
A dominant role for meiosis-specific 3′ RNA processing in controlling expression of a fission yeast cyclin gene
- Center for RNA Molecular Biology, School of Medicine, Case Western Reserve University, Cleveland, Ohio 44106-4960, USA
-
↵1 Present address: Department of Biochemistry and Molecular Biology, Thomas Jefferson University, Philadelphia, PA 19107, USA.
Abstract
Meiotic gene regulation provides a rich source of insight into mechanisms of temporal control during development. We previously reported that accumulation of many meiotic mRNAs in fission yeast is governed by changes in 3′ RNA processing and elucidated the molecular basis of this regulatory mechanism for an early meiotic gene. Here, we report that cleavage/polyadenylation is also the nexus of negative control for middle meiotic genes. Parallel profiles of splicing and polyadenylation are observed over a meiotic time course for both rem1 and spo4 but not for a constitutive control gene. Nevertheless, polyadenylation of rem1 transcripts is restricted to meiosis by a splicing-independent mechanism. Through systematic sequence substitutions, we identified a negative control region (NCR) located upstream of the rem1 transcription start site and found that it is required to block 3′ RNA processing in proliferating cells. Ablation of the NCR relieves inhibition regardless of whether the intron is present, absent, or carries splice site mutations. Consistent with the previous report of a polypeptide encoded by the first exon of rem1, we discovered a second 3′ processing site just downstream from the 5′ splice site. Polyadenylation within the intron is activated concurrent with the downstream site during meiosis, is controlled by the NCR, and is enhanced when splicing is blocked via 5′ junction or branch point mutations. Taken together, these data suggest a novel regulatory mechanism in which a 5′ element modulates the dynamic interplay between splicing and polyadenylation.
Keywords
INTRODUCTION
Meiosis is a highly conserved cellular differentiation pathway in which one round of DNA synthesis is followed by two successive rounds of division, leading to the formation of haploid cells from diploid precursors. In multicellular organisms, extrinsic cues from surrounding cells stimulate germ cells to begin meiotic differentiation, whereas in unicellular organisms such as the budding yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe, meiosis follows conjugation, which is triggered by nutrient deprivation (Harigaya and Yamamoto 2007). Like other cellular differentiation pathways, meiosis requires exquisite control of gene expression to ensure that the molecular events unfold in the proper temporal sequence. During differentiation in S. pombe, the levels of over 800 transcripts increase by a factor of four or more in three successive waves, which were initially attributed to changes in transcription (Mata et al. 2002). However, subsequent studies revealed contributions from post-transcriptional processes including splicing, polyadenylation, and nuclear RNA turnover (Averbeck et al. 2005; Malapeira et al. 2005; Harigaya et al. 2006; Moldon et al. 2008; McPheeters et al. 2009; Cremona et al. 2011).
In budding yeast, as in fission yeast, many intron-containing meiotic transcripts fail to be spliced in proliferating cells. However, the underlying regulatory mechanisms appear to be distinct. In S. cerevisiae, the MER2, MER3, SPO22, and AMA1 transcripts form a regulon controlled positively by Mer1p, a KH domain RNA binding protein expressed only during meiosis (Spingola and Ares 2000; Munding et al. 2010; Qiu et al. 2011a). These four pre-mRNAs, together with PCH2, require the U1 snRNP protein Nam8p for splicing (Qiu et al. 2011a), and PCH2 shares with SAE3 a requirement for trimethylation of the guanosine caps on spliceosomal snRNAs (Qiu et al. 2011b). In contrast to the intronic splicing enhancers that act on nearby 5′ splice sites in budding yeast, the three fission yeast transcripts in which the signals that govern meiosis-specific splicing have been mapped (mes1, crs1, and rem1) are regulated by sequences outside the coding regions, far from the introns they regulate (Shimoseki and Shimoda 2001; Averbeck et al. 2005; Moldon et al. 2008). Moreover, the factors that recognize these elements have been implicated in processes other than splicing. For early meiotic transcripts, prevention of RNA processing during proliferation is intimately linked to a specialized nuclear turnover mechanism that requires the RNA binding proteins Mmi1p and Red1p for targeting to the exosome (Harigaya et al. 2006; McPheeters et al. 2009; Yamanaka et al. 2010; Chen et al. 2011; Sugiyama and Sugioka-Sugiyama 2011). For middle meiotic genes, the forkhead transcription factor Fkh2p, expressed in both mitotic and meiotic cells, has been implicated in negative regulation, while a meiosis-specific member of this protein family, Mei4p, functions in positive control during meiosis (Malapeira et al. 2005; Mata et al. 2007; Moldon et al. 2008; Chen et al. 2012).
The discovery that meiosis-specific splicing in fission yeast is mediated by elements outside the coding regions prompted speculation that the mechanism is related to the one that modulates alternative splicing patterns in metazoans upon switching promoters (Averbeck et al. 2005; Futcher and Leatherwood 2008; Moldon et al. 2008). The analogy may well apply to the positive element required for mid-meiotic splicing of transcripts from the rem1 gene, encoding a cyclin required for meiosis in the absence of the G1/S-specific cyclin Cig2p (Moldon et al. 2008). However, the crs1 gene, also encoding a meiotic cyclin, appears to lack a positive element, and negative regulation requires both a Determinant of Selective Removal (DSR) located in the terminal exon and a noncanonical polyadenylation signal located just upstream of the major polyadenylation signal activated during meiosis (McPheeters et al. 2009).
The importance of preventing production of meiotic gene products in proliferating S. pombe cells is underscored by the deleterious effects of either expressing individual genes or relieving repression of an entire group through mutation of a regulatory factor (Averbeck et al. 2005; Malapeira et al. 2005; Harigaya et al. 2006). Whereas the molecular mechanisms that negatively regulate early meiotic genes have been intensively investigated (Harigaya et al. 2006; McPheeters et al. 2009; St-André et al. 2010; Yamanaka et al. 2010; Sugiyama and Sugioka-Sugiyama 2011), the single mechanistic study of a middle meiotic gene focused mainly on positive control of splicing during meiosis (Moldon et al. 2008). Here, we describe negative control of the same gene, rem1, extending the analysis to include polyadenylation based on our recent discovery that failure to undergo 3′ RNA processing is a widespread mechanism for preventing accumulation of meiotic transcripts in proliferating fission yeast cells (McPheeters et al. 2009; Cremona et al. 2011). Our findings demonstrate that 3′ RNA processing, rather than splicing, sits at the top of the rem1 regulatory hierarchy, as meiosis-specific polyadenylation is maintained even when the intron is deleted. However, the timing is perturbed, possibly due to elimination of the intronic polyadenylation site. Poly(A) tail addition both within the intron and downstream from the rem1 coding region is controlled by a negative regulatory element, which is physically as well as functionally distinct from the previously mapped positively acting motifs.
RESULTS
Polyadenylation and splicing are regulated in parallel for middle meiotic genes
The first goal of the present study was to determine whether splicing and polyadenylation of rem1 transcripts show the same temporal profile during meiosis. To answer this question, we took advantage of the temperature-sensitive pat1-114 mutant, as in earlier work (Averbeck et al. 2005; Cremona et al. 2011). In mitotically growing wild-type fission yeast cells, the Pat1p kinase blocks entry into meiosis via inhibitory phosphorylation of the necessary and sufficient meiotic regulator Mei2p (Iino and Yamamoto 1985; Watanabe et al. 1997). Thus, when pat1-114 mutant cells are shifted to the nonpermissive temperature, a highly synchronous gene expression program unfolds (Mata et al. 2002).
Splicing and polyadenylation of rem1 transcripts were assayed using RNA prepared from cells harvested at various times after the temperature shift (Fig. 1). In mitotically growing cells (0-h time point), the only RNA species detected in the splicing assay retains the intron. Interestingly, the level of this full-length precursor RNA increases relative to the loading control in early meiosis (1-h time point), before spliced RNA is detected (3-h time point). In addition, peak accumulation of polyadenylated rem1 mRNA over a meiotic time course coincides with peak accumulation of spliced mRNA (4- and 5-h time points). As these profiles match the steady state RNA levels determined by microarray analysis (Mata et al. 2002), changes in RNA processing may be largely responsible for determining rem1 gene output. To determine whether polyadenyation occurs in the absence of splicing, we assayed both RNA processing reactions simultaneously (Fig. 1B). No unspliced rem1 RNA carrying a poly(A) tail was detected using either a 5′ primer upstream of (top) or within (bottom) the intron. Thus, we conclude that poly(A)+ rem1 transcripts are also spliced.
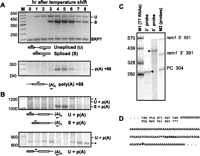
(A) Splicing (top) and polyadenylation (bottom) of rem1 transcripts peak concurrently during meiosis. RNA processing was assayed by RT-PCR as diagrammed beneath each gel image. The identities of the processed and unprocessed products, which were verified by eluting the bands and subjecting them to sequence analysis, are indicated alongside the gel image. SRP7, the small stable cytoplasmic RNA component of the signal recognition particle (Brennwald et al. 1988), was used as an internal loading control for splicing assays (Webb and Wise 2004). The 3′ primer used to assay polyadenylation (bottom) was oligo(dT) (McPheeters et al. 2009). (B) Polyadenylated transcripts that retain the intron are below the level of detection. Splicing and 3′ processing were assayed concurrently using a 5′ primer complementary to exon 1 (top) or the intron (bottom). Asterisks mark bands that did not yield readable sequence with rem1-specific primers. (C) Mapping the 5′ and 3′ termini of rem1 RNA using an RNAse protection assay. Total RNA prepared from mid-meiotic cells was incubated with uniformly labeled riboprobes extending beyond either end of the transcript. The sizes of the major digestion products, indicated by arrowheads, were determined graphically based on mobility relative to the two sets of markers. Faint bands comigrating with the undigested 5′ and 3′probes (right lane) remain in each lane. (PC) a positive control probe complementary to a mouse mRNA supplied by the manufacturer; removal of the lane displaying this assay is indicated by a white line. (D) Sequence of the region extending from the final four codons of the open reading frame through the polyadenylation site. The location of the poly(A) tail addition was determined by sequence analysis of RT-PCR products eluted from multiple gels including the one shown in panel A, with reproducible results. The site of cleavage, marked with a downward arrow, may be specified by the sequence highlighted in bold, which is a putative positioning element (see text for details).
The polyadenylation site determined by sequencing bands eluted from gels, such as the one in Figure 1A (bottom), is located at +88 relative to the stop codon. To confirm the 3′ terminus of rem1 mRNA, as well as to map the transcription initiation site, we used an RNAse protection assay on RNA isolated from mid-meiotic cells (Fig. 1C). Although signals were not observed in RNAse protection assays on RNA from vegetative cells expressing only endogenous rem1 (data not shown), transcripts produced from plasmid-borne constructs with nmt81 DNA replacing native 5′ sequences are polyadenylated at the same sites as meiotic transcripts (Figs. 4, 5, 7, see below). The position of meiotic poly(A) tail addition determined via the 3′ probe used in RNAse protection assays matches sequencing data for RT-PCR products. In agreement with a previous study (Moldon et al. 2008), we find that the 5′ untranslated region (UTR) of rem1 is 160 nt long. The rem1 3′ UTR (Fig. 1D) is very purine-rich, contains only 11 T's and lacks C's entirely; the significance of this unusual base composition is unclear, as the 3′ UTR sequences of other mid-meiotic fission yeast genes are AT-rich, similar to constitutively expressed genes (data not shown). Although the cleavage site in rem1 follows a GA dinucleotide rather than the consensus YA, a motif related to the conserved hexanucleotide (AATAAA) that serves as a positioning element in metazoan and budding yeast polyadenylation signals (Tian and Graber 2011) is found in a similar location in the rem1 3′ UTR (shown in bold type in Fig. 1D).
Another middle meiotic gene shows concurrent activation of polyadenylation and splicing
To test whether coincident peaks of spliced and poly(A)+ RNA is a general property of middle meiotic genes, we examined processing of transcripts from spo4, encoding a serine/threonine protein kinase required for initiation and progression of the second meiotic division (Nakamura et al. 2002). Like rem1, spo4 shows concurrent activation of splicing and polyadenylation in mid-meiosis (Fig. 2A), although the decline in mature mRNA levels relative to the loading control in late meiotic cells is less pronounced (Fig. 2A, top). Both introns in spo4 are removed only in meiotic cells (Fig. 2A, top). In contrast, the four introns of the constitutively expressed SPAC6F6.11c gene, encoding a predicted pyridoxine-pyridoxal-pyridoxamine kinase (http://www.pombase.org/spombe/result/SPAC6F6.11c), are spliced throughout the meiotic time course (Fig. 2B, top). As previously reported (Cremona et al. 2011), SPAC6F6.11c transcripts are polyadenylated at two sites, both of which are utilized during proliferative growth and meiotic differentiation. The biological significance of the decline in processed SPAC6F6.11c RNA levels relative to the loading control in late meiotic cells is unclear but does not appear to be a general property of constitutively expressed fission yeast genes (Mata et al. 2002). Although less marked than the decrease in rem1 mRNA (cf. Figs. 2B and 1A, 7- and 8-h time points), it is fairly comparable to the drop observed for spo4 (cf. Fig. 2A).
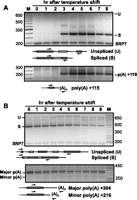
(A) Splicing (top) of both introns in the spo4 gene proceeds in parallel with polyadenylation (bottom) over a meiotic time course. RNA processing reactions were assayed as shown in the diagram beneath each gel image; the minor bands migrating between unspliced spo4 precursor and fully spliced spo4 mRNA may be derived from partially spliced products, but their abundance is insufficient for sequence analysis. (B) The constitutively expressed SPAC6F6.11c gene is fully processed throughout the meiotic time course. In the splicing assay (top), the predominant product observed at all time points lacks all four introns; the minor bands migrating more slowly than fully spliced SPAC6F6.11c RNA may reflect partial splicing, which could not be verified by sequence analysis. The major and minor 3′ RNA processing sites detected in the polyadenylation assay (bottom) were reported in a previous study (Cremona et al. 2011).
Meiosis-specific polyadenylation of rem1 transcripts is independent of splicing
The absence of polyadenylated rem1 RNA that retains the intron suggested that either the two RNA processing reactions are mechanistically linked or that splicing is required for production of stable rem1 RNA. To distinguish these possibilities, we deleted the intron from the rem1 gene and examined 3′ RNA processing over a meiotic time course. The results indicate that splicing is not absolutely required for regulation of polyadenylation, as the intronless allele produces poly(A)+ RNA only during meiosis (Fig. 3A). Interestingly, although 3′ maturation of transcripts from the Δintron gene remains meiosis-specific, polyadenylation is delayed by ∼1 h relative to wild-type rem1 (cf. Figs. 3A and 1A, bottom). To eliminate the possibility that a limiting factor might be titrated by the plasmid-borne allele, we integrated the Δintron construct at the native rem1 locus; the same ∼1-h delay was observed (data not shown).
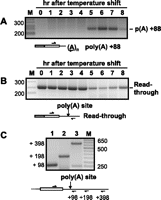
(A) Polyadenylation of rem1 transcripts is regulated independent of splicing. Cells harboring a deletion of the chromosomal rem1 gene (Averbeck et al. 2005) were transformed with a plasmid carrying an allele from which the intron had been precisely deleted. Polyadenylation was assayed using the same primers as in Figure 1A (bottom). (B) Read-through transcripts from the Δintron rem1 allele are present throughout a meiotic time course, but their levels are reduced in mid and late meiotic cells. Transcripts that extend downstream from the meiotic poly(A) site were detected using the same 5′ primer used to assay polyadenylation and a 3′ primer 10 nt downstream from the polyadenylation site. (C) Transcripts that extend well beyond the meiotic poly(A) site can be detected in proliferating cells harboring plasmid-borne wild-type rem1. Transcripts that extend beyond the polyadenylation site were assayed by RT-PCR as diagrammed, using a common 5′ primer and a series of 3′ primers (numbers are relative to the stop codon).
As 3′ processing of RNA polymerase II transcripts is mechanistically coupled to transcription termination in diverse eukaryotes (Proudfoot 2004; Richard and Manley 2009), we next examined the profile of read-through transcripts from the rem1 gene. RT-PCR with a 3′ primer complementary to sequences downstream from the 3′ UTR on the same meiotic time course assayed in Figure 3A (Δintron allele) reveals that accumulation of read-through transcripts declines at the same time that poly(A)+ RNA is detected (Fig. 3, cf. B and A). A similar reciprocal relationship between read-through and polyadenylated transcripts is observed for endogenous wild-type rem1 over a meiotic time course, and we are also able to detect unspliced read-through RNA in assays using the same RNA preparations used in Figure 1 (data not shown). In RNA from actively growing cells harboring the wild-type rem1 plasmid, robust signals reflecting read-through transcription can be detected >300 nt beyond the site of meiotic 3′ RNA processing (Fig. 3C).
Negative control of rem1 RNA processing requires an extended upstream region
In an earlier study focused on splicing, we showed that rem1 DNA flanking the coding region suffices to block removal of the heterologous azr1 intron, which is constitutively spliced in vegetative cells when expressed under control of its native flanking DNA (Averbeck et al. 2005). To determine whether the sequences that suppress splicing reside upstream of, downstream from, or in both directions from the coding region, we used the same unbiased strategy previously applied to crs1 (McPheeters et al. 2009), namely, replacement of these segments with corresponding DNA from the nmt81 vector. We then introduced these constructs into cells in which the chromosomal rem1 gene was deleted (Averbeck et al. 2005) and assayed RNA processing of the resulting transcripts (see Materials and Methods for details). Like wild-type rem1 (Fig. 4A, right, lane 1), the construct in which nmt81 sequences replace the downstream DNA produces unspliced RNA (lane 4), indicating that negative control is not mediated by sequences 3′ from the open reading frame (ORF). In contrast, replacing both the 5′ and 3′ DNA (lane 2), or the upstream region only (lane 3), relieves the block to rem1 splicing in proliferating cells. We conclude that splicing is suppressed by sequences that reside 5′ to the ORF.
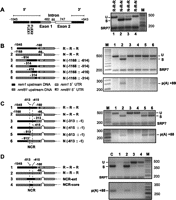
Mapping of the region that negatively controls rem1 RNA processing. (A) The element that prevents rem1 splicing in proliferating cells is located upstream of the coding region. (Left) Diagram of the wild-type insert, with noncoding DNA represented by black rectangles, the coding portions of exons 1 and 2 by gray and white rectangles, respectively, the intron by a horizontal line, and the FLEX boxes centered at −178 and −219 relative to the ATG by vertical white lines. Chimeric genes were created by replacing rem1 sequences surrounding the ORF with the corresponding segments of the nmt81 (no message on thiamine) vector, a commonly used expression system in fission yeast (see Materials and Methods). (Right) RT-PCR splicing assays on RNA isolated from cells transformed with the indicated constructs. Chimeras are labeled with a three-letter code indicating the source of the 5′, internal, and 3′ segments; (R) rem1, (N) nmt81: lane 1, wild type (R-R-R); lane 2, substitution of both 5′ and 3′ DNA (N-R-N); lane 3, substitution of 5′ DNA only (N-R-R); lane 4, substitution of 3′ DNA only (R-R-N). To eliminate endogenous signals, analyses were carried out in the rem1 gene deletion strain JLP1014 (Averbeck et al. 2005). (B,C) Splicing (top) and polyadenylation (bottom) assays on the constructs diagrammed at the left, in which increasing amounts of rem1 DNA are replaced with nmt81 sequences. The two FLEX boxes identified in a previous study (Moldon et al. 2008) are indicated by white lines. As the rem1 5′ UTR is longer than the nmt81 5′ UTR (see Materials and Methods), carets (^) are inserted into the diagrams of constructs carrying the latter to preserve ORF alignment. (D) Splicing (top) and polyadenylation (bottom) assays on chimeras in which the 5′ DNA is derived from nmt81, except for the 98-nt NCR (core) or an additional 100 nt upstream and downstream (ext).
In a study that also focused on rem1 splicing, other investigators identified two “FLEX boxes” located just upstream of the transcription start site (Moldon et al. 2008). This motif binds the positive meiotic regulator Mei4p (Horie et al. 1998), a member of the forkhead family of transcription factors, and was named for its resemblance to the binding site of the human forkhead protein FREAC-1 combined with the first fission yeast gene in which it was identified (FLEX = FREAC-like consensus element of spo six). In rem1, site-directed mutagenesis showed that the FLEX boxes are required for positive regulation of splicing during meiosis (Moldon et al. 2008). To map the region that mediates negative control, we replaced increasing amounts of rem1 upstream flanking DNA with nmt81 sequences. Analysis of chimeras with substitutions starting at the 5′ end of the insert and proceeding toward the ATG revealed that splicing inhibition is not detectably relieved by replacement of rem1 sequences between −1045 and −614 or −514 (Fig. 4B, top, lanes 3,4). However, extending the substitution to −416 or −314 (lanes 5,6) allows splicing, albeit to a lesser extent than the positive control (lane 2). As these constructs retain the FLEX boxes, the positive motifs may also play a role in negative regulation.
In light of our demonstration that polyadenylation of rem1 transcripts remains meiosis-specific even when the intron is deleted (Fig. 3A), we asked whether the same constructs that give rise to splicing also allow polyadenylation in proliferating cells. While inhibition of the two RNA processing reactions is relieved in parallel (Fig. 4B, lanes 5,6), the majority of the RNA produced from these constructs remains unspliced, and the polyadenylation signals are weak relative to the positive control (lane 2); thus, negative regulation has not been completely disrupted. Finally, replacing the rem1 sequences eliminated in construct 6 with the corresponding DNA from adh1 also partially relieves inhibition of splicing and polyadenylation (data not shown), indicating that splicing of RNA from the nmt81-rem1 chimeras is unlikely to result from positive contributions of the heterologous sequences. These results are consistent with published data indicating that similar res2 or nmt41 substitutions allow rem1 splicing in nonmeiotic cells (Moldon et al. 2008).
To determine the contribution of the region immediately upstream of the ATG to negative regulation of splicing and polyadenylation, we analyzed a complementary series of substitutions proceeding in the 3′ to 5′ direction (Fig. 4C). Importantly, processing of rem1 RNA is not observed when the region encompassing the 5′ UTR, transcription start site, and FLEX boxes is replaced (lanes 3,4). In contrast, substitutions extending further upstream lead to both splicing and polyadenylation (lanes 5,6). The segment common to the four constructs in which the block to rem1 RNA processing in vegetative cells is relieved (Fig. 4B,C, lanes 5,6) extends from −513 to −415 relative to the ATG; these 98 nt have thus been designated the negative control region (NCR). The more efficient splicing and polyadenylation of rem1 RNA produced from alleles in which DNA immediately upstream of the ORF, in addition to the NCR, is replaced by nmt81 sequences (Fig. 4, cf. B and C, lanes 5,6) is consistent with the suggestion above that the FLEX boxes may function in negative as well as positive control. However, as substitution of the region encompassing the 5′ UTR, transcription start site, and FLEX boxes alone does not relieve inhibition of RNA processing in growing cells (Fig. 4C, lanes 3,4), it most likely enhances the effect of the NCR rather than playing an autonomous role in RNA processing inhibition.
As an additional confirmation that the effect of nmt81 substitutions is due to a loss of negative control, we analyzed constructs with upstream DNA derived entirely from nmt81 except for the 98-nt NCR, either alone or in combination with an additional 100 nt of rem1 DNA on either side (Fig. 4D). Whereas the N-R-R allele, in which the entire 5′ region is replaced (lane 2), shows RNA processing comparable to the positive control (5-h time point from a meiotic time course, lane C), polyadenylation is undetectable and splicing is very weak when the only rem1 DNA present is the core or extended NCR (lanes 3,4). We conclude that the NCR suffices to block polyadenylation, and also substantially represses splicing, in proliferating cells. Splicing is suppressed to a greater extent in the NCR-ext allele than in the NCR-core allele (cf. lanes 3 and 4), suggesting that sequences immediately surrounding the NCR contribute to negative regulation. Importantly, neither NCR substitution encompasses the FLEX boxes, and MEME analysis (Gupta et al. 2007; Bailey et al. 2009) identifies no other discrete motif that is broadly conserved among genes with temporal patterns of RNA accumulation similar to rem1 (data not shown). Thus, negative control of RNA processing may not be mediated by a sequence-specific DNA binding protein (see Discussion).
The NCR regulates polyadenylation independent of splicing
As polyadenylation is restricted to meiosis even after deleting the intron (Fig. 3), we wanted to determine whether ablating the NCR relieves the block to polyadenylation in vegetative cells in the absence of splicing. To this end, we assayed RNA processing in pairs of alleles carrying 5′ DNA derived either entirely from rem1 (Fig. 5A, odd-numbered constructs) or a substitution of the 98-nt core NCR with nmt81 sequences (NCR-sub, even-numbered constructs). Transcripts from control constructs carrying native upstream DNA show no RNA processing whether the intron is present, absent, or carries an inactivating 5′ splice site mutation (Fig. 5B, lanes 1,3,5). Transcripts from the otherwise wild-type NCR-sub allele are spliced, albeit weakly (Fig. 5B, top, lane 2), indicating that replacing the NCR alone only partially relieves negative control of rem1 splicing in proliferating fission yeast cells. Although these RT-PCR assays are only semiquantitative, polyadenylation appears robust in the sample carrying the NCR substitution (Fig. 5B, bottom, lane 2). These results are consistent with the data in Figure 4D, suggesting that the NCR is sufficient to block rem1 polyadenylation, whereas additional sequences are required to completely prevent splicing in proliferating cells.
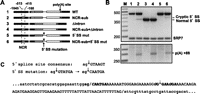
(A) Diagrams of chimeras with 5′ DNA derived solely from rem1 (odd numbers) or containing a replacement of the NCR with nmt81 sequences (even numbers). In constructs 1 and 2, the remainder of the gene is wild type. In constructs 3 and 4, the intron has been precisely deleted. In constructs 5 and 6, the nearly invariant GT at the 5′ end of the intron was mutated to CA, which generally abolishes use of a 5′ splice site (Alvarez and Wise 2001). (B) Splicing (top) and polyadenylation (bottom) assays on the chimeras diagrammed in panel A. (C) Identification of the cryptic 5′ splice site activated when the NCR is replaced in conjunction with mutating the natural exon/intron boundary. (Top) Transversions at the first two nucleotides of the intron were used to inactivate the natural 5′ splice site (Alvarez and Wise 2001). (Bottom) DNA sequence of the intron (uppercase) and surrounding exons (lowercase) with boundaries indicated by slashes. The cryptic 5′ junction, previously noted by Ayté and colleagues in an allele with nmt41 sequences replacing the entire upstream region (Moldon et al. 2008), is marked with an arrow. The octanucleotides encompassing both the natural and cryptic 5′ splice sites are highlighted in bold, with nucleotides that deviate from the fission yeast consensus (Wood et al. 2002) indicated by italics.
The most important finding from this experiment is that the NCR does indeed regulate 3′ RNA processing independent of splicing. Whereas transcripts from the mutant lacking the intron but carrying native flanking DNA are not polyadenylated (Fig. 5B, bottom, lane 3), those that carry the NCR substitution are (lane 4). Intriguingly, replacing the NCR in an allele in which the intron is present but carries inactivating mutations at the 5′ splice site does not lead to complete intron retention but rather redirects splicing to a cryptic 5′ junction (Fig. 5B, top, lane 6). Remarkably, splicing efficiency is reduced only approximately twofold (cf. lanes 2 and 6) even though the cryptic junction activated in this mutant deviates from all natural fission yeast 5′ splice sites by virtue of an A at position +2 (Fig. 5C), which normally renders a 5′ splice site nonfunctional (Alvarez and Wise 2001).
Identification of a second meiosis-specific polyadenylation site in the rem1 intron
Although the persistence of regulated polyadenylation in the absence of the rem1 intron (Figs. 3, 5) argues against splicing as the primary target of negative regulation, the Δintron allele shows a reproducible 1-h delay in 3′ RNA processing. This observation suggests a role for intronic sequences in determining the precise timing of rem1 RNA accumulation. A potential clue to what this role might be came from the previously reported finding that a polypeptide lacking the cyclin domain is encoded by exon 1 of rem1 (Moldon et al. 2008). These investigators proposed that the truncated protein, which has been detected in HA-tagged form, arises via translation of rem1 mRNA that retains the intron (Moldon et al. 2008); however, an alternative possibility is that it is produced via polyadenylation within the intron. We confirmed this hypothesis using modified RT-PCR and electrophoresis conditions (Fig. 6A). Unexpectedly, accumulation of mRNA polyadenylated within the intron peaks in mid-meiosis, similar to the full-length poly(A)+ RNA (Fig. 1A). To determine whether polyadenylation at the two sites precisely coincides over a meiotic time course, we assayed both 3′ RNA processing events concurrently. In addition to confirming that the peaks for the truncated and full-length mRNAs are indistinguishable, the data suggest preferential use of the downstream polyadenylation site, as the upper band is more intense (Fig. 6B) even though RT-PCR generally favors shorter products. Sequence analysis revealed that the cleavage event that leads to polyadenylation within the intron occurs immediately downstream from the 5′ splice site hexanucleotide (Fig. 6C) and, like the +88 site, follows a GA dinucleotide rather than the consensus YA (see Discussion).
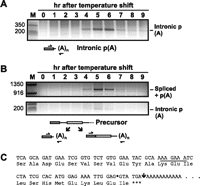
(A) Discovery of a polyadenylation site within the rem1 intron. To optimize conditions for detecting a truncated rem1 mRNA, PCR amplification was performed with a shortened extension time for each cycle. The positions of primers are diagrammed beneath the gel image. (B) Simultaneous detection of intronic and downstream polyadenylation. Once we knew its size, we were able to detect the product derived from polyadenylation within the intron using standard RT-PCR and gel conditions (see Materials and Methods); the same 5′ primer also detects the (much larger) spliced product that is polyadenylated at the downstream site as diagrammed at the bottom. Two slices from the top third and bottom quarter of the same gel are shown. (C) Sequence analysis to identify the position of intronic polyadenylation. The bands marked intronic p(A) in panels A and B were eluted from the gels, and the sequence extending into the poly(A) tail was determined. Both gave the sequence shown, in which the exon/intron boundary is marked with a diamond. As indicated by the conceptual translation, the intron lies between codons such that the unspliced RNA polyadenylated within the intron encodes a single isoleucine beyond the peptide translated from exon 1 before a termination codon is encountered immediately preceding the poly(A) tail.
Intronic polyadenylation is controlled by the NCR and enhanced by inactivating splicing signals
Given that the polyadenylation site within the rem1 intron is activated in parallel with the +88 site downstream from the ORF, we asked whether it is also regulated by the NCR. Assaying the same RNA preparations as in Figure 5, we observed no signals indicative of intronic polyadenylation for alleles carrying exclusively rem1 upstream DNA (Fig. 7A, odd lanes) nor in the Δintron sample, which lacks the site (Fig. 7A, lane 4). An intronic polyadenylation signal is almost undetectable when RNA from the NCR substitution allele is assayed (Fig. 7A, lane 2). However, far more robust polyadenylation within the intron, comparable to the 5-h meiotic control (Fig. 7A, second lane from left), is observed with the allele containing a 5′ splice site mutation in addition to the NCR substitution (Fig. 7A, lane 6). This result provides further evidence that the NCR regulates intronic polyadenylation and further suggests that the splicing and polyadenylation machinery compete for overlapping sites (see Discussion).
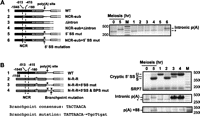
(A) The intronic polyadenylation site is regulated by the NCR. (Left) Diagrams of the relevant regions of rem1/nmt81 chimeras assayed for polyadenylation within the intron. (Right) The same RNA preparations assayed in Figure 5 were used to assay intronic polyadenylation (lanes 1–6). As negative and positive controls, the assay was also carried out on RNA extracted from vegetative (0-h time point) and meiotic (5-h time point) cells, respectively. Sequence analysis indicates that the product marked with an asterisk is derived from rpl2001/2002. It is absent from the meiotic sample because the transcript accumulates only in vegetative cells (Mata et al. 2002). (B) Effects of splice site mutations on rem1 RNA processing. The alleles assayed are diagrammed at the left. The results of RNA processing assays are shown at right, with splicing in the top panel, intronic polyadenylation in the middle, and polyadenylation at the +88 site at the bottom.
To explore the dynamic interplay between splicing and polyadenylation at the intronic and +88 sites, we analyzed all three RNA processing events in alleles carrying either native 5′ DNA or an nmt81 substitution, with and without splicing signal mutations (Fig. 7B). Notably, whereas unspliced rem1 RNA produced from the wild-type chromosomal gene (Fig. 7B, top, left-most lane, 0-h meiotic time point) is barely detectable, the signal from plasmid-borne rem1 is more robust (Fig. 7B, top, lane 1), suggesting that the capacity of the pathway responsible for turnover may be exceeded (see Discussion). Intronic polyadenylation is not observed in the wild-type allele (Fig. 7B, middle, lane 1), nor when splicing is efficient due to a complete 5′ nmt81 substitution (Fig. 7B, middle, lane 2). Conversely, the signal reflecting polyadenylation within the intron is dramatically enhanced relative to the meiotic control in alleles carrying splicing signal mutations (Fig. 7B, middle, cf. lanes 3 and 4 to the 5-h sample, second lane from left). The similar results for the allele that shows no splicing and the one that shows cryptic splicing suggests that the distance between the intronic polyadenylation site and the cryptic 5′ junction (22 nt) is sufficient to prevent steric interference between the splicing and 3′ processing machinery. Alternatively, intronic polyadenylation may occur only on the subpopulation of rem1 transcripts that did not assemble a spliceosome. Interestingly, the strongest band reflecting polyadenylation at the +88 site is observed in the double splicing signal mutant (Fig. 7B, bottom, lane 4), bolstering the conclusion that downstream 3′ RNA processing is not dependent on or enhanced by splicing.
DISCUSSION
The central conclusion of this study, that polyadenylation serves as the actuator to temporally control expression of the rem1 gene, significantly changes our view of how this meiotic cyclin, and most likely other middle meiotic genes, are regulated in fission yeast. In a broader context, our results add to the growing body of evidence that 3′ RNA processing is an important target of eukaryotic gene regulatory mechanisms. To date, alternative polyadenylation (for review, see Di Giammartino et al. 2011) has received far more attention than on-off regulation, largely due to the bias introduced by analyzing poly(A)+ RNA. Our results highlight the importance of studying read-through as well as 3′ processed RNA to gain a complete picture of how the decision about when and where to polyadenylate a given transcript impacts gene output.
Distinct mechanisms regulate polyadenylation of early and middle meiotic transcripts
The regulatory mechanism described here for rem1 shares two features with the one we described earlier for crs1 (McPheeters et al. 2009): (1) The level of unspliced transcripts increases before spliced RNA is detected; and (2) the peaks in accumulation of spliced and polyadenylated RNA during meiosis coincide (Fig. 1). Despite these superficial similarities, the molecular mechanisms appear to be distinct. Early meiotic genes, including crs1, are negatively regulated via selective nuclear RNA turnover, and the central player in the targeting mechanism is Mmi1p, a member of the YTH family of RNA binding proteins (Harigaya et al. 2006). In contrast, middle meiotic genes are regulated both positively and negatively by members of the forkhead family of transcription factors, which control cotranscriptional RNA processing decisions (Moldon et al. 2008). For early genes, “determinants of selective removal” are generally located near the 3′ end of the ORF and are recognized at the RNA level (Harigaya et al. 2006; McPheeters et al. 2009), while the cis-acting signals that control middle meiotic gene output are located upstream of the transcription start site and recognized at the DNA level (Horie et al. 1998; Moldon et al. 2008; this work).
A striking feature of the rem1 regulatory mechanism is that meiosis-specific polyadenylation is independent of splicing (Fig. 3). The resulting conclusion that 3′ processing rather than splicing is the primary target of regulation is bolstered by the absence of introns in approximately half of the middle meiotic genes with RNA accumulation profiles similar to rem1 (Mata et al. 2002); thirteen of these were shown in our previous study to accumulate polyadenylated RNA only during meiosis (Cremona et al. 2011). In spo4, both introns are spliced exclusively during meiosis, similar to the multi-intronic crs1 transcript (Averbeck et al. 2005; McPheeters et al. 2009) but distinct from another early gene, rec8, in which only the terminal intron displays meiosis-specific splicing (Chen et al. 2011). We previously proposed that coincident peaks of polyadenylation and splicing might reflect communication between the splicing and polyadenylation machinery across the 3′ exon (McPheeters et al. 2009), as in mammals (Niwa and Berget 1991; Cooke et al. 1999; Kyburz et al. 2006). However, the persistence of meiosis-specific polyadenylation in the absence of the intron, together with the large size of rem1 exon 2 (835 nt), argues against a requirement for terminal exon definition. Finally, the location of the cryptic 5′ splice site downstream from the natural 5′ junction supports an intron definition mechanism for splice site pairing (Romfo et al. 2000; Alvarez and Wise 2001).
Shifting paradigms for negative regulation of middle meiotic genes
Whereas the signals and factors that positively regulate rem1 and other middle meiotic genes in fission yeast have been extensively investigated (Horie et al. 1998; Abe and Shimoda 2000; Nakamura et al. 2000; Watanabe et al. 2001; Okuzaki et al. 2003; Nakamura et al. 2004; Malapeira et al. 2005; Nakamura et al. 2005; Ogino and Masai 2006; Mata et al. 2007; Murakami-Tonami et al. 2007; Moldon et al. 2008; Nakase et al. 2009), only three published reports have focused on negative regulation (Shimoseki and Shimoda 2001; Averbeck et al. 2005; Chen et al. 2012), and none of these examined polyadenylation. In the earliest, splicing of transcripts from the mes1 gene was shown to occur even in vegetative cells when a secondary structure in the 5′ UTR was disrupted (Shimoseki and Shimoda 2001). Although mes1 and rem1 show similar temporal patterns of RNA accumulation, we do not find a stable secondary structure in the rem1 5′ UTR. Furthermore, in that the cis-acting signal in rem1 lies upstream of the transcription start site, the mechanism we have uncovered must operate at the DNA level. The second study (which we coauthored) showed that sequences flanking the rem1 coding region are necessary and sufficient to block splicing of a heterologous transcript in proliferating cells (Averbeck et al. 2005). Here, we have extended this investigation by mapping the negative regulatory region and showing that its primary target in its native context is polyadenylation rather than splicing.
The most recent study reported that anti-sense transcripts that overlap many middle meiotic genes are present in vegetatively growing fission yeast cells (Chen et al. 2012). These authors proposed that transcripts complementary to the mRNA were the source of signals attributed to unspliced RNA from middle meiotic genes in earlier studies (Averbeck et al. 2005; Moldon et al. 2008; Chen et al. 2011). This inference was based on the use of protocols to assay splicing that did not preserve information about the strand from which the original RNA was transcribed. Because the splicing assays employed here are strand-specific (see Materials and Methods for details), the proposed scenario does not apply. Why, then, were unspliced rem1 and spo4 RNAs not detected by Chen et al. (2012) using a strand-specific assay? The apparent discrepancy is due to the fact that the anti-sense study was limited to poly(A)+ RNA, whereas the unspliced rem1 and spo4 transcripts we detect here in proliferating and early meiotic cells are not properly cleaved and polyadenylated. The analysis of total vs. oligo(dT)-primed cDNA as the substrate for splicing assays also provides an explanation for why unspliced mes1 RNA could be detected in some studies (Kishida et al. 1994; Shimoseki and Shimoda 2001) but not others (Averbeck et al. 2005; Malapeira et al. 2005). Finally, we note that our strand-specific assays of total RNA allowed the identification of two transcripts that are spliced in vegetative and meiotic cells but polyadenylated only during meiosis (Cremona et al. 2011).
How do 5′ sequences control events at the 3′ end of the gene?
The most unprecedented finding reported here is that DNA sequences located upstream of the transcription start site of the rem1 gene regulate polyadenylation to produce both truncated and full-length mRNAs, in addition to contributing to splicing regulation (Figs. 4–7). Although there is ample precedent for communication between the two ends of a gene to positively regulate gene expression (see below), negative control of 3′ RNA processing by a 5′ element has not, to our knowledge, been previously reported in any organism. A complete picture of how the NCR functions in proliferating cells to block the molecular events that lead to proper temporal expression of rem1 during meiosis has not yet emerged. However, important clues are provided by comparing processing of the RNA in cells harboring mutant alleles with the pattern observed during meiosis. The most parsimonious explanation for our results is a cotranscriptional mechanism consisting of two bifurcating pathways in which a choice is made between splicing and polyadenylation at the intronic site and between polyadenylation and read-through transcription at the downstream site (Fig. 8).

(Top) The salient features of the rem1 gene elucidated in this study. (Bottom) Cotranscriptional models to account for the fates of mutant rem1 transcripts. The RNA processing events that unfold as RNA polymerase II (white on black stippling) traverses the wild-type rem1 gene in mid-meiotic cells (left, based on Fig. 1A, bottom, 5-h sample and Fig. 7A, 5-h meiotic control) or the mutant rem1 gene (black box = NCR substitution; x = 5′ splice site mutation) in proliferating cells (right, based on Fig. 5B, bottom, lane 6 and Fig. 7A, lane 6) are depicted. The size of each arrow indicates the relative yield of cryptically spliced and unspliced RNA.
In light of the dominant role for polyadenylation in the rem1 regulatory mechanism, it is notable that the 3′ processing of wild-type rem1 RNA in mid-meiotic cells (Fig. 8, bottom left panel) is similar to that of the allele with an NCR substitution and a mutant 5′ splice site in proliferating cells (Fig. 8, bottom right panel). On the other hand, there is a dramatic difference in the splicing patterns: whereas little unspliced RNA is detected in meiotic cells, the mutant mRNA is inefficiently spliced, presumably due to the unusual nature of the cryptic site (Fig. 5C). The accumulation of unspliced precursor may reflect production of RNA from the plasmid-borne allele that exceeds the capacity of the pathway responsible for destroying middle meiotic transcripts in mitotically growing cells. Although we have not yet investigated this mechanism, it is tempting to speculate that a newly discovered nuclear surveillance pathway that selectively targets intron-containing RNAs in fission yeast (Lemieux et al. 2011) may be involved. As RNA from the intron deletion allele would not be a substrate for this pathway, this may explain the apparently higher levels of unprocessed read-through RNA in proliferating cells (Fig. 3B), which may, in turn, lead to the reproducible delay in 3′ processing during meiosis (Fig. 3A).
The findings reported here necessitate a substantive revision of models for middle meiotic gene regulation in fission yeast. Based on experiments that did not consider polyadenylation as a variable, previous investigators proposed that the forkhead transcription factor Mei4p, which is produced only during meiosis, displaces the negative regulator Fkh2p, which is expressed in both mitotic and meiotic cells, from the FLEX boxes (Moldon et al. 2008). Our mapping of the negative control region (Fig. 4) is difficult to reconcile with this simple exchange model, as the block to polyadenylation and splicing was partially relieved even when the FLEX region was present, and replacement of the region that encompasses these motifs alone did not allow RNA processing. As MEME analysis (Gupta et al. 2007; Bailey et al. 2009) revealed no discrete motifs conserved between the rem1 NCR and other similarly regulated middle meiotic genes, it is unlikely that a transcription factor other than Fkh2 holds the key. We can envision two general models through which the NCR might regulate polyadenylation and splicing. First, juxtaposition of the two polyadenylation sites in the rem1 gene with the promoter is an attractive scenario in light of evidence that gene looping requires interactions between factors that function in 3′ processing and transcription initiation (Ansari and Hampsey 2005; Mukundan and Ansari 2011), which, in turn, might promote recycling of the transcription/RNA processing machinery (Mapendano et al. 2010; Shandilya and Roberts 2012). Second, we were unable to further delimit the NCR by scanning mutagenesis (data not shown), and sequences extending downstream are required to completely block RNA processing in proliferating cells. Thus, this element may nucleate a repressive chromatin structure incompatible with recruitment or deposition of splicing and polyadenylation factors in proliferating cells. Importantly, these mechanisms are not mutually exclusive; additional work will be required to determine whether either or both function in regulation of rem1 and other middle meiotic genes.
Is alternative polyadenylation an evolutionarily conserved mechanism for regulating meiotic transcripts?
In light of the general rule that poor matches to consensus motifs are associated with regulated polyadenylation (Lutz 2008; Di Giammartino et al. 2011), it is notable that the cleavage site for both intronic and downstream polyadenylation follows a GA dinucleotide (Figs. 1, 6). Although a GA cleavage site is found in at least six other transcripts that show rem1-like regulation (Cremona et al. 2011), this feature is not ubiquitous among middle meiotic genes (KD Potter and JA Wise, unpubl.). In contrast, the site of poly(A) tail addition in crs1 and other early meiotic genes is preceded by the consensus dinucleotide, YA (McPheeters et al. 2009; Chen et al. 2011; Cremona et al. 2011). The presumptive positioning elements upstream of both cleavage sites in rem1 (Figs. 1D, 6C) also deviate from the consensus hexanucleotide (AAUAAA) (Tian and Graber 2011), whereas the major polyadenylation signal of crs1 contains an exact match to the mammalian consensus (McPheeters et al. 2009). Finally, analogous to rem1, testis-specific mouse transcripts often contain deviant hexanucleotides and are inefficiently polyadenylated in somatic cells (McMahon et al. 2006; Liu et al. 2007).
In contrast to mammals, where tissue and developmental-stage-specific regulation of the choice between splicing and polyadenylation is long-established and quite commonplace (Early et al. 1980; Maki et al. 1981; Rosenfeld et al. 1983; Di Giammartino et al. 2011), our discovery of a polyadenylation site within the rem1 intron is the first example of this phenomenon in fission yeast. The polypeptide encoded solely by the first exon of rem1 was previously implicated in meiotic recombination (Moldon et al. 2008), but this effect was not shown to be direct. In light of the autoregulatory mechanism elucidated for the Arabidopsis FCA gene (Quesada et al. 2003), encoding an RNA binding protein that helps determine whether polyadenylation occurs at a site within an intron or in the 3′ UTR (Simpson et al. 2003), it is tempting to speculate that the truncated rem1 protein may make a similar contribution to autoregulation. One difference is that 3′ processing of rem1 transcripts within the intron and downstream from the second exon show indistinguishable peaks during meiosis (Fig. 6B), whereas the two polyadenylation sites in FCA are utilized in temporally distinct patterns. However, this may, at least in part, reflect analysis of total RNA here vs. poly(A)+ RNA in the plant studies (Simpson et al. 2003). Another potential parallel is that alternative polyadenylation in Arabidopsis is regulated by anti-sense transcripts (Hormyik et al. 2010), as is middle meiotic gene expression in fission yeast (Chen et al. 2012).
Although the role of cytoplasmic polyadenylation and translational control has dominated the investigation of meiotic gene regulatory mechanisms in animals, nuclear polyadenylation also plays a crucial role (McMahon et al. 2006; Liu et al. 2007). Given the important contributions of changing polyadenylation patterns during meiosis in organisms as diverse as fission yeast, plants, and mammals, it is plausible to suggest that further investigation might reveal a single evolutionary origin.
MATERIALS AND METHODS
S. pombe manipulations
Complete genotypes for strains used in this study are as follows:
-
F90: h- leu1-32 ura4-D18 pat1-114 (Figs. 1, 2, 6, 7; Averbeck et al. 2005).
-
JLP1014: h- ade6-M210 ura4-D18 his7-366 leu1-32 rem1::his7+ (Figs. 3C, 4, 5, 7; Averbeck et al. 2005).
-
JAW-FY2: h- ade6-M210 ura4-D18 rem1::ura4+ pat1-114 (Fig. 3A,B).
Fission yeast media and growth conditions, as well as the protocol for inducing ectopic meiosis, were described in a previous publication (Averbeck et al. 2005).
Construction and analysis of chimeric and mutant plasmids
Plasmids were constructed using standard or overlap-extension PCR as previously described (Averbeck et al. 2005; McPheeters et al. 2009). The sequences of oligonucleotides employed in these manipulations are available upon request. In most experiments, we used the nmt81 vector as the source of segments for replacing rem1 flanking DNA (Basi et al. 1993). This vector contains mutations in the TATA box of the nmt1 (no message on thiamine) promoter (Maundrell 1990), which dramatically reduce transcription relative to the full-strength promoter commonly used to drive high-level expression in fission yeast (Basi et al. 1993; Forsburg 1993). The selection of segments for substitution took into account the different 5′ UTR lengths of nmt81 and rem1 transcripts (66 vs. 160 nt) (Fig. 1; Maundrell 1990), i.e., corresponding fragments came from the same location relative to each transcription start site.
RNA processing assays
For meiotic time courses, total RNA was isolated from the pat1-114 strain F90 at 1-h intervals after induction of ectopic meiosis. Total RNA was also used in RNA processing assays of chimeric or mutant genes. Splicing of rem1, spo4, and SPAC6F6.11c transcripts was assayed by semiquantitative RT-PCR using gene- and strand-specific oligonucleotides as previously described (Webb et al. 2005; McPheeters et al. 2009); amplicons are shown in the relevant figures. Only the 3′ primer was present during initial cDNA synthesis with MuLV reverse transcriptase (Applied Biosystems, Life Technologies), which has extremely low RNAse H activity and is, therefore, suitable for first-strand cDNA synthesis. Reverse transcription was carried out for a limited time to avoid second-strand synthesis, and the 5′ primer was then added for subsequent rounds of PCR amplification. In contrast, the protocols utilized in several published studies that reported meiosis-specific splicing in fission yeast (e.g., Figs. 1–3 in Averbeck et al. 2005; Malapeira et al. 2005; Moldon et al. 2008) employed double-stranded cDNA primed with oligo(dT) as the starting material. In addition to losing information about strand-specificity, restricting the analysis to RNA that had undergone polyadenylation precluded discovery of the regulatory mechanism elucidated here. Conversely, analysis of poly(A)+ rather than total RNA explains why mes1 RNA, the first reported example of meiosis-specific splicing in S. pombe (Kishida et al. 1994) appeared to be constitutively spliced in these studies (Averbeck et al. 2005; Malapeira et al. 2005). As mes1 lacks an annotated anti-sense transcript (see http://www.pombase.org/spombe/result/SPAC5D6.08c), the polyadenylated species detected was presumably mature mRNA that had escaped the negative regulatory mechanism.
Despite considerable effort, we were unable to devise primers or conditions to accurately assay rem1 splicing using quantitative real-time PCR. Using the same design principles that yielded useful primers for qPCR assays of multiple other meiotic genes (McPheeters et al. 2009; Cremona et al. 2011), the numerous combinations tested yielded multiple bands under all conditions, rendering the results unreliable. Although qPCR could not be used, an internal loading control (SRP7 RNA) was included in all splicing assays, and care was taken to minimize cycle numbers and maintain consistency throughout all experiments.
Polyadenylation of rem1, spo4, and SPAC6F6.11c transcripts downstream from the ORFs was assayed using gene-specific 5′ primers complementary to the terminal exon and oligo(dT) as the 3′ primer (McPheeters et al. 2009). Polyadenylation at the site within the rem1 intron was detected using a 5′ primer complementary to exon 1 and oligo(dT) as the 3′ primer. Read-through transcription of rem1 was assayed using a 5′ primer complementary to exon 2 and multiple gene-specific 3′ primers located downstream from the polyadenylation site at +88 as shown in the relevant figures. The next gene downstream from rem1 is transcribed from the opposite strand; however, despite their convergent orientations, it seems unlikely that they exhibit transcriptional interference due to the long distance (591 nt) between the two polyadenylation sites (http://genomebrowser.pombase.org/Schizosaccharomyces_pombe/Location/View?db=core;g=SPBC16E9.20;r=II:1951398-1952828;t=SPBC16E9.20.1). More importantly, the protocol we use to assay read-through transcription is strand-specific and thus would not detect transcripts from either the downstream gene or from the two anti-sense transcripts currently annotated at the rem1 locus (see http://www.pombase.org/spombe/result/SPBC16E9.17c), one of which extends several hundred nucleotides beyond the 5′ mRNA boundary we mapped (Fig. 1C).
All RNA processing assay data reported here have been reproduced 2–5 times.
ACKNOWLEDGMENTS
We thank Dr. David McPheeters for contributions to the early stages of this work, with assistance from Ms. Laura Paszkowski. We also thank our colleagues Hua Lou and Helen Salz for helpful discussions and critical reading of the manuscript. This research was supported by NIGMS.
Footnotes
-
↵2 Corresponding author
E-mail jaw17{at}case.edu
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.033423.112.
- Received March 22, 2012.
- Accepted May 15, 2012.
- Copyright © 2012 RNA Society








