 1-, 2-, 4-, 8-cell, or blastocyst stage embryos. Zygotes were isolated by puncturing oviducts ampulla in hyaluronidase solution (Sigma-Aldrich) in 300 μg/mL PBS to dissociate cumulus cells. Other embryos were isolated by flushing oviducts and uterus with M2 medium (Sigma-Aldrich). After isolation, all embryos were rinsed in an M2 medium and used for further procedures. Postimplantation embryos were isolated from unstimulated females 6 or 7 days after mating with males.
All embryos were cultured in a cell culture incubator with 5% CO2 in the atmosphere and 38°C. Embryos were placed in drops of either M2 (for short-term culture) or M16 (for multiday culture; Sigma-Aldrich) medium under mineral oil (Sigma-Aldrich) on 35-mm Falcon Easy-Grip tissue culture dishes (Thermo Fisher Scientific).
Generation and in vitro culture of ES cells and EBs
For the derivation and culture of ES cells, feeder cells—inactivated mouse embryonic fibroblasts (MEFs)—were prepared according to Robertson (1987). Blastocysts collected 96 h after hCG injection were transferred to single wells of culture plates coated with 0.2% gelatin (Sigma-Aldrich) with a feeder layer of inactivated MEFs. The composition of the derivation medium was designed based on the 2i method (Tamm et al. 2013) (full composition in Table 1). After 3–4 days of culture in a cell culture incubator at 38°C and with 5% CO2 in the atmosphere, blastocysts formed outgrowths, which were disaggregated by 5 min incubation in 0.25% trypsin-EDTA (Gibco) followed by mechanical pipetting. The resulting cell suspensions were transferred into the separate culture plate wells onto a fresh layer of inactivated MEFs and inspected daily for the appearance of primary colonies. Cultures containing ES cells were expanded, processed for genotyping, and frozen for further investigation. Established ES cell lines were cultured in ES cell medium composed of KO Dulbecco's modified Eagle's medium (KnockOut DMEM, Gibco) supplemented with 15% heat-inactivated FBS (Performance Plus, Gibco), with the addition of nonessential amino acids, L-glutamine, β-mercaptoethanol, penicillin and streptomycin, and 500 IU/mL LIF (Gibco).
Medium composition for ES cell derivation
To gather a pure ES cell population, MEFs were removed from cultures by preplating; i.e., incubating cells with 0.05% trypsin-EDTA (Sigma-Aldrich) for 3–5 min and seeding of suspended cells onto a culture dish covered with gelatin for 20 min, allowing MEFs to attach to the dish. MEF-free suspension of ES cells was cultured on a new gelatin-covered culture dish until reaching confluency. ES cells were then detached by incubation in 0.05% trypsin-EDTA for 3–5 min and washed in PBS (BioShop). Centrifugated dry pellets were frozen at −80°C until further processing.
For subcellular protein fractionation, ES cells were cultured on plates coated with 0.2% gelatin without feeder cells in ESGRO-2i medium (Sigma-Aldrich).
To induce differentiation of ES cells, EBs were generated. First, ES cell colonies were disaggregated using trypsin and preplated for 20 min (as described above). Part of the collected cell suspension (day 0 of EB culture) was kept for RNA isolation by freezing a dry pellet after a wash in PBS. The second part of cell suspension was centrifuged, and cells were cultured in a differentiation medium (ES cell culture medium without LIF) in the concentration of 800 cells per 30 μL in hanging drops to stimulate EB formation. After 2 days of culture in hanging drops, EBs were either collected for analysis or washed and placed in low-adhesive dishes (Medlab) allowing for their culture in suspension. After 5 days of differentiation, the rest of the EBs were collected for analysis.
Immunoprecipitation
Livers from 10–20 week old Dis3lGFP/GFP and Dis3lwt/wt knock-in mice were homogenized in lysis buffer (10 mM Tris-HCl at pH 8, 125 mM NaCl, 1 mM DTT, 1 mM PMSF, 0.02 μM pepstatin A, 0.02 µg/mL chymostatin, 0.006 μM leupeptin, 20 µM benzamidine hydrochloride), processed as described previously (Tomecki et al. 2010; Mroczek et al. 2017) and analyzed by mass spectrometry by the Mass Spectrometry Laboratory, IBB PAS. Proteins identified in the WT sample were treated as nonspecifically bound to the anti-GFP resin and were used to normalize GFP samples. Identified proteins that were considered nonspecific contaminants (hemoglobin subunits, immunoglobulins, keratins, and trypsinogens) were excluded from the final analysis of enriched proteins.
Protein isolation and western blotting
To isolate proteins from subcellular fractions, ES cells were detached from plates using 0.25% Trypsin-EDTA and suspended in ice-cold PBS. Fractionation was then performed using the Subcellular Protein Fractionation Kit for Cultured Cells (Thermo Scientific), following the manufacturer's protocol. Obtained individual fractions were snap-frozen in liquid nitrogen and stored at −80°C. Before further use, samples were thawed and mixed with loading Laemmli buffer, and afterward stored in −20°C.
Proteins from Dis3l−/−; Rosa26(hDIS3L) tissue samples were isolated by homogenization in NP40 buffer (0.1% NP40, 1 mM PMSF, protease inhibitors, and chymostatin in PBS) using glass Dounce homogenizers. Two microliters of viscolase (A&A Biotechnology) was added per every homogenized sample, and samples were incubated with 600 rpm shaking for 30 min at 37°C following centrifugation at maximum settings for 5 min at 4°C. The supernatant was then mixed with loading Laemmli buffer and stored at −20°C.
For western blotting, samples were thawed and denatured by heating at 99°C for 10 min and resolved by SDS-PAGE. Proteins were then transferred from polyacrylamide gel onto Amersham Protran nitrocellulose membrane (Cytiva) and stained with Ponceau S solution to verify appropriate protein transfer. Unspecific binding sites were blocked by membrane incubation in 5% milk for 1 h, and membranes were immunostained by incubation in blocking solution with appropriate primary antibody (Table 3) overnight at 4°C with gentle shaking. Then, membranes were washed for 15 min three times with PBST solution and stained with secondary antibodies (Table 3) in blocking solution for 1.5 h, followed by another three washes as previously. Stained membranes were developed using either Clarity Western ECL Substrate (Bio-Rad) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific), depending on previous experimental assessments of signal strength. Staining was documented using the iBright 1500 system (Thermo Fisher Scientific).
DNA isolation and genotyping
For genotyping of mice, postimplantation embryos, and ES cells, adjusted HotShot method was used (Truett et al. 2000). DNA from fixed and stained embryos was isolated using the protocol previously described (Artus et al. 2005). Preimplantation embryos were repeatedly rapidly frozen and thawed several times in a minimal amount of RNase and DNase-free water in PCR tubes. Obtained DNA was used for genotyping by PCR reaction using Phusion High-Fidelity DNA polymerase (Thermo Fisher Scientific). Reactions of 20 μL volume were set up according to the manufacturer's protocol, with no DMSO and 10 μM primers used. Each genotyping used one set of primers, except for the Rosa26(hDIS3L) line, where the hDIS3L transgene was genotyped by two sets of primers to amplify products of upstream fusion site and within hDIS3L transgene (primer sequences in Table 2). Additionally, blastocysts from which RNA was extracted for sequencing and RT-qPCR experiments, were genotyped as described for gDNA above, with cDNA generated during those experiments used as a genotyping PCR template.
List of primers
RNA isolation and processing
RNA from ES cells was extracted by lysis of 1 million cells using TRI Reagent (Sigma-Aldrich), following the manufacturer's protocol. RNA from EBs was isolated using the High Pure RNA Isolation Kit (Roche), following the manufacturer's protocol. RNA from living embryos was extracted using the PicoPure RNA Isolation Kit (Thermo Fisher Scientific), following the manufacturer's protocol with the following modifications: single embryos were incubated for 30 min at 42°C in the extraction buffer and then mixed with ethanol in 1:1 ratio. All isolated RNA samples were stored at −80°C until further use. DNA was removed from all RNA samples using the TURBO DNA-free Kit (Thermo Fisher Scientific), following the manufacturer's protocol for routine DNase treatment, with an additional 2 μL of RiboLock RNase Inhibitor (Thermo Fisher Scientific) added to every reaction. For single blastocyst RNA-seq and RT-qPCR, SuperScript III Reverse Transcriptase (Thermo Fisher Scientific) and oligo(dT)20 primer was used to generate cDNA from total RNA, following the modified manufacturer's protocol. Reverse transcription reactions were cleaned up using AMPure XP Reagent magnetic beads (Beckman Coulter). For EBs RT-qPCR, 250 ng of total RNA was used for reverse transcription reaction with RevertAid First-Strand cDNA Synthesis kit (Thermo Fisher Scientific) using oligo(dT)20 primer, following manufacturer's protocol. For ES cell sequencing, cDNA was prepared using random primers as a step in the library preparation protocol described in the “RNA-sequencing libraries” section.
Chimeric embryos
To produce tetraploid (4n) WT embryos, 2-cell diploid (2n) WT embryos were isolated from superovulated females 30 h after hCG administration. Early 2-cell embryos were electroporated using a homemade electroporation chamber and a programmable square pulse generator. Embryos were rinsed several times and placed in electroporation buffer (100 mM MgSO4·7H2O, 100 mM CaCl2·2H2O, 0.045 g/mL glucose in H2O) warmed to 38°C in a glass petri dish between two straight electrodes of platinum wire attached to the dishes bottom ∼100 μm apart, with the embryo cleavage plane parallel to the electrodes. Two 40 V pulses lasting 40 μsec in 100 μsec intervals were generated to induce fusion of the blastomeres. Two cell embryos were washed once in M2 medium and placed in culture for 24 h. Within the first hour from electroporation, embryos were inspected for survival and blastomere fusion and sorted adequately to trace their development back to the 2-cell stage.
Simultaneously to 4n WT embryos reaching the 2-cell stage, 4- to 8-cell 2n embryos were isolated from Dis3l+/− females previously mated with males. All embryos had their zona pellucida removed by short (∼10 sec) rinsing in Tyrode's acidic solution (Sigma-Aldrich) warmed to 38°C. Two 4n and one 2n embryos were rinsed and then moved to fresh aggregation solution: 0.3 mg/mL phytohemaglutynin (Sigma-Aldrich) in M2 medium without BSA (homemade). Embryos were manually attached together using a mouth pipette. Chimeric embryos prepared this way were rinsed with M2 medium and cultured overnight in the M16 medium. Chimeric embryos were then transferred to pseudopregnant females on the first day of pseudopregnancy, following the same procedure as for the mice line generation.
Embryo fixation and staining
Preimplantation embryos were fixed in 4% PFA (Thermo Fisher Scientific) for 30 min and permeabilized in 0.5% Triton X-100 (Sigma-Aldrich) for 20 min. After fixation, embryos were placed in drops of blocking solution (3% BSA [Santa Cruz Biotechnology] in PBST) on plastic dishes overnight before immunostaining or were stored that way until further procedures.
All immunofluorescence labeling was performed by incubating fixed embryos in a blocking solution with appropriate primary antibody dilution (Table 3) overnight at 4°C. Embryos were washed once in PBS and twice in blocking solution and incubated with a secondary antibody (Table 3) for 2 h at room temperature (RT). After 30 min washing in PBS, blastocysts were ready for imaging or additional staining. Methionine analog labeling was performed by incubating embryos in 25 μM Click-iT AHA (L-Azidohomoalanine, Thermo Fisher Scientific) solution in M16 medium for 2 h and then fixing them as described above. AHA incorporated in proteins was detected using a dedicated Click-iT Cell Reaction Buffer Kit (Thermo Fisher Scientific) and Alexa Fluor 594 Alkyne (Thermo Fisher Scientific) in 1 μM final concentration, according to the manufacturer's protocol. After brief washing in 3% BSA and chromatin staining, embryos were ready for imaging. Chromatin in embryos was stained with Hoechst 33343 (1:5000 dilution in 2 μg/mL PBS; Sigma-Aldrich) for 15 min.
List of antibodies
All stained embryos were imaged on uncoated 35 mm plastic dishes with glass bottoms (MatTek) in drops of M2 medium using an LSM 800 Confocal Laser Scanning microscope (Zeiss). Obtained images were analyzed using ImageJ and Python software. The cell number in blastocysts was counted in confocal sections manually using ImageJ built-in tools. SLC37A2 and AHA intensities were gathered from the maximum projection of individually scanned blastocysts using Python and ImageJ, respectively.
RNA-sequencing libraries
For ES cell lines RNA-seq, total DNA-free RNA was ribodepleted using a Ribo-Zero Gold rRNA Removal Kit (Illumina). rRNA-free RNA samples were cleaned up by precipitation with 3 M sodium acetate. Sequencing libraries were prepared using the KAPA Stranded RNA-seq Library Preparation Kit (KAPA Biosystems). Between each step, samples were cleaned up using AMPure XP Reagent magnetic beads. Library quality and fragment size distribution were verified by electrophoresis in the Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.).
For RNA-seq libraries from single blastocysts, DNA-free RNA samples were prepared by tagmentation reaction following published protocols (Picelli et al. 2014; Hennig et al. 2018), with various steps and amount of enzyme optimized for the use of a home-made batch of Tn5 transposase produced in our laboratory. Briefly, Tn5 (0.25 mg/mL) was loaded with linker oligonucleotides Tn5ME-A/Tn5Me-rev and Tn5ME-B/Tn5Me-rev by mixing 10 μL Tn5 with 0.5 μL of both linkers (0.35 μM) and incubating with shaking at 350 rpm for 45 min at 23°C. Right before the reaction setup, loaded Tn5 was diluted 10 times with nuclease-free water. Ten microliters of freshly prepared tagmentation buffer (20 mM Tris-HCl at pH 7.5, 20 mM MgCl2, 50% dimethylformamide [Sigma-Aldrich]) were mixed with 5 μL of diluted Tn5 and 5 μL of cDNA, incubated for 3 min at 55°C in a preheated thermocycler and cooled for 1 min to 10°C. The reaction was inactivated by the addition of 5 μL of 0.2% SDS (Sigma-Aldrich) and incubation for 5 min at RT. Tagmented cDNA was purified with AMPureXP Reagent magnetic beads using a 1:1.25 cDNA to beads ratio. For library amplification, the KAPA HiFi HotStart ReadyMix PCR Kit (Roche) with the addition of 5% DMSO was used. The number of cycles yielding the best results was determined experimentally and was further individually adjusted for each batch of prepared cDNA. Library quality and fragment size distribution were verified by electrophoresis using Agilent 2100 Bioanalyzer.
ES cell line RNA libraries were sequenced on a NextSeq 500 instrument (Illumina) in 2 × 75 cycles paired-end mode in the Next-Generation Sequencing Unit of the International Institute of Molecular and Cell Biology in Warsaw. Single blastocyst RNA libraries were sequenced on a NovaSeq 6000 (Illumina) instrument in 2 × 100 cycles paired-end mode in Genomics Core Facility of the Centre of New Technologies, University of Warsaw.
RNA-seq bioinformatic analysis
For ES cell line RNA-seq, the Illumina sequencing reads were quality filtered using BBDuk, a part of BBTools (http://sourceforge.net/projects/bbmap/) to remove Illumina adapter sequences and remove reads that were <20 nt after trimming. Processed reads were mapped to the mouse genome (GRCm39) using STAR v2.7.11b (Dobin and Gingeras 2015). For downstream analysis, only uniquely mapped reads were used. The feature assignments of reads were performed with featureCounts v2.06, a part of the Subread package (Liao et al. 2014), using whole main gene annotation on reference chromosomes (vM35 from Gencode), choosing genes as representative features. The normalization and differential gene expression analysis were performed with DESeq2 (v1.44.0) Bioconductor R package (Love et al. 2014).
For blastocyst RNA-seq, the Illumina sequencing reads were mapped to the reference mouse genome (GRCm38) using the STAR v2.7.10a (Dobin and Gingeras 2015). Quality control, read processing and filtering, and visualization of the results were performed using custom scripts and elements of the RSeQC, BEDtools, and SAMtools packages. Reads were counted to the Gencode v M6 basic annotation using featureCounts v2.0.1 a part of the Subread package (Liao et al. 2014). Differential expression analyses were performed using the DESeq2 v1.34.0 Bioconductor R package (Love et al. 2014). RNA-seq reads were aligned to the murine ribosomal DNA consensus sequence using the STAR short-read aligner, using custom-prepared indices. These indices included the complete ribosomal DNA repeating unit (GenBank accession: BK000964.3), 5S rRNA (accession: NR_023363.1) , and mitochondrial 12S rRNA (mt-R) and 16S rRNA (RNR2), which served as control sequences. On average, 5%–7% of the total library is mapped to rRNA. Final results were analyzed using the feature annotation of the complete rDNA repeating unit from GenBank. Read pairs were counted using featureCounts (Liao et al. 2014) to the GenBank annotation, adjusted for feature size, and normalized to protein-coding genes using library size factors from the DESeq2 differential expression analysis described above. Error bars indicate standard deviation.
For both RNA-seq, the analysis was performed on three biological replicates for each genotype. The thresholds for identification of significantly different gene expression levels are given in the plot's descriptions.
Functional enrichment analysis was performed with g:Profiler using gprofiler2 package for R (database version: e106_eg53_p16_65fcd97) with the following settings: g:SCS multiple testing correction; 0.05 significance threshold (Raudvere et al. 2019).
RT-qPCR
RT-qPCR experiments on ES cells and blastocysts were performed using Platinum SYBR Green qPCR SuperMix-UDG (Invitrogen) in LightCycler 480 Instrument (Roche) according to the manufacturer's protocol. All ES cell cDNA samples were diluted 10 times before using as a reaction template. All primers used are described in Table 2. All biological samples were run in technical triplicates. The number of biological samples is denoted in the Results section.
RT-qPCR experiment on EBs was performed using TaqMan Gene Expression Master Mix (Life Technologies) in LightCycler 96 instrument (Roche), according to the manufacturer's protocol. All RNA samples were diluted four times. All samples were run in technical duplicates. Specific TaqMan probes against four different transcripts were used: Mm00443081_m1 (Pax6), Mm01976556_s1 (Foxa2), Mm01318252_m1 (Tbxt), and Mm01205647_g1 (Actb).
Cp values of all technical replicates of one biological sample results were averaged. These results were analyzed using the 2−ddCp method (the final formula used was 2−ddCp = 2−[(CpGOI – Cphousekeeping)KO – (CpGOI – Cphousekeeping)WT]). Obtained Cp values of genes of interest from every sample were normalized to either actin b (EBs) or GAPDH (ES cells and blastocysts) Cp value. For blastocyst RT-qPCR analysis, only samples with a technical replicate standard deviation <1 were chosen.
Statistical analysis
All statistical analyses were performed using RStudio. All data plots present individual data points (where applicable), as well as mean value with standard error of the mean (SEM) indicated as error bars (where applicable). All experiments were performed in at least two individual repetitions (except the immunoprecipitation experiment with one repetition) and in each experiment, biological samples were obtained from at least three different animals. Distribution ratios were compared by the Fisher exact test for 2 × 2 contingency tables (viability of embryos) or the χ2 test for 2 × 3 tables (genotype distributions). Mean values were compared using either a parametric two-sided t-test for sample sets with normal distribution or a nonparametric two-sided Mann–Whitney–Wilcoxon test for sample sets without normal distribution. Distribution normality was tested with the Shapiro–Wilk test. Value n in figures and figure descriptions denotes the number of individual embryos analyzed. P-values for every test are reported on plots.
DATA DEPOSITION
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al. 2022) partner repository with the data set identifier PXD038745, with access available via an anonymous reviewer account (username: reviewer_pxd038745@ebi.ac.uk, password: AR7t8RdV). RNA-seq data have been deposited in NCBI's Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE220800 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE220800) using private reviewer access token (qrabsqganbkzncb). Other numerical data underlying results presented are available in the article and its online supplemental materials (Supplemental Table S2).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Andrzej Dziembowski laboratory members for their support and fruitful discussions, Anna Ciemerych for support in the experiments involving ES cells and postimplantation embryos, Olga Gewartowska for multiple suggestions and critical reading of the manuscript, Aleksandra Brouze for critical and proofreading of the manuscript, and all members of the Genome Engineering Unit of the International Institute of Molecular and Cell Biology in Warsaw for maintenance of animal colony and animal genotyping. NGS was performed thanks to Genomics Core Facility CeNT UW (RRID:SCR_022718), using the NovaSeq 6000 platform financed by the Polish Ministry of Science and Higher Education (decision no. 6817/IA/SP/2018 of 2018-04-10). RNA-seq analysis of ES cell lines was performed at the IIMCB Bioinformatics facility (IN-MOL-CELL Infrastructure) funded by the European Union—NextGenerationEU under the National Recovery and Resilience Plan. IN-MOL-CELL Infrastructure was also funded by the European Union under Horizon Europe (Project 101059801 - RACE) and by the RACE-PRIME project carried out within the IRAP programme of the Foundation for Polish Science cofinanced by the European Union under the European Funds for Smart Economy 2021–2027 (FENG). This work was supported by the National Science Centre (grant numbers UMO-2013/10/M/NZ4/00299; UMO-2016/22/A/NZ4/00380), Foundation for Polish Science (grant number TEAM TECH CORE FACILITY/2017-4/5) and European Union's Horizon 2020 research and innovation program (grant agreement no. 810425).
Author contributions: M.B. performed all embryo genotyping, postimplantation embryo analysis, blastocyst, and ES cell qPCR experiments, all western blot experiments, GO analyses of RNA-seq results, prepared RNA-seq libraries and prepared all figures. M.B. and M.S. constructed and transferred chimeric embryos, A.C. derived ES cells and performed EB experiments, M.B and W.A. performed blastocyst cell lines staining and analysis and SLC37A2 and AHA labeling experiments, O.G. and M.S. performed rescue experiments, S.M. performed IP experiments, A.H.-O. and T.K. analyzed RNA-seq results, D.C. performed and analyzed mass spectrometry, D.A. performed sequencing experiments, J.G. and E.B. produced all mice lines, M.B. and A.D. wrote the manuscript, A.D. conceived the study.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080350.124.
-
Freely available online through the RNA Open Access option.
- Received December 9, 2024.
- Accepted January 11, 2025.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Michał Brouze is the first author of this paper, “DIS3L, cytoplasmic exosome catalytic subunit, is essential for development but not cell viability in mice.” Michał is a research assistant completing his PhD in the Laboratory of RNA Biology led by Professor Andrzej Dziembowski, International Institute of Molecular and Cell Biology. The main focus of the laboratory is RNA processing and modification, recently the role of TENT5 poly(A) polymerases and the biology of mRNA therapeutics. Michał's focus is RNA biology in the context of mammalian development.
What are the major results described in your paper and how do they impact this branch of the field?
DIS3L was the last of the known catalytic subunits of the RNA exosome complex of unknown function. The other two ribonucleases, nuclear DIS3 and RRP6, were both identified as essential for mouse embryo development. We confirmed this is also the case for cytoplasmic-exosome bound exoribonuclease DIS3L. After implantation, the condition of embryos with DIS3L knockout mutation begins to deteriorate, leading to their death after 7 days of development. However, even before implantation, those embryos displayed changes in the transcriptome leading to a decrease in global protein production—possibly the effect of translation stress response pathways triggered by accumulation of aberrant transcripts in the absence of DIS3L functionality.
What led you to study RNA or this aspect of RNA science?
I completed my bachelor's and master's degrees in developmental biology, and it always remained in the center of my research interests. After graduating, I wanted to pursue an academic career. Projects initiated by Prof. Dziembowski allowed me to expand my research interests to molecular aspects of mammalian development. At that time I was a stranger to the world of RNA biology, and studying how novel RNA processing mechanisms can impact the development of the oocyte and embryo proved to be a fascinating and fulfilling endeavor.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
Studying embryo lethality poses numerous significant obstacles, which I and my co-workers were learning to navigate as we progressed. The challenge of studying molecular mechanisms of RNA metabolism in a preimplantation embryo, which consists of up to 120 cells, prompted us to seek and experiment with different angles of approaching the problem—from stem cell derivation, which provides more material for high-throughput analyses, to single embryo dissections and cell lineages selection to be able to look into the biology of individual embryos affected by the mutation of DIS3L exoribonuclease.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
I can name two individuals whose influence undeniably steered my scientific development. Firstly, Dr. Anna Ajduk from the University of Warsaw, Faculty of Biology. As my supervisor for both my bachelor's and master's theses, she imparted essential knowledge in developmental biology and, more importantly, gave me invaluable lessons in results analysis, critical thinking, and scientific integrity, which have been fundamental to my growth as a researcher and guided me toward an academic career.
Secondly, Prof. Andrzej Dziembowski, supervisor of my PhD thesis and my longtime mentor. He expanded my perspective on possibilities in research work and showed me the importance of applying different, out-of-the-box approaches to be able to truly understand the nature of the studied phenomenon, while at the same time, he taught how to focus on the goals at hand and prioritize the tasks to achieve long-term goals.
What are your subsequent near- or long-term career plans?
During my work in Prof. Dziembowski's lab, I had an opportunity to participate in many adjacent projects and initiatives. One of them was introducing several new methods and solutions in the Mouse Genome Engineering Facility established by Prof. Dziembowski within the TEAM-TECH Core Facility Project. In the past years this Core Facility, now operating as a Genome Engineering Unit, became not only a vital part of research conducted in the International Institute of Molecular and Cell Biology (IIMCB) but also a provider of many services to the broader scientific community in Europe. These services now include transgenic mice derivation, cloning, Nanopore sequencing, and more. As the current head of the facility, Dr. Olga Gewartowska will move on to lead the newly established Technology Development Unit within our Institute, which will support, integrate, and enhance the operations of Core Facilities within IIMCB. With this, I will take over as the head of the Genome Engineering Unit, hoping to further lead its development, beginning with extending our offer of transgenic animal generation to new models: Danio rerio and Caenorhabditis elegans.








