
KREPB6, KREPB7, and KREPB8 are important for editing endonuclease function in Trypanosoma brucei
- 1Seattle Biomedical Research Institute, Seattle, Washington 98109, USA
- 2Department of Global Health, University of Washington, Seattle, Washington 98195, USA
Abstract
Three distinct editosomes are required for the uridine insertion/deletion editing that creates translatable mitochondrial mRNAs in Trypanosoma brucei. They contain KREPB6, KREPB7, or KREPB8 proteins and their respective endonucleases KREN3, KREN2, or KREN1. RNAi knockdowns of KREPB6, KREPB7, and KREPB8 variably affect growth and RNA editing. KREPB6 and KREPB7 knockdowns substantially reduced in vitro insertion site cleavage activity of their respective editosomes, while KREPB8 knockdown did not affect its editosome deletion site cleavage activity despite inhibition of growth and editing. KREPB6, KREPB7, and KREPB8 knockdowns disrupted tagged KREN3, KREN2, or KREN1 editosomes, respectively, to varying degrees, and in the case of KREN1 editosomes, the deletion editing site cleavage activity shifted to a smaller S value. The varying effects correlate with a combination of the relative abundances of the KREPB6-8 proteins and of the different insertion and deletion sites. Tagged KREPB6-8 were physically associated with deletion subcomplexes upon knockdown of the centrally interactive KREPA3 protein, while KREN1-3 endonucleases were associated with insertion subcomplexes. The results indicate that KREPB6-8 occupy similar positions in editosomes and are important for the activity and specificity of their respective endonucleases. This suggests that they contribute to the accurate recognition of the numerous similar but diverse editing site substrates.
Keywords
INTRODUCTION
The mitochondrial genome of trypanosomes is composed of dozens of maxicircles and thousands of minicircles, all of which are intercatenated forming the complex DNA network called kinetoplast DNA (kDNA). The maxicircles contain 18 genes that encode proteins of the oxidative phosphorylation system and the two mitochondrial rRNAs. Twelve of the 18 maxicircle-encoded mRNAs undergo uridine (U) insertion/deletion RNA editing, a post-transcriptional process that is directed by minicircle-encoded guide RNAs (gRNAs) and that creates the open reading frames (ORFs) of the functional mRNAs (Hajduk and Ochsenreiter 2010; Stuart et al. 2005; Aphasizhev and Aphasizheva 2011). In Trypanosoma brucei, nine mRNAs are extensively edited with hundreds of U's inserted and tens of U's deleted throughout their length, while other transcripts are edited to a limited extent. Apocytochrome b (CYb) and maxicircle unidentified reading frame 2 (MURF2) mRNAs are edited at their 5′ termini, and cytochrome oxidase subunit II (COII) is edited internally by the insertion of four U's. Mitochondrial RNA editing is differentially regulated between the different life stages of T. brucei. For example, edited CYb and COII mRNAs are abundant in the insect procyclic form (PF) but are essentially absent in the slender mammalian bloodstream form (BF) (Schnaufer et al. 2002). Reciprocally, fully edited ND7 RNAs are abundant in the BF, while ND7 mRNAs that are only edited in the 5′ terminal domain are abundant in the PF. Edited cytochrome oxidase subunit III (COIII) and ATPase subunit 6 (A6) RNAs are similarly abundant in both stages. The mechanism that regulates this differential RNA editing is unknown.
The editing of each editing site (ES) occurs through a series of sequential catalytic steps beginning with endonucleolytic cleavage of the mRNA followed by either U insertion or deletion, as specified by the gRNA, and finally mRNA ligation. The enzymes that can catalyze these steps have been identified in multi-protein complexes, the ∼20S editosomes, that can catalyze a full round of insertion or deletion RNA editing cycle in vitro. Three types of ∼20S editosomes have been identified, each of which has a common set of 12 proteins and one of three related endonucleases, KREN1, KREN2, or KREN3 (Fig. 1). A general structural organization of the three types of editosomes has emerged from a series of studies of the compositions, protein interactions, and functions of purified editosomes and subcomplexes thereof (Schnaufer et al. 2003, 2010; Panigrahi et al. 2006; Carnes et al. 2011). Each endonuclease-specific editosome also contains one of three related U1-like zinc finger proteins, KREPB6, KREPB7, or KREPB8, that are the focus of this study (Fig. 1B). KREPB6 is found in editosomes with KREN3, KREPB7 is in editosomes with KREN2, and KREPB8 is in editosomes with KREN1. Only KREN1/KREPB8 editosomes contain the KREX1 U-specific exonuclease.
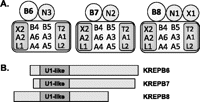
Diagrams of (A) three types of ∼20S editosomes and (B) KREPB6, KREPB7, and KREPB8 proteins that are each uniquely associated with one type of editosome. The KREPB6, KREPB7, and KREPB8 proteins (B6, B7, and B8) are aligned by their U1 zinc finger motif and show their relative sizes. The three editosomes have either KREN3 (N3) or KREN2 (N2) insertion site or KREN1 (N1) deletion site endonucleases. The KREX1 exoUase (X1) is uniquely associated with B8/N1 editosomes. All three editosomes have a common set of 12 proteins that are organized into the KREPA1, KRET2, and KREL2 insertion subcomplex (A1, T2, L2); the KREPA2, KREX2, and KREL1 deletion subcomplex (A2, X2, L1); the KREPA3, KREPA4, KREPA5, and KREPA6 interacting proteins (A3, A4, A5, A6); and KREPB4 and KREPB5 that have degenerate RNase III motifs (B4, B5).
Six editosome proteins, KREPA1-6, each of which has a C-terminal OB-fold motif, form a network of interactions that appears to provide a structural scaffold for the editosome (Schnaufer et al. 2010). Indeed, knockdown of expression of KREPB5, KREPB4, KREPA3, KREPA4 (KREPA5 has not been tested), or KREPA6 results in editosome disruption and ultimately complete loss of editosomes (Wang et al. 2003; Salavati et al. 2006; Babbarwal et al. 2007; Guo et al. 2008; Tarun, Jr. et al. 2008). This network of proteins interacts with the insertion and deletion heterotrimeric subcomplexes to form a common set of 12 proteins found in all ∼20S editosomes. In the insertion subcomplex, KREPA1 directly and stably interacts with the KRET2 3′-terminal uridylyl transferase and the KREL2 RNA ligase and enhances their catalytic activities (Schnaufer et al. 2003). Similarly, in the deletion subcomplex, KREPA2 directly and stably interacts with the KREX2 3′ U-specific exonuclease and the KREL2 RNA ligase and enhances their catalytic activities. These two heterotrimeric subcomplexes can catalyze the U addition/ligation or U removal/ligation steps that follow the endonucleolytic cleavage step that initiates the coordinated catalytic reactions of editing. The relative functions of the two RNA ligases, KREL1 and KREL2, and the two exoUases, KREX1 and KREX2, have not yet been fully determined. Binary protein–protein interactions between the endonucleases and any of the uniquely associating editosome partner proteins have not yet been observed (Schnaufer et al. 2010).
Gene inactivation, RNase III motif mutation, and in vitro editing assays show that KREN1, KREN2, and KREN3 are catalytically active RNase III endonucleases (Carnes et al. 2005, 2008; Trotter et al. 2005; Kang et al. 2006). In addition, each of these editosomes has a distinct ES specificity, presumably to enable accurate recognition and cleavage of numerous diverse ES sequences. The editing of most mRNAs requires multiple gRNAs, each of which specifies multiple cycles of the cleavage, U insertion or deletion, and ligation steps. Indeed, some gRNAs have both insertion and deletion sites. Hence, KREN1, KREN2, and KREN3 editosomes must in some way collaborate to edit multiple sites and use multiple gRNAs to completely edit an mRNA. The process by which editing progresses from one ES to the next and from one gRNA to the next is not yet known.
To date, our laboratory has only detected endonuclease activity in ∼20S editosomes. Cleavage of a synthetic substrate by recombinant KREN1 has been reported but with low specific activity that was only partially diminished upon deletion of the entire RNase III motif (Kang et al. 2006). In contrast, cleavage activity was essentially eliminated in purified editosomes in which the RNase III motif had a single nucleotide mutation (Carnes et al. 2005, 2008; Trotter et al. 2005). This result implies that efficient catalytic activity of KREN1-3 may require other editosome components. Thus, the composition and organization for efficient catalysis by these enzymes are not yet known. Editing endonuclease function likely requires formation of a dimeric RNase III domain as do all characterized RNase III enzymes. The RNase III motifs of KREPB4 and KREPB5 are divergent and appear to be catalytically inactive (A Schnaufer, J Carnes, R Proff, and K Stuart, in prep.). While most RNase IIIs cleave both strands of double-stranded RNA substrate, editing endonucleases have been proposed to form a heterodimeric domain with KREPB4 or KREPB5, thereby resulting in cleavage of the mRNA but not gRNA (Carnes et al. 2011). If such heterodimeric interactions exist, then the interaction between KREPB5 and the KREPA3 scaffold protein provides a physical link between endonuclease and the common set of editosome proteins (Schnaufer et al. 2010).
The U1-like zinc finger motif of KREPB6, KREPB7, and KREPB8 (Fig. 1B) resembles that in the U1C spliceosomal protein that functions in both RNA and protein interactions (Heinrichs et al. 1990; Nelissen et al. 1994). This similarity suggests that these editosome proteins are involved in interactions with the RNA substrate and/or other editosome proteins. Sequence identity/similarity between KREPB6 and KREPB7 is 25.5%/38.4%, between KREPB7 and KREPB8 is 21.8%/35.1% and between KREPB6 and KREPB8 is 21.4%/31.7% (Supplemental Fig. 1). Orthologs are present in Trypanosoma cruzi and Leishmania major, suggesting that they have important roles in editosome structure and/or function. KREPB6, KREPB7, and KREPB8 have been shown to associate preferentially with the deletion subcomplex when these TAP-tagged proteins were isolated from cells in which the partner endonuclease was repressed (Carnes et al. 2011). Thus, while the three different editosomes have a similar structural organization, the specific association of KREPB6, KREPB7, and KREPB8 with KREN3, KREN2, and KREN1 complexes, respectively, implies a role in endonuclease activity and/or substrate (ES) specificity.
We report here that KREPB6, KREPB7, or KREPB8 have differential effects on cell growth, editosome integrity, and editing in vivo. In addition, RNAi knockdown studies show that KREN1, but not KREN2 or KREN3, can have in vitro endonuclease cleavage activity in the absence of its partner protein. Taken together, these results suggest that KREPB6, KREPB7, and KREPB8 are accessory proteins that affect editing endonuclease activity and perhaps specificity.
RESULTS
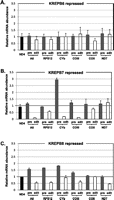
RNAi knockdowns of KREPB7 and KREPB8 but not KREPB6 resulted in growth inhibition in PF T. brucei (Fig. 2). RNAi-mediated repression of KREPB7 resulted in detectable growth inhibition by day 4 and almost complete cessation by day 6 (Fig. 2B), although real-time PCR at this time indicated no reduction of KREPB7 mRNA (Fig. 2D). KREPB8 RNAi inhibited growth to a lesser extent (Fig. 2C) but only had a ∼20% knockdown of the mRNA by day 6 (Fig. 2D). In contrast, a 40% knockdown of KREPB6 mRNA by RNAi did not affect growth (Fig. 2A). TAP-tagged KREPB6, KREPB7, or KREPB8 were inserted into the β-tubulin locus of their respective RNAi cell line and expressed constitutively under the control of the endogenous promoter to assess protein expression since antibodies specific to these proteins are unavailable. Western analysis showed that RNAi resulted in a substantial knockdown of these tagged proteins, especially KREPB6 (Fig. 2E). Simultaneous probing of cell lysates with tagged proteins using antibodies against editosome components of KREPA1, KREPA2, KREL1, and KREPA3 showed that the overall abundance of these four editosome components was essentially unaffected by the RNAi knockdowns. These data indicate that KREPB7 and KREPB8 proteins are essential for cell viability. They further suggest that KREPB6 may be either required in amounts below that resulting from the RNAi knockdown or it may not be essential in PF T. brucei.

RNAi repression of KREPB6, KREPB7, or KREPB8 in procyclic form T. brucei. Growth of transgenic cells in the absence of tet (squares) and following tet induction of RNAi (triangles), which resulted in KREPB6 (A), KREPB7 (B), and KREPB8 (C) knockdown. (D) Real-time analysis of the relative abundance of KREPB6, KREPB7, or KREPB8 mRNA after 6 d with or without tet-induced RNAi. β-Tubulin (black bars) or 18S rRNA (gray bars) was used as an internal control. The thick black line at 1 indicates no relative change in mRNA level. (E) Western analyses of cell lysates following 6 d of RNAi knockdown of TAP-tagged KREPB6, KREPB7, or KREPB8. The tagged proteins were detected using the rPAP reagent, which is specific to the Protein A–tagged component of the tag, while the KREPA1, KREPA2, KREL1, and KREPA3 proteins were detected using a mixture of MAbs as indicated. E indicates expressed, and R indicates repressed by RNAi.
Effects of KREPB6, KREPB7, or KREPB8 repression on in vivo editing
The abundance of edited and pre-edited RNAs in cells in which KREPB6, KREPB7, or KREPB8 was repressed was determined relative to cells in which they were expressed using real-time PCR (Fig. 3). After KREPB6 repression, pre-edited mRNA levels remained mostly unchanged as were those of edited CYb, COIII, and ND7, and those of edited A6, RPS12 appeared reduced, although not beyond standard deviation (Fig. 3A). However, edited COII mRNA was reduced by ∼30%, which is relevant since KREN3 editosomes specifically cleave COII (Carnes et al. 2008). After KREPB7 repression, the levels of most pre-edited mRNAs remained essentially unchanged, except that the amount of pre-edited CYb mRNA level was increased threefold (Fig. 3B). This may reflect accumulation of the unprocessed pre-mRNA. The levels of most edited mRNAs were substantially reduced, except edited ND7, which was unchanged or perhaps slightly increased. The levels of edited A6, CYb, and COIII were reduced by ∼80%–90%, RPS12 by ∼50%, and COII mRNA by ∼30%. After KREPB8 repression, the levels of pre-edited mRNAs and edited CYb and COII mRNAs were either unchanged or increased somewhat, while those of edited A6, RPS12, COIII, and ND7 mRNAs were decreased by ∼50% (Fig. 3C). Because CYb and COII only have insertion editing, the lack of an editing defect after KREPB8 repression is consistent with the previous finding that KREN1–KREPB8 editosomes specifically cleave at deletion sites. Never-edited ND4 mRNA abundance was unaffected by the repression of KREPB6, KREPB7, or KREPB8, which is similar to previous results following knockdown of other editosome components (Carnes et al. 2005, 2008; Trotter et al. 2005). The differential effects of RNA editing after repression of KREPB6, KREPB7, or KREPB8 likely reflect a combination of the distinct functions of each type of editosome as well as the differential extent of knockdown for each target.
Effect of RNAi repression of KREPB6, KREPB7, or KREPB8 on in vivo RNA editing. Real-time PCR analyses of mitochondrial RNAs from KREPB6 RNAi (A), KREPB7 RNAi (B), or KREPB8 RNAi (C) cell lines after 6 d of RNAi repression. Pre-edited (gray), edited (white), and control never edited ND4 (black) mRNA levels from the RNAi cells are shown relative to those of the same cell lines in which expression was not repressed by RNAi knockdown. The means and standard deviations from three independent experiments are shown. The thick black line at 1 indicates no relative change in mRNA level. β-Tubulin was used as the internal control.
Effects of KREPB6 or KREPB7 repression on in vitro editing
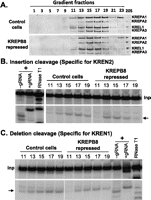
KREPB6 and KREPB7 are essential for the in vitro endonuclease activity of their respective editosomes. RNAi-mediated repression of KREPB6 or KREPB7 did not alter the abundance or sedimentation of total ∼20S editosomes but significantly reduced the in vitro cleavage activity (Fig. 4). Cleared lysates of crude mitochondria from an equal number of KREPB6 RNAi or KREPB7 RNAi cells after 6 d of induction and from KREPB7 RNAi cells without induction (control cell) were fractionated on 10%–30% glycerol gradients and analyzed by Western blots probed using antibodies specific for editosome components KREPA1, KREPA2, KREL1, and KREPA3 (Fig. 4A). The fractions from the uninduced KREPB7 control cells have the typical ∼20S editosome sedimentation profile (Fig. 4A, top panel). The gradient profiles from cells after repression of KREPB6 or KREPB7 are essentially indistinguishable from that of the control cells (Fig. 4A, middle and bottom panels). The lack of a change in total editosome sedimentation likely results from a specific effect on only one type of editosome that leaves the other editosomes intact (see below).

Effects of KREPB6 or KREPB7 repression on editosome sedimentation and in vitro endoribonucleolytic cleavage activity. (A) Western analysis of glycerol gradient fractions from crude mitochondrial lysates comparing KREPB6 RNAi or KREPB7 RNAi cells repressed for 6 d with nonrepressed KREPB7 RNAi cells as the control. A mixture of MAbs against the editosome components was used as indicated on the right. In vitro endoribonucleolytic cleavage activities in fractions 11–19 that are specific for sites cleaved by KREN3 (B), KREN2 (C), or KREN1 (D) endoribonucleases. Note the loss of cleavage characteristic of KREN3 upon KREPB6 knockdown, of cleavage characteristic of KREN2 upon KREPB7 knockdown, but no effect on cleavage characteristic of KREN1 by the knockdown of either KREPB6 or KREPB7 as indicated by arrows. Open and closed triangles indicate KREN2 and KREN1 cleavage sites, respectively, in COII-derived substrate in (B). Input substrate RNA (Inp) digested with RNase T1 (T1) or alkaline hydrolysis (OH) was used for size markers, pooled ∼20S editosomes from wild-type cells were used for positive reaction controls (+) and with gRNA omitted (−gRNA) as a negative control. See Materials and Methods for details.
The effect of KREPB6 or KREPB7 repression on endonuclease function was examined by in vitro cleavage assays of gradient fractions corresponding to the ∼20S editosome (Figs. 4B–D). KREN3-specific cleavage activity was assayed by using an insertion site substrate modeled on COII mRNA, KREN2-specific cleavage activity with an A6-derived insertion site substrate, and KREN1-specific cleavage activity using an A6-derived deletion site substrate. Fractions from uninduced KREPB7 RNAi cells were used as a control and showed the typical endonuclease activity for all three substrates. KREPB6 repression resulted in a substantial reduction of KREN3 insertion site cleavage activity (Fig. 4B) and a modest increase in KREN2 insertion site cleavage activity (Fig. 4C) but little or no effect on KREN1 deletion site cleavage activity (Fig. 4D). The COII-derived substrate in Figure 4B also shows cleavage at distinct KREN1 and KREN2 sites and shows a decrease in KREN2 cleavage following repression of KREPB7. Repression of KREPB7 also resulted in a moderate increase of KREN3 insertion site cleavage activity (Fig. 4B), a substantial reduction of KREN2 insertion site cleavage activity (Fig. 4C), and an increase in KREN1 deletion site cleavage activity (Fig. 4D). The increases in activities may reflect relative increases in the abundance of the editosomes whose components were not targeted by RNAi. Pre-cleaved assays show no change in editing activities following repression of KREPB6 or KREPB7 (data not shown). The loss of activities indicates that KREPB6 and KREPB7 are critical for KREN3 and KREN2 insertion site-specific cleavage activities, respectively.
Effect of KREPB8 repression on in vitro editing
KREPB8 repression had little effect on ∼20S editosome sedimentation and in vitro deletion site endonuclease activity as assessed by glycerol gradient fractionation and in vitro endonuclease assays (Fig. 5). Western analysis of glycerol gradient fractions from lysates of crude mitochondria from similar numbers of cells showed that 6 d of KREPB8 repression did not alter either editosome sedimentation or the amount of the KREPA1, KREPA2, KREL1, and KREPA3 editosome proteins relative to unrepressed cells (Fig. 5A). Pre-cleaved assays show no change in editing activities following repression of KREPB8 (data not shown). Analyses of endonuclease activities in peak editosome fractions showed that neither KREN2 insertion nor the KREN1 deletion cleavage was affected by KREPB8 repression (Fig. 5B,C). Thus, either KREPB8 is not essential for KREN1 deletion site cleavage activity, or the knockdown was insufficient to result in a detectable reduction in activity.
Effects of KREPB8 repression on editosome sedimentation and in vitro endoribonucleolytic cleavage activity of editosomes. (A) Western analysis of glycerol gradient fractions from crude mitochondrial lysates from KREPB8 RNAi cells comparing KREPB8 RNAi cells repressed for 6 d with nonrepressed cells as a control. The MAb mixture was used as described for Figure 4. Gradient fractions 11–19 were assayed for endonuclease activities that are specific for an insertion site cleaved by KREN2 (B) or deletion site cleaved by KREN1 (C). See legend for Figure 4 or Materials and Methods for details.
Effect of KREPB6, KREPB7, or KREPB8 repression on individual editosome structure
Analysis of TAP-isolated KREN3, KREN2, or KREN1 editosomes showed that RNAi knockdown of KREPB6, KREPB7, or KREPB8, respectively, affected complex integrity, even though total editosome sedimentation was unchanged (cf. Fig. 6 and Figs. 4, 5). TAP-tagged KREN1, KREN2, or KREN3 was constitutively expressed from the β-tubulin locus in KREPB8, KREPB7, or KREPB6 RNAi cell lines, respectively. These cells exhibited the same growth phenotype upon RNAi knockdown as cells with no tag (data not shown). The tagged editosomes were purified from the resultant KREPB6 RNAi-N3 TAP, KREPB7 RNAi-N2 TAP, and KREPB8 RNAi-N1 TAP cell lines and examined by Western analysis of glycerol gradient fractions as well as by SYPRO-Ruby staining (Fig. 6). Editosomes purified from cells expressing KREPB6, KREPB7, and KREPB8 contained all four editosome proteins in normal relative amounts. However, RNAi knockdown in each case resulted in substantial loss of the specific editosomes in the ∼20S region, a shift in sedimentation, and a proportional change in the four editosome proteins assayed by Western. Simultaneous probing for the four editosome proteins permits relative comparisons using the intensity of one band to normalize the signal of the others. KREPB6 repression resulted in substantial loss of KREN3 editosomes and preferential loss of KREPA2 and KREL1 relative to KREPA1 and KREPA3 as seen by both Western and SYPRO-Ruby staining (Fig. 6A). These results indicate that KREPB6 is essential for the integrity of ∼20S KREN3 editosomes, and KREN3 predominantly associates with the insertion subcomplex in the absence of KREPB6. KREPB7 repression resulted in a dramatic loss of editosome components, but the relative abundance of the four editosome proteins that remained was similar to that before repression. Hence, KREPB7 is essential for the integrity of KREN2 editosomes. KREPB8 repression resulted in a small reduction in the amount of KREN1 editosomes and a minor differential loss among the four proteins detected by Western blot. These results are consistent with the effect of KREPB6, KREPB7, and KREPB8 RNAi knockdown on their respective endonuclease activity observed in Figures 4 and 5.
Effects of KREPB6, KREPB7, or KREPB8 repression on tagged KREN3, KREN2, or KREN1 editosomes, respectively. (A) Western analysis using an MAb mixture (upper panel) and SYPRO-Ruby stained gels (lower panel) of editosomes that were purified via the TAP-tag from total cell lysates. Editosomes were purified by sequential IgG Sepharose and calmodulin affinity chromatography from equal numbers of KREPB6 RNAi-N3TAP, KREPB7 RNAi-N2TAP, KREPB8 RNAi-N1TAP cells in which the KREPB protein expression was repressed by RNAi for 6 d (+) or were nonrepressed (−). (B–D) Western analysis of the glycerol gradient fractions of the complexes purified via IgG affinity chromatography and TEV protease cleavage from these cells.
Sedimentation analysis of the TAP-isolated KREN3, KREN2, and KREN1 editosomes revealed editosome subcomplexes upon KREPB6, KREPB7, and KREPB8 RNAi knockdown, respectively (Fig. 6B–D). Endonuclease-specific editosomes isolated by IgG affinity were eluted by TEV cleavage, fractionated on 10%–30% glycerol gradients, and analyzed by Western blot. While some editosomes sediment in the ∼20S region (fractions 11–17; see Fig. 4A), a substantial proportion sedimented at lower S values. The complexes with the lower sedimentation had a greater relative abundance of KREPA1 and KREPA3 proteins compared with KREPA2 and KREL1, which is indicative of preferential association with insertion subcomplexes. Hence, editosome complexes isolated via tagged endonuclease include a significant proportion of subcomplexes, but the relative amount differs depending on the editosome. RNAi repression of KREPB6 in cells with tagged KREN3 resulted in editosome loss and an increase in subcomplexes relative to ∼20S, while RNAi knockdown of KREPB7 in cells with tagged KREN2 resulted in substantial overall loss of editosome material. RNAi knockdown of KREPB8 in cells with tagged KREN1 increased the proportion of insertion subcomplexes, but a substantial amount of ∼20S editosomes were retained. Hence, the loss of KREPB6 and KREPB8 resulted in some disruption of the structure of ∼20S KREN3 and KREN1 editosomes, respectively, while the loss of KREPB7 resulted in substantial disruption of KREN2 editosomes. The loss in each case results in preferential, perhaps initial, loss of deletion subcomplex proteins and may reflect their lack of integration into complexes or destabilization of complexes followed by protein turnover. Taken together, these results indicate that KREPB6, KREPB7, and KREPB8 are each important for editosome structural integrity. Although the extent of editosome disruption differed, the fact that the loss of KREPB6, KREPB7, or KREPB8 altered sedimentation of their respective editosomes suggests that these related proteins play similar structural roles.
The effect of KREPB6, KREPB7, or KREPB8 repression on in vitro cleavage activity of purified TAP-tagged KREN1, KREN2, and KREN3 editosomes was assessed. No cleavage activity was detected in glycerol gradient fractions of TEV eluates from cells with tagged KREN2 (fractions 9–15 tested on A6-derived insertion and deletion substrates) or KREN3 (fractions 9–15 also tested on A6-derived insertion and deletion substrates, and fractions 7–15 tested on COII-derived substrate) in which expression of KREPB7 and KREPB6, respectively, was repressed (data not shown). This may reflect the substantial loss of the tagged KREN2 editosomes upon KREPB7 knockdown and of KREN3 editosomes to a lesser extent, as well as fragmentation upon KREPB6 knockdown since, to date, cleavage has been only observed with ∼20S–40S complexes (see Fig. 6B,C). Thus, the insertion site cleavage catalyzed by each of these editosomes was lost upon knockdown of their partner protein. Knockdown of KREPB8 expression reduced the amount of tagged KREN1 editosomes to a lesser extent and was less disruptive than the KREPB6 and KREPB7 knockdowns since ample ∼20S and less smaller fragments resulted (see Figs. 6D, 7A). These tagged KREN1 editosomes and ∼15S subcomplexes catalyzed both insertion and deletion pre-cleaved editing with the greatest activity in fraction 9, which contained the most editosome protein (Fig. 7B,C). This is consistent with enrichment of insertion and deletion subcomplexes in fraction 9 and the presence of ∼20S editosomes (i.e., in fractions 11–15) from partial disruption of these editosomes (Fig. 7A). KREN1-specific cleavage was detected after KREPB8 repression in the ∼20S fractions, which is indicative of intact editosomes. However, relatively increased KREN1-specific cleavage product was also detected in fraction 9 compared with cells expressing KREPB8 (Fig. 7D). This implies the release of a fragment of the editosome that retains the KREN1 cleavage-specific activity. This is the first time that such activity was seen in complexes smaller than ∼20S.
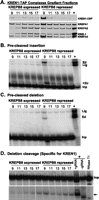
Activities of KREN1 tagged editosomes from cells in which KREPB8 expression has been knocked down. (A) Western analyses of IgG purified editosomes from KREPB8 RNAi-N1TAP cells with KREPB8 expressed or repressed. The blots were probed with the editosome MAb mixture (bottom panel) or a MAb against calmodulin binding peptide (CBP) on the tagged KREN1 protein (top panel). In vitro pre-cleaved insertion (B), deletion (C) assays and full-round deletion editing assays (D). The input RNAs (Inp) are edited by addition of two U's (+2U) in the insertion assay, removal of four U's (−4U) in the deletion assay, and result in ligated products of unprocessed 5′ and 3′ input RNAs (Lig) and edited (Ed) products. Full round assays assess deletion cleavage activity (denoted by arrow). Other designations are as in Figure 4.
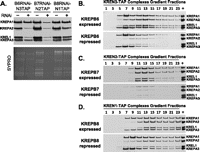
The effect of KREPA3 repression on the association of TAP-tagged endonucleases or their partner B proteins with other editosome proteins was assessed. The three endonucleases each interact similarly with other editosome proteins, as do their partner B proteins. Previous studies have indicated that the endonucleases primarily associate with the insertion subcomplex, while their KREPB partners associate with the deletion subcomplex. Editosome protein interactions with the endonucleases and their partner B proteins were examined after KREPA3 repression, since this protein mediates interactions between components of the deletion and insertion subcomplexes. Western analysis of the total lysates of cells in which one of the three endonucleases was tagged showed that KREPA3 repression for 6 d had no effect on the levels of editosome proteins except for KREPA3 (data not shown). Western analyses of TEV eluates showed that the RNAi knockdown essentially eliminated KREPA3 from the tagged editosomes and resulted in a slight reduction in the abundance of the tagged proteins, although the level of tagged KREN2 protein was slightly elevated (Fig. 8A). KREPA3 knockdown in cells with tagged KREN1 or KREN3 resulted in substantial loss of these tagged editosomes, especially KREN3 editosomes, and reduced levels of KREPA2 and KREL1 relative to KREPA1 (Fig. 8B,D, left panels). This indicates a preferential association of these endonucleases with the insertion subcomplex. KREPA3 knockdown in cells with tagged KREN2 did not substantially affect the overall levels of KREPA1, KREPA2, and KREL1, but glycerol gradient analysis revealed a shift toward the lower S region of the gradient and somewhat reduced levels of KREPA2 and KREL1 relative to KREPA1 (Fig. 8C, left panel).
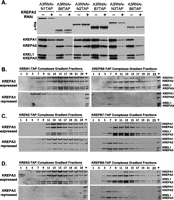
Association of KREPB6-8 and KREN1-3 proteins with deletion and insertion subcomplexes, respectively. Expression of KREPA3 was repressed by RNAi (+) in PF cells in which either KREPB6, 7, or 8 or KREN1, 2, or 3 were TAP-tagged. Western analyses of (A) complexes after purification by IgG sepharose chromatography and TEV protease cleavage and (B–D) subsequent glycerol gradient fractionation. The blots were probed with α-His MAb against tagged proteins (A, top panel) or a mixture of MAbs against KREPA1, KREPA2, KREL1, and KREPA3 as indicated.
Inactivation of KREPA3 in cells in which KREPB8, KREPB7, or KREPB6 was tagged resulted in essentially a reciprocal pattern of protein loss/retention compared with those in which the endonuclease was tagged. KREPA3 repression resulted in loss of KREPA1, albeit less extensive in cells with tagged KREPB7 (Fig. 8A). The retention of KREPA2 and KREL1 is indicative of preferential association of KREPB6, KREPB7, and KREPB8 with the deletion subcomplex. KREPA3 knockdown resulted in greater loss of tagged KREPB8 and tagged KREPB6 editosomes compared with tagged KREPB7 editosomes. This resembles the lesser effect of KREPA3 knockdown on tagged KREN1 and KREN3 editosomes compared with KREN2 editosomes. The data from these KREPA3 knockdown studies indicate that KREPB6, KREPB7, and KREPB8 primarily associate with the deletion subcomplex and that KREN1, KREN2, and KREN3 primarily associate with the insertion subcomplex.
DISCUSSION
The KREPB6, KREPB7, and KREPB8 proteins that associate with KREN3, KREN2, or KREN1 editosomes, respectively, are differentially important for the structural integrity of their respective editosomes, endonuclease activity, and editing in vivo. In addition, KREPB7 and KREPB8 appear to be essential for survival of PF T. brucei. KREPB6 may also be essential, because the lack of a growth phenotype may result from insufficient knockdown by RNAi. The specific roles of these proteins are uncertain, but their U1 zinc finger motif and associations with specific types of editosomes suggest that they are accessory factors that function in discrimination among the numerous editing sites to ensure recognition and cleavage by the endonuclease.
Gene essentiality
The results show that KREPB7 and KREPB8 are essential for PF parasite viability, and the knockdown of KREPB6 may have been insufficient to reveal its essentiality. The minimal knockdowns of the KREPB7 and KREPB8 mRNA and protein levels inhibited growth, while greater knockdowns of KREPB6 mRNA and protein did not (Fig. 2). The differences in the phenotypic outcomes of these RNAi knockdowns may reflect a combination of the distinct functions of the different editosomes, the abundance of their target ESs, and the abundance of the respective editosomes needed for the essential editing. Both KREPB6/KREN3 and KREPB7/KREN2 editosomes cleave insertion ESs in vitro but with different specificity. The KREPB6/KREN3 editosomes appear to process all three of the COII insertion editing sites in vivo (Carnes et al. 2008). This is consistent with the reduction in COII editing in vivo (Fig. 3A) and significant loss of cleavage of a COII-derived substrate in vitro upon repression of KREPB6 expression (Fig. 4B). The specificity in vivo of KREPB6/KREN3 versus KREPB7/KREN2 editosomes among the hundreds of different insertion ESs is unknown. However, if KREPB6/KREN3 editosome function is restricted to COII or a relatively small number of editing sites, then fewer of these editosomes would be needed compared with KREPB7/KREN2 editosomes. Hence, KREPB6 levels may not have been sufficiently reduced to inhibit growth. In addition, the specificity for different ESs in vivo may not be absolute. KREPB7/KREN2 editosomes may compensate for the loss of KREPB6/KREN3 editosomes, but not vice versa. Similarly, KREPB7 or KREPB8 may partially substitute for KREPB6 after its loss; however, such a substitution has not been observed.
KREPB6, KREPB7, and KREPB8 abundance
Mass spectrometry data compiled from 363 independent editosome purifications revealed strikingly fewer peptides for KREPB6 relative to those corresponding to either partner endonuclease or core editosome proteins, even when the number of potential tryptic peptides for each protein is considered (M Lerch, J Carnes, N Acestor, X Guo, A Schnaufer, and K Stuart, in prep.). Hence, KREPB6 may interact transiently with editosomes. This suggests a dynamic process consistent with editosomes encountering different editing sites during the process. The low abundance of KREPB6 compared with KREPB7 and KREPB8 editosomes may reflect specificity for COII pre-mRNA. We recently discovered two additional related proteins, KREPB9 and KREPB10, that also appear to be in lower abundance and interact with editosomes in a similar fashion to KREPB6, KREPB7, and KREPB8 albeit not in a mutually exclusive manner (M Lerch, J Carnes, N Acestor, X Guo, A Schnaufer, and K Stuart, in prep.). An intriguing possibility is that these proteins may be involved in regulating editing during the life cycle.
Editosome structure
KREPB6, KREPB7, and KREPB8 contribute to the structural integrity of their respective editosomes and appear to occupy a similar structural location in the ∼20S complex. RNAi repression of these genes did not alter overall editosome sedimentation on glycerol gradients, because only one of the three types of editosome was affected; however, examination of the respective endonuclease isolated editosomes showed that repression generated subcomplexes that lacked some proteins (Figs. 4A, 5A, 6). The composition of the subcomplexes generated after RNAi consists of editosome proteins that interact with the tagged protein. All three tagged KREPB proteins associate directly or indirectly with components of the deletion subcomplex. This conclusion is supported by the relative enrichment for KREPA2 and KREL1 in subcomplexes that are isolated by these tagged proteins. These preferential interactions are enhanced by knockdown of the KREPA3 protein that bridges the insertion and deletion subcomplexes (Figs. 6, 8; Guo et al. 2008; Schnaufer et al. 2010). Hence, all three KREPB proteins associate with the editosome in a similar fashion, namely, with a primary association with the deletion subcomplex. Reciprocally, all three tagged KREN proteins associate directly or indirectly with specific components of the insertion subcomplex. Tagged KREN subcomplexes are relatively enriched for the KREPA1 component of the insertion subcomplex that is enhanced upon knockdown of KREPA3 expression. These results are consistent with the finding that repression of endonuclease expression leads to the loss of insertion subcomplex proteins from tagged KREPB6, KREPB7, and KREPB8 complexes (Carnes et al. 2011). Hence, there is a reciprocal or structurally complementary arrangement in which KREPB6-8 proteins are primarily associated with the deletion subcomplex and KREN1-3 proteins are primarily associated with the insertion subcomplex. Overall, it seems likely that these specific endonuclease and KREPB pairs interact directly or indirectly.
Structural differences among KREN3/KREPB6, KREN2/KREPB7, and KREN1/KREPB8 editosomes
The similarities in sequence and physical associations in editosomes suggest that KREPB6, KREPB7, and KREPB8 have similar roles, but the distinct cleavage specificities of each editosome imply that they have some functional differences. For example, editosomes isolated via tagged endonuclease with partner B protein expressed or repressed gave somewhat different gradient profiles for each endonuclease (Fig. 6). Such differences are evident in the ratios of ∼20S and smaller complexes, as well as the relative amounts of editosome proteins. Similarly, the gradient profiles differ between the endonuclease specific complexes following inactivation of the bridging KREPA3 protein (Fig. 8). These differences may be due to the distinct specificity, affinity, and/or stability of the interactions of each protein with their respective editosome, and the consequences to editosome assembly, stability, and dynamic interactions. We cannot exclude the possibility that some observed differences might also reflect the impact of the tag on the different proteins or differential effects of RNAi. Thus, while KREN1, KREN2, and KREN3 each preferentially interact with the insertion subcomplex, there are differences in their interactions with other proteins in the editosome. Such structural differences between editosomes were previously noted after expression of KREN1 and KREN2 containing similar mutations in the RNase III motifs resulted in different associations with editosome proteins (Panigrahi et al. 2006). The differences in protein interactions with each endonuclease may reflect an accommodation for multiple different substrates.
Role of KREPB6, KREPB7, and KREPB8 in endonuclease function
The pairwise associations of the KREPB proteins with their partner KREN proteins imply that their functions relate to endonuclease activity. The loss of specific cleavage activity upon KREPB6 or KREPB7 knockdown confirms such a role for these two proteins (Fig. 4). In vitro cleavage was not lost upon knockdown of KREPB8, although growth and in vivo editing were inhibited. Notably, the loss of KREPB8 did not affect in vivo editing of CYb and COII, both of which lack deletion sites. Loss of cleavage appears specific to endonuclease activity, because pre-cleaved assays revealed no differences in ligation, U insertion, or U deletion. Loss of cleavage could also result from defects in editing site recognition, but experiments dissecting site recognition from site cleavage are not currently feasible. Interestingly, KREPB8 knockdown resulted in a lower S value fraction with enhanced pre-cleaved insertion, pre-cleaved deletion activity, and, importantly, enhanced deletion site cleavage. Indeed, this is the first observation of such cleavage by an editosome subcomplex rather than ∼20S editosomes. Hence, the endonuclease activity, at least for KREN1, does not require a complete ∼20S editosome. Analysis of KREN1–TAP complexes isolated from cells expressing KREPB8 RNAi by mass spectrometry revealed the presence of KREPB10, which is closely related to KREPB8 (M Lerch, J Carnes, N Acestor, X Guo, A Schnaufer, and K Stuart, in prep.). KREPB10 may partially compensate for the loss of KREPB8 and prevent complete loss of KREN1 activity. Thus, the functions of KREPB6, KREPB7, and KREPB8 may not be essential for endonuclease catalytic activity per se and suggests that they are endonuclease accessory factors.
Mechanism of KREPB6, KREPB7, and KREPB8 functions
The mechanisms by which KREPB6, KREPB7, and KREPB8 affect endonuclease functions remain to be elucidated, but the conserved U1-like zinc finger motif provides some hints (Heinrichs et al. 1990; Nelissen et al. 1994). The U1-like zinc finger implies coordinated interactions with both RNA and protein and suggests these proteins play a role in substrate interaction and editosome structure, including stabilizing the association of the deletion subcomplex with ∼20S editosome. The KREPB proteins may promote the association and proper positioning of the RNA substrates with the editosomes, and their interactions with other proteins may properly position the catalytic domains, especially those of the endonucleases. Simultaneous modulation of RNA and protein interactions by the KREPB proteins may function to affect the specificity of the endonuclease at numerous different ESs.
Endonuclease substrate recognition in vivo
An ongoing question about RNA editing in vivo is how three different editosomes function together to edit the hundreds of insertion editing sites and tens of deletion editing sites. These editing sites are fundamentally similar substrates since they have an mRNA/gRNA duplex 3′ to cleavage sites (relative to mRNA) and incomplete base-pairing and the oligo (U) tail of the gRNA to the 5′ side. However, the different in vitro specificities of the different endonucleases (KREN1 for deletion sites and KREN2 and KREN3 for different insertion sites) suggest that editosome diversity is essential for the recognition of numerous editing sites with distinct mRNA/gRNA duplexes. How the different editosomes discriminate between the different editing sites, especially KREN2 and KREN3, which distinguish between different insertion sites, is not known but presumably is critical for accurate editing given the essentiality of these endonucleases. Overall, the data in this paper suggest that the KREPB6, KREPB7, and KREPB8 proteins are important for endonuclease activity and specificity and thus may contribute to the accurate recognition of the numerous similar but diverse substrates.
MATERIALS AND METHODS
Plasmid constructs
The plasmids expressing tet-inducible double-stranded RNA for RNAi targeting KREPB6 (Tb927.3.3990), KREPB7 (Tb09.160.4130), or KREPB8 (Tb927.8.5690) were generated by inserting 803 bp, or 789 bp, or 504 bp of the respective gene into the pZJM vector (Wang et al. 2000). Gene fragments were PCR-amplified from T. brucei strain 427 genomic DNA using 5′-CCGCTCGAGTACAAGCGGCGTTTGGAG-3′ and 5′-CCCAAGCTTGACAATCTCTTCGGAGCC-3′ for KREPB6, 5′-CCGCTCGAGAAGCACTTCGTCGACTGC-3′ and 5′-CCCAAGCTTTGAAGCGGTTGAACTCGC-3′ for KREPB7, and 5′-ATACTCGAGGCGAGTACGAAACTACCA-3′ and 5′-ATAAGCTTATGCAAGTGGTCCCACGTTG-3′ for KREPB8. (Restriction sites are italicized.) PCR products were first cloned into pGEM-T Easy (Promega), digested with XhoI and HindIII and subcloned into similarly digested pZJM to create pZJM-KREPB6, pZJM-KREPB7, and pZJM-KREPB8. The pHD1344tubulin vector, a kind gift from Achim Schnaufer, integrates into the β-tubulin locus and was used to constitutively express tagged proteins in the background of RNAi cell lines. Plasmids pHD1344tub-KREPB6-TAP, pHD1344tub-KREPB7-TAP, pHD1344tub-KREPB8-TAP, pHD1344tub-KREN1-TAP (named pN1-WT in Trotter et al. 2005), pHD1344tub-KREN2-TAP (named pWT-β in Carnes et al. 2005), and pHD1344-KREN3 (named pWTN3-β in Carnes et al. 2008) express KREPB6, KREPB7, KREPB8, KREN1, KREN2, and KREN3, respectively, with a C-terminal myc/his tag followed by a TAP tag. The method for generating these constructs was described elsewhere (Carnes et al. 2005; Trotter et al. 2005).
Transfection and RNAi induction
T. brucei 29-13 procyclic cells were grown in SDM-79 medium containing 10% FBS, 15 μg/mL G418, and 25 μg/mL hygromycin. RNAi cell lines were generated in 29-13 cells by transfection with ∼12 μg of NotI-linearized pZJM-KREPB6, pZJM-KREPB7, or pZJM-KREPB8 individually as described previously (Guo et al. 2008). The stable transfectants were designated KREPB6 RNAi, KREPB7 RNAi, and KREPB8 RNAi, respectively, and grown in the medium supplemented with 15 μg/mL G418, 25 μg/mL hygromycin, and 2.5 μg/mL phleomycin. To assess the RNAi efficiency, pHD1344-KREPB6, pHD1344-KREPB7, and pHD1344-KREPB8 were NotI-linearized and transfected into the cell lines KREPB6 RNAi, KREPB7 RNAi, and KREPB8 RNAi, respectively. The stable transfectants were designated KREPB6 RNAi-B6TAP, KREPB7 RNAi-B7TAP, and KREPB8 RNAi-B8TAP and grown in the medium supplemented with 15 μg/mL G418, 25 μg/mL hygromycin, 2.5 μg/mL phleomycin, and 1 mg/mL puromycin. To assess the RNAi efficiency, pHD1344tub-KREPB6-TAP, pHD1344tub-KREPB7-TAP, and pHD1344tub-KREPB8-TAP were NotI-linearized and transfected into the cell lines KREPB6 RNAi, KREPB7 RNAi, and KREPB8 RNAi, respectively. The stable transfectants were designated KREPB6 RNAi-B6TAP, KREPB7 RNAi-B7TAP, and KREPB8 RNAi-B8TAP and grown in the medium supplemented with 15 μg/mL G418, 25 μg/mL hygromycin, 2.5 μg/mL phleomycin, and 1 mg/mL puromycin. To assess the effect of RNAi knockdown on editosome structure, the plasmids pHD1344tub-KREN3-TAP, pHD1344tub-KREN2-TAP, and pHD1344tub-KREN1-TAP were linearized with NotI and transfected into the cell lines KREPB6 RNAi, KREPB7 RNAi, and KREPB8 RNAi, respectively, and selected with 1 mg/mL puromycin, and the resulted resistant clones were designated KREPB6 RNAi-N3TAP, KREPB7 RNAi-N2TAP, and KREPB8 RNAi-N1TAP. The cell line KREPA3 RNAi, previously derived from T. brucei 29-13 procyclic cells, was used to assess editosome associations in the absence of KREPA3. The six pHD1344-derived plasmids expressing TAP-tagged KREPB6, KREPB7, KREPB8, KREN1, KREN2, or KREN3 were linearized with NotI and individually transfected into KREPA3 RNAi cells, and resulting cell lines were designated KREPA3 RNAi-B6TAP, KREPA3 RNAi-B7TAP, KREPA3 RNAi-B8TAP, KREPA3 RNAi-N1TAP, KREPA3 RNAi-N2TAP, and KREPA3 RNAi-N3TAP, respectively. All RNAi cell lines expressing TAP-tagged proteins from pHD1344tubulin-derived plasmids were grown in the SDM-79 supplemented with 15 μg/mL G418, 25 μg/mL hygromycin, 2.5 μg/mL phleomycin, and 1 mg/mL puromycin. RNAi was induced with 1 μg/mL tet, and the uninduced and induced cells were counted daily using a Coulter counter (Beckman).
RNA isolation and real-time PCR
Total RNAs were isolated from cells with RNAi either uninduced or induced using TRIzol reagent following the manufacturer's protocol. Real-time PCR was performed to assess the levels of the pre-edited, edited, and never edited mRNAs as previously described (Carnes et al. 2005). Briefly, 10 μg of RNA was treated with DNase I using the DNA-free kit (Ambion), and cDNA was generated by using random hexamers with TaqMan Reverse Transcription Reagents (ABI). Control reactions without reverse transcriptase were used to confirm the absence of contaminating genomic DNA. cDNA reactions were diluted by fivefold and amplified in 25-μL reactions containing 2.5 μL of each cDNA, 5 μL of each 1.5 μM forward and reverse primer, and 12.5 μL of SYBR Green PCR Master Mix in 96-well plates using the ABI Prism 7500 Sequence detection System (Applied Biosystems). The cDNA for the β-tubulin control reactions was further diluted by 50-fold to ensure that the Ct value for this more abundant RNA was in the same range as the other targets. The real-time PCR primers were 5′-CGCTGGTCACCATCGACAT-3′ and 5′-ATGGCGACGCAGCAAAGT-3′ for KREPB6; 5′-TGATGCTCTTGGGTGAGATTTG-3′ and 5′-CACAGGCGTGCCGCTATTA-3′ for KREPB7; and 5′-CGCTTCTTTTCCATCTGACACA-3′ and 5′-GCTGCGACCCTGAAGCA-3′ for KREPB8; all amplicons were sequenced to confirm targets. Other primers used here were described previously (Carnes et al. 2005). For each real-time PCR analysis, three independently generated samples of total RNA were analyzed, with triplicate wells for each mRNA assessed. Error bars represent the standard deviation of the three independent samples. Analysis of real-time PCR data used the Pfaffl method, with PCR efficiencies calculated by linear regression with LinRegPCR software (Pfaffl 2001; Ramakers et al. 2003). Relative changes for the target amplicon were determined after normalization to β-tubulin mRNA and were expressed as relative mRNA abundance from the respective control cells.
Mitochondria isolation, TAP-tag purification, and glycerol gradient sedimentation
Crude mitochondrial preparations were isolated from ∼1 × 1010 cells, with RNAi either uninduced or induced, as previously described (Carnes et al. 2005). Mitochondria were lysed in IPP150 buffer (10 mM Tris-HCl at pH 8.0, 150 mM NaCl, 0.1% NP-40) plus 1% Triton X-100, clarified by centrifugation at 13,000 rpm for 10 min at 4°C, and the cleared lysates were loaded onto 10%–30% glycerol gradients. Gradient samples were centrifuged at 38,000 rpm for 9 h at 4°C (SW40 rotor; Beckman). Twenty-four 500-mL fractions were collected from the top, flash-frozen in liquid nitrogen, and stored at −80°C. TAP-tagged complexes were isolated via IgG Sepharose affinity from lysates of ∼1.6 × 1010 uninduced or induced RNAi cells in which TAP-tagged proteins were constitutively expressed. Complexes were eluted from IgG Sepharose by TEV protease cleavage and then fractionated on 10%–30% glycerol gradients as above. Similarly, 24 500-μL fractions were collected from the top, flash-frozen in liquid nitrogen, and stored at −80°C. Tagged complexes were also purified from 2 × 109 uninduced and induced RNAi cells by sequential IgG affinity and calmodulin affinity purification (tandem affinity purification, TAP) as described (Rigaut et al. 1999; Panigrahi et al. 2006), and calmodulin eluates were analyzed by Western blot. Tagged proteins were detected using rPAP reagent (Sigma-Aldrich), which recognizes the protein A moiety of the tag.
Western blot analysis and SYPRO Ruby staining
Whole cell lysate samples were prepared by dissolving cells 1 × 107 cells directly in 200 μL of SDS-PAGE loading buffer. SDS-PAGE loading buffer was added to glycerol gradient fractions and TAP purified samples for electrophoresis. Protein samples were resolved on SDS-PAGE gels and transferred to PVDF membranes, then probed either with a cocktail of monoclonal antibodies (MAbs) against four editosome components KREPA1, KREPA2, KREL1, and KREPA3 (Panigrahi et al. 2001) or with individual MAb specific for His tag or calmodulin binding peptide (CBP) (Carnes et al. 2011). HRP-conjugated goat-anti-mouse secondary antibody was diluted 1:2000, and blot visualized by ECL chemiluminescence kit (Pierce). Where relevant, densitometric analysis of signal intensities were quantified using ImageQuant TL 7.0 (GE Healthcare) to determine relative amounts of proteins within a sample. For SYPRO Ruby staining, the protein samples were resolved on SDS-PAGE gel, then stained with SYPRO Ruby stain (Molecular Probes) and detected by UV light.
In vitro RNA editing assays
In vitro full-round deletion and insertion editing assays based on ATP synthase subunit 6 (A6) or insertion editing assays based on COII were performed as described previously (Kable et al. 1996; Seiwert et al. 1996; Carnes et al. 2005, 2011). The A6-derived deletion editing assay used the A6short/TAG.1 pre-mRNA and the D34 gRNA in the presence of 1 mM ADP to enhance KREN1 cleavage. The A6-derived insertion editing assays used A6-eES1 pre-mRNA and gA6[14] gRNA in the absence of ATP to enhance KREN2 cleavage. The COII-derived cis-guided insertion editing assay used Triple substrate RNA to assess KREN3 cleavage (Carnes et al. 2011). Pre-cleaved deletion and insertion assays were performed as previously described (Igo et al. 2000, 2002) using 5′-labeled U5-5′, U5-3′, and the gA6[14]PC-del RNAs and 5′-labeled 5′CL18, 3′CL13pp, and gPCA6-2A RNAs, respectively.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Achim Schnaufer (University of Edinburgh) for the gift of the pHD1344tub plasmid, and Melissa Lerch and Achim Schnaufer for sharing unpublished data. This work was supported by National Institutes of Health (NIH) Grant AI014102 (to K.S.).
Footnotes
-
Abbreviations: Kinetoplastid RNA Editing Protein A1: KREPA1, A2, A3, A4, A5, A6; Kinetoplastid RNA Editing Protein B4: KREPB4, B5, B6, B7, B8, B9, B10; Kinetoplastid RNA Editing eNdonuclease 1: KREN1, N2, N3 (formerly KREPB1, B3, B2); Kinetoplastid RNA Editing eXoUase 1: KREX1, 2; Kinetoplastid RNA Editing TUTase 2: KRET2; Kinetoplastid RNA Editing Ligase 1: KREL1, 2; editing site: ES.
-
↵4 Corresponding author.
E-mail ken.stuart{at}seattlebiomed.org.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.029314.111.
- Received July 14, 2011.
- Accepted October 31, 2011.
- Copyright © 2012 RNA Society








