
Comparative analyses of disease-linked missense mutations in the RNA exosome modeled in budding yeast reveal distinct functional consequences in translation
- Maria C. Sterrett1,2,
- Lauryn A. Cureton3,4,
- Lauren N. Cohen4,
- Ambro van Hoof5,
- Sohail Khoshnevis4,
- Milo B. Fasken1,
- Anita H. Corbett1 and
- Homa Ghalei4
- 1Department of Biology, Emory University, Atlanta, Georgia 30322, USA
- 2Biochemistry, Cell and Developmental Biology Graduate Program, Emory University, Atlanta, Georgia 30322, USA
- 3Genetics and Molecular Biology Graduate Program, Emory University, Atlanta, Georgia 30322, USA
- 4Department of Biochemistry, Emory University School of Medicine, Atlanta, Georgia 30322, USA
- 5Department of Microbiology and Molecular Genetics, The University of Texas Health Science Center at Houston, Houston, Texas 77030, USA
- Corresponding authors: acorbe2{at}emory.edu, hghalei{at}emory.edu
-
Handling editor: Eric Phizicky
Abstract
The RNA exosome is a multisubunit, evolutionarily conserved ribonuclease complex that is essential for processing, decay, and surveillance of many cellular RNAs. Missense mutations in genes encoding the structural subunits of the RNA exosome complex cause a diverse range of diseases, collectively known as RNA exosomopathies, often involving neurological and developmental defects. The varied symptoms suggest that different mutations lead to distinct in vivo consequences. To investigate these functional consequences and distinguish whether they are unique to each RNA exosomopathy mutation, we generated a collection of in vivo models by introducing pathogenic missense mutations in orthologous Saccharomyces cerevisiae genes. Comparative RNA-seq analysis assessing broad transcriptomic changes in each mutant model revealed that three yeast mutant models, rrp4-G226D, rrp40-W195R, and rrp46-L191H, which model mutations in the genes encoding EXOSC2, EXOSC3, and EXOSC5, respectively, had the largest transcriptomic differences. While some transcriptomic changes, particularly in transcripts related to ribosome biogenesis, were shared among mutant models, each mutation also induced unique transcriptomic changes. Thus, our data suggest that while there are some shared consequences, there are also distinct differences in RNA exosome function by each variant. Assessment of ribosome biogenesis and translation defects in the three models revealed distinct differences in polysome profiles. Collectively, our results provide the first comparative analyses of RNA exosomopathy mutant models and suggest that different RNA exosome gene mutations result in in vivo consequences that are both unique and shared across each variant, providing further insight into the biology underlying each distinct pathology.
Keywords
INTRODUCTION
The steady-state levels of cellular RNAs are regulated through a delicate balance of transcription and decay. This balance is fine-tuned through co- and post-transcriptional events that include precise processing, decay, and quality control/surveillance of the RNA (Corbett 2018; Cramer 2019; Wolin and Maquat 2019). Beyond their impact on the transcriptome, the post-transcriptional regulatory events are critical to define the proteome in both time and space. The RNA exosome is an abundant, essential cellular machine that is a critical mediator of both RNA processing and decay. This ring-like, macromolecular complex is composed of nine structural subunits and a catalytic 3′–5′ exo-endoribonuclease (DIS3 or DIS3L in humans and Dis3/Rrp44 in budding yeast) (Mitchell et al. 1997; Makino et al. 2013). The subunits of the RNA exosome are highly conserved and were initially identified in Saccharomyces cerevisiae through a genetic screen for ribosomal RNA processing (rrp) mutants and biochemical analyses (Mitchell et al. 1996, 1997; Allmang et al. 1999b). The structural components of the RNA exosome include three S1/K homology (KH) cap subunits and a lower ring of six PH-like core subunits, hereafter referred to as “cap” and “core” subunits, respectively. The three-subunit cap is composed of EXOSC1/Csl4 (Human/S. cerevisiae), EXOSC2/Rrp4, and EXOSC3/Rrp40. The six-subunit core is composed of EXOSC4/Rrp41, EXOSC5/Rrp46, EXOSC6/Mtr3, EXOSC7/Rrp42, EXOSC8/Rrp43, and EXOSC9/Rrp45. The structural subunits form a barrel-like structure through which RNA can be threaded in a 5′–3′ orientation. The DIS3, DIS3L, or Dis3/Rrp44 catalytic subunit is located at the bottom of the barrel and can process or degrade the RNA targets (Fig. 1A). Structural studies of both yeast and human RNA exosome complexes have revealed conservation in the organization of the RNA exosome (Supplemental Fig. S1A,B; Liu et al. 2006; Bonneau et al. 2009; Makino et al. 2013; Wasmuth et al. 2014; Zinder et al. 2016), beyond the evolutionary sequence conservation of the subunits.
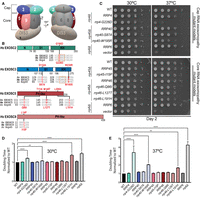
Overview of pathogenic amino acid substitutions in the human structural subunits of the RNA exosome. (A) Schematic view of the human RNA exosome with nine structural subunits (EXOSC1–9), denoted as 1–9, and one catalytic subunit (DIS3). (B) Domain maps of EXOSC2, EXOSC3, EXOSC5, and EXOSC9. EXOSC2 and EXOSC3 are composed of three domains: an N-terminal domain, a central putative RNA-binding S1 domain, and a C-terminal putative RNA-binding K homology (KH) domain. The “GxNG” motif in the KH domain of cap subunits is boxed in orange. EXOSC5 and EXOSC9 are composed of a singular PH-like domain. The positions of the RNA exosomopathy disease-linked amino acid substitutions in the human subunits are depicted above the domain structures in red. Sequence alignments of the orthologs from Homo sapiens (Hs), Mus musculus (Mm), and S. cerevisiae (Sc) reveal the high degree of conservation of the residues altered in disease (in red) and the sequences flanking these residues (in gray). The amino acid substitutions generated in the budding yeast Rrp orthologs for this study that correspond to the pathogenic amino acid substitutions are shown below the sequence alignments in red. The rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant yeast cells, modeling EXOSC2-G198D, EXOSC3-W238R, and EXOSC5-L206H variants, respectively, exhibit (C) impaired growth in a solid media assay and (D,E) increased doubling times calculated from a liquid media assay at (D) 30°C and (E) 37°C. For these assays, the growth of RNA exosome deletion strains solely containing wild-type or mutant RNA exosome plasmid was analyzed by serial dilution and spotting of cells onto SD -Leu media and in liquid media, at 30°C and 37°C. BY4741 cells were used as wild-type isogenic controls (labeled WT). The rrp6Δ deletion containing an empty vector or wild-type RRP6 plasmid was used as a control. Growth measurements in liquid media used three biological replicates.
The RNA exosome plays a pivotal role in the processing, degradation, and surveillance of nearly every class of RNA in both the nucleus and cytoplasm (Schneider and Tollervey 2013; Kilchert et al. 2016; Morton et al. 2018). First discovered as a crucial complex required for proper maturation of ribosomal RNA (Mitchell et al. 1997; Allmang et al. 1999a), the RNA exosome has subsequently been shown to contribute to the processing of small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), and transfer RNAs (tRNAs) (Allmang et al. 1999a; Chekanova et al. 2007; Gudipati et al. 2012; Schneider et al. 2012; Pefanis et al. 2014; Kilchert et al. 2016). In addition, the RNA exosome is critical for RNA homeostasis within the nucleus through targeting and degrading highly unstable species, such as cryptic unstable transcripts (CUTs) in S. cerevisiae and promoter upstream transcripts (PROMPTs) in human cells (Wyers et al. 2005; Preker et al. 2008; Kiss and Andrulis 2010; Parker 2012; Schneider et al. 2012; Belair et al. 2018). The RNA exosome also plays a crucial role in RNA surveillance in both the nucleus and cytoplasm, degrading aberrant RNAs (Belair et al. 2018). In addition to surveillance of misprocessed endogenous RNA species, the RNA exosome has been implicated in targeting foreign RNA through antiviral surveillance pathways (Molleston et al. 2016).
Though the RNA exosome is essential (Mitchell et al. 1997; Lorentzen et al. 2007; Hou et al. 2012; Lim et al. 2013; Pefanis et al. 2014; Brouze et al. 2025), recent clinical studies have identified pathogenic missense mutations in the structural subunit genes that result in distinct tissue-specific defects comprising a growing family of diseases termed RNA exosomopathies (Fasken et al. 2020). Pathogenic missense mutations have been identified in the cap subunit genes EXOSC1/2/3 and core subunit genes EXOSC4/5/8/9 (Wan et al. 2012; Biancheri et al. 2013; Boczonadi et al. 2014; Eggens et al. 2014; Di Donato et al. 2016; Burns et al. 2017, 2018; Schottmann et al. 2017; Bizzari et al. 2020; Slavotinek et al. 2020; Somashekar et al. 2021; Fasken et al. 2024). Missense mutations in the genes encoding the EXOSC1 and EXOSC3 cap subunits and EXOSC8 and EXOSC9 core subunits cause forms of pontocerebellar hypoplasia type 1 (PCH1), a severe disease characterized by early onset atrophy of the pons and cerebellum (Wan et al. 2012; Biancheri et al. 2013; Eggens et al. 2014; Halevy et al. 2014; Burns et al. 2017, 2018; Schottmann et al. 2017; Bizzari et al. 2020; Sakamoto et al. 2021; Somashekar et al. 2021; Damseh et al. 2023). Missense mutations in the gene encoding the EXOSC5 core subunit are linked to a disease characterized by cerebellar atrophy, spinal muscular atrophy (SMA)-like motor delays and hypotonia (Slavotinek et al. 2020). In contrast to most of the other mutations that primarily cause neurological defects, missense mutations in the gene encoding the EXOSC2 cap subunit are linked to a novel syndrome termed SHRF (short stature, hearing loss, retinitis pigmentosa, and distinctive facies) (Di Donato et al. 2016). While diverse in their clinical manifestations, typically, RNA exosomopathy missense mutations result in single amino acid substitutions in conserved domains of the structural subunits of the RNA exosome.
Several recent studies have begun to investigate the molecular consequences of the different pathogenic amino acid substitutions that occur in exosomopathies (summarized in Supplemental Table S1; Morton et al. 2018; Fasken et al. 2020). Expression levels of EXOSC3-G31A and EXOSC3-W238R variants in a mouse neuronal line were reduced compared to wild-type mouse EXOSC3, suggesting that these amino acid substitutions could affect the stability of the subunit (Fasken et al. 2017). Additionally, analyses of PCH patient fibroblasts and skeletal muscle cells homozygous for the EXOSC9-L14P mutations revealed that the variant protein levels are decreased compared to EXOSC9 levels in control samples, suggesting the EXOSC9-L14P substitution impacts the stability of the subunit (Burns et al. 2018). Similarly, analyses of the EXOSC8-S272T variant in myoblasts and fibroblasts showed that the steady-state EXOSC8 level is significantly decreased compared to EXOSC8 in wild-type control cells (Boczonadi et al. 2014). In addition, in patient fibroblasts with mutations in EXOSC3 and EXOSC8, the EXOSC9 protein level was reduced, suggesting that reduced levels of one RNA exosome subunit can destabilize the RNA exosome complex (Burns et al. 2018). However, reconciling the diverse clinical pathologies seen in RNA exosomopathies cannot be solely explained by reductions in levels of individual essential subunits and/or the level of the RNA exosome complex. Thus, modeling these missense mutations and performing functional in vivo studies is critical to reveal the biology underlying RNA exosomopathy diseases.
Analyses of some of these RNA exosomopathy mutations in genetic model systems reveal distinct molecular and functional consequences resulting from the different pathogenic amino acid substitutions (Fasken et al. 2017; Gillespie et al. 2017; Morton et al. 2020; Slavotinek et al. 2020; Sterrett et al. 2021). These studies suggest that both complex integrity and interactions with known RNA exosome cofactors may be differentially impacted by specific RNA exosomopathy mutations (Fasken et al. 2017; Gillespie et al. 2017; Morton et al. 2020; Slavotinek et al. 2020). Nuclear RNA exosome cofactors have been extensively characterized in the budding yeast system and include the exonuclease Rrp6, its obligate binding partner Rrp47, the essential RNA helicase Mtr4, and Mpp6 (de la Cruz et al. 1998; LaCava et al. 2005; Schneider and Tollervey 2013; Zinder and Lima 2017). Structural studies of both the budding yeast and mammalian RNA exosome show conservation in the composite sites through which these cofactors interact with the complex and suggest similar roles in vivo across the eukaryotic systems (Schuch et al. 2014; Falk et al. 2017; Wasmuth et al. 2017; Schuller et al. 2018; Weick et al. 2018). Any alteration in the RNA exosome levels or key cofactor interactions resulting from these amino acid substitutions would ultimately have an impact on the ability of the complex to process, degrade, or survey RNA targets in a cell. Changes in RNA target levels could profoundly impact certain tissues if key RNA classes or specific RNAs are misprocessed, defective RNA accumulates, and/or RNA homeostasis is dysregulated. While previous studies of these RNA exosomopathy mutations provide valuable characterization in vivo, there has not yet been a direct comparison of the defects in RNA exosome function across multiple cap and core RNA exosomopathy mutant models. A comparative assessment of how these exosomopathy amino acid substitutions affect the ability of the RNA exosome to process, degrade, and survey aberrant RNAs in vivo is critical to further understand the molecular consequences underlying each distinct exosomopathy disease pathology.
Here, we take advantage of the budding yeast model system to explore and compare the functional and molecular consequences of a set of pathogenic amino acid substitutions within the RNA exosome. Given that the RNA exosome was initially identified and has been most extensively studied in S. cerevisiae (Mitchell et al. 1997; Allmang et al. 1999b; Sloan et al. 2012) and the high conservation in overall complex structure between the human and budding yeast RNA exosomes (Makino et al. 2013, 2015; Kowalinski et al. 2016; Zinder et al. 2016; Wasmuth et al. 2017; Gerlach et al. 2018), a budding yeast system provides a robust platform to comparatively assess the in vivo consequences of exosomopathy mutations. In this study, we generated and analyzed S. cerevisiae models of the exosomopathy amino acid changes identified in EXOSC2, EXOSC3, EXOSC5, and EXOSC9 genes by mutating the orthologous budding yeast genes RRP4, RRP40, RRP46, and RRP45. We analyzed yeast cell growth and employed an unbiased RNA-seq approach to explore the consequences of these missense mutations. From these approaches, we detect the greatest functional defects in three of our mutant models, rrp4-G226D, rrp40-W195R, and rrp46-L191H. Comparative analyses of transcriptomes across these three models revealed some shared changes, particularly in coding and noncoding transcripts required for rRNA processing and ribosome biogenesis, suggesting potential defects in translation. We also identified differentially expressed genes unique to each of the three mutant models, suggesting that while there are some shared consequences, there are also distinct differences in RNA exosome function. Assessment of ribosome biogenesis and translation defects in the three models revealed shared defects in rRNA processing but distinct differences in polysome profiles. Our results represent an unbiased approach to comparatively characterize the molecular defects in the function of the RNA exosome across a collection of RNA exosomopathy mutant models and suggest that distinct translational defects may underlie the unique molecular pathology of RNA exosomopathies.
RESULTS
RNA exosomopathy mutations modeled in S. cerevisiae cause different growth phenotypes
We used the budding yeast model to assess the effects of each RNA exosomopathy mutation. As shown in Figure 1B, the residues that are substituted in RNA exosome subunits of individuals with RNA exosomopathy often lie within evolutionarily conserved regions of the proteins, allowing the variant to be readily modeled in S. cerevisiae. The EXOSC2 amino acid substitution Gly198Asp (G198D) (Di Donato et al. 2016) and the EXOSC3 amino acid substitutions Asp132Ala (D132A) and Trp238Arg (W238R) (Rudnik-Schoneborn et al. 2013; Eggens et al. 2014; Schottmann et al. 2017) occur in highly conserved domains of both cap subunits in similar regions. EXOSC2-G198D and EXOSC3-W238R affect a conserved structural “GxNG” motif within the RNA-binding KH domain. The RNA exosomopathy-linked missense mutations identified in the core subunit genes EXOSC5 and EXOSC9 also result in amino acid substitutions in conserved domains of each protein. Distinct mutations in the EXOSC5 gene result in amino acid changes Thr114Ile (T114I), Met148Thr (M148T), and Leu206His (L206H), which are located throughout the PH-like domain of the EXOSC5 protein. The RNA exosomopathy mutation in the EXOSC9 gene results in the amino acid change Leu14Pro (L14P) located near the N-terminus of the EXOSC9 protein. Structural analysis of each RNA exosomopathy amino acid substitution suggests that these changes could affect intersubunit-binding interfaces or the conformation of the subunits themselves (Liu et al. 2006; Fasken et al. 2017, 2020; Slavotinek et al. 2020; Sterrett et al. 2021).
We modeled the RNA exosomopathy mutations found in EXOSC2/3/5/9 in the corresponding S. cerevisiae genes RRP4/40/46/45. The SHRF-linked EXOSC2-G198D mutation is modeled by the rrp4-G226D yeast cells. The PCH-linked EXOSC3-D132A, EXOSC3-W238R, and EXOSC9-L14P mutations are modeled by the rrp40-S87A, rrp40-W195R, and rrp45-I15P yeast cells, respectively. The EXOSC5 RNA exosomopathy mutations EXOSC5-T114I, EXOSC5-M148T, and EXOSC5-L206H are modeled by the rrp46-Q86I, rrp46-L127T, and rrp46-L191H yeast cells, respectively. We first examined the functional consequences of rrp4/40/45/46 yeast mutants. RNA exosome deletion strains (rrp4Δ, rrp40Δ, rrp45Δ, and rrp46Δ) solely containing the RRP4/40/45/46 wild-type gene or rrp4/40/45/45 mutant gene were analyzed in both solid and liquid media growth assays (Fig. 1C–E). The parental wild-type budding yeast strain (BY4741; WT) was included as an isogenic control for the RNA exosome deletion strains.
Previous studies have shown that the rrp4-G226D, rrp40-W195R, and rrp46-L191H yeast mutants are viable but exhibit growth defects compared to the corresponding wild-type control cells (Fasken et al. 2017; Gillespie et al. 2017; Slavotinek et al. 2020; Sterrett et al. 2021). Consistent with these results, the rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells show impaired growth at 37°C, and rrp4-G226D and rrp46-L191H cells exhibit impaired growth at 30°C on solid media compared to corresponding control cells (Fig. 1C). The rrp40-S87A, rrp46-Q86I, rrp46-L127T, and rrp45-I15P mutant cells show no growth defects compared to corresponding wild-type control cells (Fig. 1C). As a comparative control for cells disrupted for RNA exosome function, we also examined the growth of rrp6Δ cells, which lack the RNA exosome cofactor Rrp6 and exhibit growth defects (Briggs et al. 1998). The Rrp6 exonuclease is nonessential in budding yeast; however, Rrp6 assists the RNA exosome in the targeting and degradation of several key transcript RNAs (Briggs et al. 1998; Schuch et al. 2014; Wasmuth et al. 2014; Marin-Vicente et al. 2015). As expected, the rrp6Δ cells show extremely poor growth at 37°C compared to control cells (Fig. 1C). In liquid media, the rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells display significantly increased doubling times compared to wild-type RRP4/40/45 control cells at both 30°C and 37°C (Fig. 1D,E). Notably, the rrp46-L191H cells have the longest doubling time at 30°C and the rrp4-G226D cells have the longest doubling time at 37°C, similar in intensity to that of rrp6Δ cells.
Overall, the comparative growth analyses suggest that RNA exosomopathy mutations that alter different subunits of the RNA exosome have varied functional consequences in vivo, with the most significant growth defects observed for rrp4-G226D, rrp40-W195R, and rrp46-L191H mutants. These three modeled mutations have previously been shown to have varying impacts on the protein levels of the individual yeast RNA exosome subunits (Fasken et al. 2017; Slavotinek et al. 2020; Sterrett et al. 2021), with the Rrp40-W195R variant showing the most significant protein instability at both 30°C and 37°C (Fasken et al. 2017). Given the rrp40-W195R cells show the mildest growth defect compared to the rrp4-G226D and rrp46-L191H cells, these data suggest that the observed growth defects are not simply due to varying levels of loss of the essential subunits and subsequent loss of the complex. Thus, these S. cerevisiae models can be used to assess the molecular consequences that may arise in the processing and/or degradation of RNA from these RNA exosomopathy mutations.
Broad transcriptomic changes observed in rrp4-G226D, rrp40-W195R, and rrp46-L191H mutants
To perform an unbiased analysis of the molecular consequences of the modeled RNA exosomopathy pathogenic amino acid substitutions, we performed RNA-seq analysis on the rrp4-G226D, rrp40-S87A, rrp40-W195R, rrp45-I15P, rrp46-Q86I, rrp46-L127T, and rrp46-L191H mutants and the corresponding wild-type controls. Differential transcript expression analysis of each mutant compared to its corresponding wild-type control revealed that rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells exhibit a large number of differentially increased or decreased transcripts compared to the corresponding wild-type RRP4/40/46 control cells (Fig. 2A). In contrast, the rrp40-S87A, rrp45-I15P, rrp46-Q86I, and rrp46-L127T transcriptomes show only minor changes (Supplemental Fig. S2). Unbiased principal component analyses (PCA) of the RNA-seq data revealed reproducibility among the RNA-seq biological replicates and confirmed that the rrp4-G226D, rrp40-W195R, and rrp46-L191H transcriptomes are distinct from those of their wild-type controls (Supplemental Fig. S3).
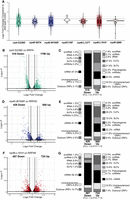
RNA-seq analysis of RNA exosomopathy mutant yeast models reveals large and distinct transcriptomic changes in the rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells. (A) Violin plots showing the distribution of transcripts identified in differential analysis as significant (P < 0.05) in each mutant compared to the corresponding wild-type control. The y-axis shows the log2 fold change (LFC) for each transcript. The solid gray line demarcates a FC of +1.5 or −1.5 (LFC = 0.585 or −0.585). The dotted gray line marks a fold change (FC) of +2 or −2 (LFC = 1 or −1). (B, D, F) Volcano plots of the differentially expressed transcripts and classification of RNA types in rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells as labeled. (C, E, G) Stacked bar graph of the percentages of different RNA classes within the differentially expressed transcripts in rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells as indicated. RNA classes are shown as percentages and include messenger RNAs (mRNA), small nuclear RNAs (snRNA), small nucleolar RNAs (snoRNA), transfer RNAs (tRNA), cryptic unstable transcripts (CUTs), stable unannotated transcripts (SUTs), other noncoding RNA (ncRNA; e.g., TLC1), pseudogenes, and uncharacterized or dubious open reading frames (ORFs). Vertical lines mark FC values of ±1.5 (straight line) and ±2 (dotted line).
RNA-seq analysis provides insight into the transcripts altered but does not provide any insight into which changes result directly from RNA exosome-mediated decay as compared to more systemic effects. Given the role of the RNA exosome in RNA decay, transcripts that show an increase in steady-state levels are more likely to be direct targets than transcripts that show a decrease in steady-state levels, so our analysis considers first the increased transcripts and then the decreased transcripts. Differential gene expression analysis of the rrp4-G226D cells reveals 1196 increased transcripts (FC ≥ 1.5, P < 0.05) and 516 decreased transcripts (FC ≤ −1.5, P < 0.05) compared to control cells (Fig. 2B). Increased transcripts are predominantly ncRNAs (Fig. 2C), especially cryptic unstable transcripts (CUTs) and stable unannotated transcripts (SUTs), and the two most significantly increased mRNAs include PIR3, encoding a protein required for cell wall stability, and DDR2, encoding a multi-stress response protein (Fig. 2C). In contrast, nearly 90% of decreased transcripts are mRNAs (Fig. 2C), and the most significantly decreased transcripts include SSA1, SSA2, HSC82, and HSP60, encoding chaperones, as well as RPS3 and RPL15A, encoding ribosomal proteins. In rrp40-W195R cells, 569 increased (FC ≥ 1.5, P < 0.05) and 426 decreased (FC ≤ −1.5, P < 0.05) transcripts were identified (Fig. 2D). Increased transcripts in rrp40-W195R cells compared to control (Fig. 2E) are mostly ncRNAs, such as CUTs (29%), SUTs (15%), snoRNAs (5%), and tRNAs (6%). In contrast, over 80% of the decreased transcripts are mRNAs (Fig. 2E), including HIS4, URA1, URA4, and MDH2, involved in metabolism, as well as RPS13 and RPS7B, encoding ribosomal proteins. In rrp46-L191H cells, 724 increased transcripts (FC ≥ 1.5, P < 0.05) and 487 decreased transcripts (FC ≤ −1.5, P < 0.05) were identified (Fig. 2F). The increased transcripts in rrp46-L191H cells also include CUTs (30%), SUTs (15%), and mRNAs (30%) (Fig. 2G), with RPL18B, encoding a ribosomal protein, and CBT1, encoding a protein involved in 5′ RNA end processing of mitochondrial cytochrome b mRNA that is linked to processing of rRNA, among the most significantly increased mRNAs. Approximately 60% of decreased transcripts in rrp46-L191H cells are mRNAs (Fig. 2G), including HIS4, URA1, URA4, BIO3, BIO4, and RIB4, involved in metabolism, as well as RPS3 and RPL15A mRNAs, encoding ribosomal proteins.
The overall comparison across the rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells reveals distinct changes in RNA categories affected (Fig. 2C,E,G), particularly in the distribution of increased ncRNAs and decreased mRNAs across these models. All three models show increases in CUTs and SUTs. As CUTs and SUTs are stabilized in RNA exosome mutants and crosslink to the RNA exosome (Wyers et al. 2005), these transcripts are likely direct targets of the RNA exosome, indicative of defective complex function. To compare changes in CUTs and SUTs across the three mutant models, we generated a heat map of normalized fragments per kilobase of transcript per million mapped (FPKM) expression estimates (Fig. 3). We included FPKM estimates of the rrp6Δ deletion strain as CUTs and SUTs were first identified in cells deleted for RRP6 and Rrp6 activity is important for degradation of the CUTs (Wyers et al. 2005; Davis and Ares 2006; Xu et al. 2009). The rrp6Δ cells show a broad, indiscriminate increase in all CUTs and SUTs (Fig. 3). The rrp4-G226D cells show a broad increase in CUTs and SUTs, while rrp40-W195R and rrp46-L191H cells also exhibit increases, but to a lesser extent than rrp4-G226D (Fig. 3). Interestingly, not all the same CUTs and SUTs are changed in rrp4-G226D, rrp40-W195R, and rrp46-L191H cells. The rrp4-G226D cells exhibit the broadest increase in CUTs and SUTs among the three mutants, though certain groups remain unaffected. Similarly, rrp40-W195R and rrp46-L191H cells show both shared and unique changes in CUTs and SUTs (Fig. 3).
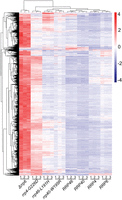
Heat maps of RNA exosomopathy yeast mutants reveal an accumulation in CUTs/SUTs. Transcript level estimates of all cryptic unstable transcripts (CUTs) and stable unannotated transcripts (SUTs) are scaled for heat map visualization. Coloring is a gradient of higher (red) to lower (blue) scaled values.
While most decreased transcripts in all three mutant models are mRNAs, the degree by which mRNAs, among other transcripts, are affected varies in each mutant. In rrp46-L191H cells, expressing a core subunit variant, ∼60% of the significantly reduced transcripts are mRNAs, compared to 80%–90% in the rrp4-G226D and rrp40-W195R cells, expressing cap subunit variants. Additionally, in rrp46-L191H cells, ∼20% of total significantly decreased transcripts are tRNAs, CUTs, and SUTs, while these transcripts are significantly less affected in the other two mutants. These differences highlight the differential impact of cap versus core RNA exosome variants on the affected transcript types. These observations suggest that within the rrp4-G226D, rrp40-W195R, and rrp46-L191H cells, RNA exosome targeting and degradation are impacted, yet in distinct ways, as all the direct RNA targets are not indiscriminately elevated across all three mutant models. These data strongly suggest that the broader differences in transcriptome profiles seen in these models (Figs. 2, 3) are unlikely to merely reflect the variable penetrance of the variants.
In summary, the diverse differentially increased/decreased transcripts observed across rrp4-G226D, rrp40-W195R, and rrp46-L191H cells highlight the distinct changes resulting from each RNA exosome variant, suggesting differential functional impacts of the variants in vivo.
Comparative assessment of differentially expressed transcripts within rrp4-G226D, rrp40-W195R, and rrp46-L191H mutants suggests shared impacts on metabolic pathways and rRNA modification and processing
To compare the molecular impacts of the modeled RNA exosomopathy mutations, we analyzed shared increased and decreased transcripts across the three models (rrp4-G226D, rrp40-W195R, and rrp46-L191H) using UpSet plots (Fig. 4). We identified 209 significantly increased transcripts (FC ≥ +1.5, P < 0.05) across all three mutant models (Fig. 4A), mostly CUTs and SUTs (Fig. 4B), consistent with the trend observed for each individual mutant model. GO analysis revealed significant enrichment in rRNA modification (GO:0000154) (Fig. 4C), possibly driven by the increased snoRNAs, which are required for rRNA modifications. We note that the total RNA-seq approach used here is not ideal for capturing all snoRNA changes in an unbiased manner as that was not the goal of this study and we did not employ a thermostable reverse transcriptase to accurately capture highly structured RNAs like snoRNAs (Boivin et al. 2018). Nonetheless, we observe a fraction of significantly differentially expressed snoRNA transcripts, which are known to be direct targets of the RNA exosome (Webster and Ghalei 2023). Overall, the significant enrichment among the three mutant models in rRNA modification and processing (GO:0000154; GO:00031167; GO:0006364) (Fig. 4C) strongly suggests significant impacts on ribosome biogenesis within these models.
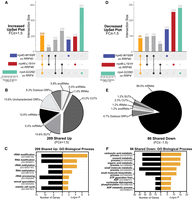
UpSet plots of differentially expressed transcripts in rrp4-G226D, rrp40-W195R, and rrp46-L191H cells reveal shared targets involved in metabolism and rRNA processing. The UpSet plot of (A) significantly increased (FC ≥ +1.5) transcripts or (D) significantly decreased (FC ≤ −1.5) transcripts within the rrp4-G226D, rrp40-W195R, and rrp46-L191H data sets. Pie chart of the RNA classes that comprise the intersection of shared (B) increased (Up) transcripts or (E) decreased (Down) transcripts (Up). Gene ontology (GO) analysis for the biological process of the shared (C) increased transcripts or (F) decreased transcripts. The sets of samples are color-coded: transcripts identified with FC ≤ −1.5 or FC ≥ +1.5 from differential expression analysis of rrp40-W195R versus RRP40 cells are colored blue, those of rrp46-L191H versus RRP46 cells are colored red, and those of rrp4-G226D versus RRP4 cells are colored teal. Black bars in C and F represent the number of transcripts that are linked to each biological process category, whereas orange bars represent the −log of the associated P-value for each GO term. GO analyses were performed on coding RNAs (mRNA) and noncoding RNAs (tRNAs, snoRNAs, and snRNAs).
We also detected 86 transcripts shared that are significantly decreased (Fig. 4D) among the rrp4-G226D, rrp40-W195R, and rrp46-L191H cells. Of these decreased transcripts, ∼90% are mRNAs (Fig. 4E). GO analysis on these shared decreased transcripts revealed metabolic and biosynthetic biological processes as the top significantly impacted categories (GO:0019752; GO:0043436; GO:0006082) (Fig. 4F). GO analyses on the identifiable human homologs of the shared increased and decreased transcripts (Supplemental Documentation S1) show that while most increased transcripts are yeast-specific CUTs and SUTs, those with human homologs are enriched in synaptic vesicle priming and fusion biological processes (GO:0016082; GO:0031629; GO:0099500). Similarly, the shared decreased transcripts with human homologs are enriched in metabolic and biosynthetic processes (GO:0019752; GO:0043436; GO:0006082). This analysis suggests that the modeled RNA exosomopathy mutations result in changes in highly conserved metabolic and biosynthetic pathways.
Comparing mutant pairs, we identified shared significantly increased transcripts (Supplemental Fig. S4A). Most increased transcripts shared between rrp40-W195R and rrp46-L191H are mRNAs (Supplemental Fig. S4B). GO analysis of these transcripts revealed significant enrichment in rRNA and ncRNA processing and ribosome biogenesis (GO:0016072; GO:0034470; GO:0006364; GO:0042254) (Supplemental Fig. S4C). However, shared increased transcripts in rrp40-W195R and rrp4-G226D cells mostly consist of CUTs and SUTs, with no significant biological process enrichment (Supplemental Fig. S4B). Similarly, shared increased transcripts in rrp4-G226D and rrp46-L191H cells are mostly CUTs and SUTs (Supplemental Fig. S4B), with GO analysis revealing a significant enrichment in telomere maintenance and mitotic recombination (GO:0000722; GO:0006312) (Supplemental Fig. S4D). GO analyses of the shared increased transcripts between rrp4-G226D and rrp40-W195R, and rrp40-W195R and rrp46-L191H reveal significant enrichment of biosynthetic processes (GO:0043604; GO:1901566; GO:0034645).
We also identified shared significantly decreased transcripts among the pairs of models (Supplemental Fig. S4E). Most decreased transcripts are mRNAs (Supplemental Fig. S4F), but a notable percentage of shared transcripts in the rrp4-G226D and rrp46-L191H cells are from the tRNA genes (Supplemental Fig. S4F). GO analyses on the shared decreased transcripts between paired groups reveal enrichment in different biological processes related to translation. Specifically, a significant number of the shared significantly decreased transcripts in rrp4-G226D and rrp40-W195R cells impact cytoplasmic translation (GO:0002181) (Supplemental Fig. S4G) and those in rrp4-G226D and rrp40-L191H cells show translation elongation (GO:0006414) (Supplemental Fig. S4H). This is consistent with the large percentage of tRNAs that are decreased in rrp4-G226D and rrp46-L191H cells. We found no significant enrichment of any specific biological process for the shared significantly decreased transcripts in the rrp40-W195R and rrp46-L191H cells.
In summary, transcripts involved in ribosome biogenesis are significantly enriched across all three mutants, as expected given the major role of the RNA exosome in rRNA processing. The rrp40-W195R and rrp4-G226D cells, modeling mutations in the cap subunit genes, share different targets from rrp46-L191H cells, modeling a mutation in a core subunit gene. These data strongly suggest that the type of RNA exosome subunit that is altered influences the specific RNA classes affected by each mutant.
Comparative assessment of differentially expressed transcripts specific to rrp4-G226D, rrp40-W195R, or rrp46-L191H mutant suggests impacts on translation and ribosome biogenesis
In examining unique changes caused by each variant, we identified 154 transcripts increased specifically in the rrp40-W195R cells, 193 in the rrp46-L191H cells, and 567 in the rrp4-G226D cells (Fig. 5A). Analysis of these specific transcript sets reveals a divergent pattern between the three mutant models (Fig. 5B). In the rrp40-W195R cells, nearly 25% map to snoRNA, snRNA, or tRNA genes, another 25% map to CUTs and SUTs, another 25% map to mRNAs, and the rest map to dubious or uncharacterized open reading frames (ORFs). In contrast, rrp46-L191H-specific increased transcripts are predominantly mRNAs, while the rrp4-G226D-specific increased RNAs are mostly CUTs and SUTs. GO analysis of the rrp40-W195R-specific transcripts revealed significant enrichment in biological processes involved in gene expression (GO:0010467), rRNA modification (GO:00000154), and translation elongation (GO: 0006414) (Fig. 5C). For the rrp46-L191H cells, GO analysis revealed significant enrichment in processes related to ncRNA processing (GO:0034470) and ribosome biogenesis (GO:0042254) (Fig. 5D). No significant enrichment was found for the rrp4-G226D-specific increased transcripts, likely due to the large percentage of CUTs and SUTs.
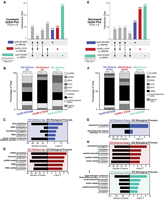
UpSet plots of differentially expressed transcripts in rrp4-G226D, rrp40-W195R, and rrp46-L191H cells reveal RNA targets that are uniquely impacted. UpSet plots were generated as in Figure 4. The intersections assessed here are transcripts significantly increased or decreased by 1.5-fold or more only in the rrp40-W195R data set (blue), the rrp46-L191H data set (red), or the rrp4-G226D data set (teal). The UpSet plot of significantly (A) increased (FC ≥ +1.5) transcripts or (E) decreased (FC ≤ −1.5) transcripts occurring solely in the rrp40-W195R, rrp46-L191H, or rrp4-G226D data set. Stacked bar percentages of the RNA types that comprise the (B) increased (Up) transcripts or (F) decreased (Down) transcripts identified only in the rrp40-W195R, rrp46-L191H, or rrp4-G226D data set. GO analysis for biological process of the (C) increased transcripts or (G) decreased transcripts occurring only in the rrp40-W195R data set; (D) increased transcripts or (H) decreased transcripts occurring only in the rrp46-L191H data set, and the (I) decreased transcripts occurring only in the rrp4-G226D data set. In C,D and G–I, black bars represent the number of transcripts that are linked to each biological process category, whereas colored bars represent the −log of the associated P-value for each GO term. All GO analyses were performed on coding RNAs (mRNA) and noncoding RNAs (tRNAs, snoRNAs, and snRNAs).
We identified 143 transcripts significantly decreased specifically in the rrp40-W195R cells, 254 in the rrp46-L191H cells, and 280 in the rrp4-G226D cells (Fig. 5E). Distinct patterns emerge from the analysis of these transcripts, with the cap mutants (rrp4-G226D and rrp40-W195R) primarily affecting mRNAs, while the rrp46-L191H core mutant impacts a more diverse range of transcripts, including tRNA gene transcripts, CUTs and SUTs (Fig. 5F). GO analysis of the 143 decreased transcripts in the rrp40-W195R cells revealed significant enrichment in biological processes related to cytoplasmic translation (GO:0002181) and ribosomal small subunit assembly (GO:0000028) (Fig. 5G). The 254 decreased transcripts in rrp46-L191H and 280 in rrp4-G226D are enriched in translation (GO:0006412), amide and peptide biosynthesis, and metabolic processes (Fig. 5H,I).
Overall, the GO analyses of transcripts uniquely changed in each mutant model revealed enrichment in several similar biological processes, particularly ribosome biogenesis, translation, and biosynthesis. However, the transcripts that produce the shared GO terms are distinct within each mutant cell model. These data suggest that while there may be overlapping impacts on key biological processes, the overall consequences are in part due to distinct target changes.
RNA exosomopathy yeast mutant models exhibit defects in ribosome biogenesis, impact global translation, and alter translation fidelity
The RNA exosome is required for the processing of rRNAs (Schmid and Jensen 2008; Fernández-Pevida et al. 2015). In line with this function, GO analyses of the RNA-seq data from rrp4-G226D, rrp40-W195R, and rrp46-L191H cells revealed enrichment in terms related to translation and ribosome biogenesis (Figs. 4C, 5C,D). In addition, differential expression analysis revealed that some of the most significantly decreased transcripts in all three mutant models are RPS and RPL mRNAs, which encode ribosomal proteins. To broadly compare impacts on ribosomal protein genes across the rrp4-G226D, rrp40-W195R, and rrp46-L191H cells, we generated heat maps of normalized FPKM expression estimates specifically for the RPS and RPL genes (Fig. 6), using FPKM estimates and including data from rrp6Δ cells collected in the same RNA-seq experiment. Consistent with previous work, there is a decrease in transcript levels for most ribosomal protein genes in rrp6Δ cells (Fox et al. 2015). Our analysis also included mitochondrial ribosomal protein genes (MRPS and MRPL), which in large part show a different transcriptomic pattern change, compared to RPS and RPL transcripts, in rrp6Δ cells. Furthermore, we detect an overall reduction of ribosomal protein gene transcripts in all three RNA exosomopathy models with the biological triplicates clustering together. However, the magnitude of the decrease is less than the decrease observed in rrp6Δ cells. To assess the effect of RNA exosome variants on ribosome biogenesis, we examined rRNA processing by northern blotting of total RNA (Fig. 7). For these assays, we generated CRISPR/Cas9-edited rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells to be able to compare them to each other directly and parent wild-type cells, instead of comparing each deletion with a point mutant plasmid to the same deletion with a wild-type plasmid. The temperature-sensitive (ts) growth of the rrp4/40/46 CRISPR mutants was confirmed compared to the wild-type parental cells in a solid media growth assay, and rescue of the growth of the mutants by wild-type RRP4, RRP40, or RRP46 plasmid was confirmed (Supplemental Fig. S5). Consistent with previously published data for rrp4-G226D, rrp40-W195R, and rrp46-L191H (Gillespie et al. 2017; Slavotinek et al. 2020; Sterrett et al. 2021), northern blots show that all RNA exosome variants cause significant accumulation of 7S pre-rRNA, a precursor of mature 5.8S rRNA that is processed by the RNA exosome (Fig. 7A,B). The magnitude of 7S accumulation is higher for rrp4-G226D than for the other models (Fig. 7C), consistent with the more severe growth defect of rrp4-G226D cells compared to the other mutants (Fig. 2), resulting in a more significant reduction of mature 5.8S rRNA in these cells (Fig. 7D). In addition, cap mutant cells (rrp4-G226D and rrp40-W195R) show a clear accumulation of 27SA2, which is not seen in the core mutant cells (rrp46-L191H) (Fig. 7A,B). In contrast, rrp46-L191H cells show significant accumulation of 23S pre-rRNA, a precursor of 18S rRNA, accompanied by a reduction in levels of 20S rRNA (Fig. 7A), resulting in a significant reduction of mature 18S rRNA (Fig. 7D). The rrp46-L191H rRNA processing defects are similar to those of another core mutant (rrp41-L187P), which we recently analyzed (Fasken et al. 2024). Overall, the ribosome biogenesis defects in the cap and core mutant cells exhibit both shared and distinct characteristics. While all mutants result in accumulation of 7S pre-rRNAs, the cap mutants have a more significant impact on the biogenesis of the 60S subunit, which is also reflected in a reduced ratio of 25S/18S rRNA in these cells (Fig. 7D).

Heat maps of RNA exosomopathy yeast mutants reveal broad changes in ribosomal protein gene transcript levels. Heat maps were generated with FPKM estimates of all annotated ribosomal protein RPS and RPL genes. Cytoplasmic and mitochondrial subunit genes are shaded in yellow and green, respectively. Gene expression estimates are scaled for heat map visualization, and coloring is a gradient of higher (red) to lower (blue) scaled values. The gene name for each RPS or RPL transcript is listed on the right.
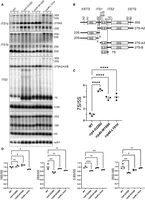
Ribosome biogenesis defects in rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells. (A) Northern blot analysis of steady-state levels of precursor rRNA levels in the indicated cells. Three biological replicates were grown at 30°C and analyzed. (B) Schematic of the yeast precursor rRNAs and the binding site of the probes used in A. Probes used to detect 18S, 25S, 5.8S, 5S, and scR1 bind to an internal region within the sequence of the mature RNA. (C) Quantification of the ratio of 7S precursor rRNA to mature 5S rRNA for data shown in A. (D) Quantification of 18S, 25S, and 5.8S mature rRNA levels relative to 5S rRNA, and the ratio of 25S to 18S, in the RNA exosomopathy mutant yeast models compared to wild-type control cells for three biological replicates shown in A.
Polysome profiling using RNA exosomopathy mutant yeast models reveals translation differences that suggest distinct molecular consequences in ribosome pools between cap and core RNA exosome variants
To further explore how specific ribosome biogenesis defects caused by RNA exosomopathy variants affect cellular translation, we performed polysome profiling on mutant and wild-type control cells. As expected from cells with severe ribosome biogenesis defects, all three mutant cells show a significant decrease in the level of polysomes compared to wild-type control cells (Fig. 8A). In addition, the rrp4/40 cap subunit mutants show an accumulation of halfmer polysomes, evident as shoulders on the 80S monosome peak in the rrp4-G226D cells and on the monosome, disome, and trisome peaks in the rrp40-W195R cells, not found in the profiles from the wild-type cells or rrp46-L191H core subunit mutant (Fig. 8A) or another recently analyzed core subunit mutant (rrp41-L187P) (Fasken et al. 2024). The halfmers could form due to inefficient ribosome subunit joining during translation initiation because of defects/reduction in biogenesis or stability of 60S subunits compared to 40S. Indeed, the 25S/18S rRNA ratio is decreased in the cap mutant cells (Fig. 7D). Additionally, the polysome profiles of the cap subunit mutants exhibit a distinct pattern of free 40S and 60S peaks when compared to those of the core subunit mutant described here and another core subunit mutant that was recently analyzed (rrp41-L187P) (Fasken et al. 2024), in which the free 60S peak shows a pronounced spike and free 40S subunits are reduced (Fig. 8A). Thus, the polysome profiles in the cap versus core mutant yeast cells appear to be distinctly different.
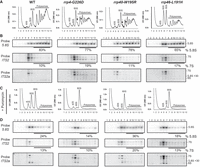
Distinct translation defects in rrp4-G226D, rrp40-W195R, and rrp46-L191H mutant cells. (A,C) Sucrose density gradients of wild-type, rrp4-G226D, rrp40-W195R, and rrp46-L191H cells that were grown at 30°C are shown. Clarified cell extracts were resolved on a 10%–50% sucrose gradient and scanned at 260 nm. Arrows indicate halfmers. (B,D) Northern blots of gradient fractions indicating the distribution of 5.8S rRNA and 7S pre-rRNA. Samples in C and D were treated with 2.5 mM puromycin after lysis.
The Rrp4-G226D variant has decreased association with the essential RNA helicase Mtr4 (Sterrett et al. 2021). The helicase Mtr4 (budding yeast ortholog of human MTR4/MTREX) aids the RNA exosome in targeting the 5.8S rRNA precursor (7S rRNA) among other target RNA transcripts and makes direct contact with the RNA exosome cap subunit Rrp4 and cofactors Rrp6 and Mpp6 (de la Cruz et al. 1998; LaCava et al. 2005; Vaňáčová et al. 2005; Houseley and Tollervey 2008, 2009; Lubas et al. 2011; Stuparevic et al. 2013; Schuch et al. 2014; Rodríguez-Galán et al. 2015; Kilchert et al. 2016; Falk et al. 2017; Zinder and Lima 2017; Weick et al. 2018). Previous work has shown that a ts mutant of MTR4 in budding yeast results in escape of 7S pre-rRNA-containing ribosomes into the pool of translating ribosomes (Rodríguez-Galán et al. 2015). To assess whether and how RNA exosomopathy yeast mutants affect the quality of translating ribosomes, we assayed the distribution of 7S pre-rRNA in polysome fractions of mutant cells compared to control wild-type cells (Fig. 8B). In wild-type cells, 7S pre-rRNA peaks with the free 60S fraction, where precursor 60S subunits would co-migrate. In contrast, in rrp4-G226D mutant cells, a fraction of 7S pre-rRNA co-migrates with polysomes. Overall, in rrp4-G226D and rrp40-W195R cap mutants, the spread of 7S pre-rRNA in polysome fractions is broader and across all polysome fractions, compared to the 7S pre-rRNA distribution in the rrp46-L191H core mutant and another recently described core variant (rrp41-L187P) (Fasken et al. 2024). This effect is pronounced at 37°C where the cells have a more significant growth defect (Supplemental Fig. S6A,B).
To determine whether the 7S pre-rRNA containing complexes migrating in polysome fractions in rrp4-G226D mutant cells are simply aggregated complexes, we prepared cell lysates under polysome run-off conditions by omitting cycloheximide and adding 2.5 mM puromycin, which dissociates the 80S ribosomes into 40S and 60S subunits (Blobel and Sabatini 1971). Under these conditions, a portion of 7S pre-rRNAs and 5.8S rRNAs in rrp4-G226D cells was shifted to the 80S and 60S fractions in all tested strains (Fig. 8C). Again, this effect was more pronounced at 37°C (Supplemental Fig. S6C,D). Northern blots also showed that the pattern of 7S pre-rRNA processing/degradation was distinct between the rrp4-G226D, rrp40-W195R, and rrp46-L191H mutants (Fig. 8B; Supplemental Fig. S6B). Together, these data further corroborate the differential molecular consequences of the RNA exosomopathy mutants.
To test how exosomopathy mutants impact the quality of protein synthesis, we assayed translation fidelity (Fig. 9A–C), using previously established dual-luciferase reporters (Harger and Dinman 2003; Salas-Marco and Bedwell 2005; Cheung et al. 2007). In these reporter plasmids, Renilla luciferase is constitutively expressed, whereas the production of firefly luciferase is dependent on a translation fidelity defect, such as a programmed frameshifting event, recognition of an alternative start site, or miscoding. Interestingly, the rrp4-G226D and rrp40-W195R cells show a statistically significant decrease in decoding the H245R near-cognate mutant firefly luciferase mRNA compared to wild-type controls (Fig. 9A,B). Together, the polysome profiles combined with dual luciferase reporter assays indicate a severe defect in translation in RNA exosomopathy yeast mutant models. At least some of these defects, e.g., formation of halfmers, appear to correlate with the type of RNA exosome subunit that is changed, i.e., cap versus core subunit, in the tested variants.
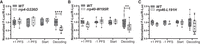
Translation fidelity defects in the RNA exosomopathy mutant yeast models compared to wild-type control cells. Dual-luciferase reporter assays were used to measure translation fidelity. The expression of firefly and Renilla luciferase was measured in rrp4-G226D (A), rrp40-W195R (B), and rrp46-L191H (C) mutant cells, compared to the corresponding wild-type control cells. The ratio of firefly luciferase to Renilla luciferase was normalized to control plasmids. Four to 12 biological replicates were analyzed.
DISCUSSION
This work represents the first in vivo comparative study of a collection of RNA exosomopathy mutant models. Comparison of the cellular RNAs that show changes in steady-state levels across the modeled RNA exosomopathy mutations in yeast provides intriguing results regarding the shared and distinctly altered transcripts and the potential pathways—metabolic and biosynthetic processes, rRNA processing/modification, and ribosome biogenesis—impacted within the models analyzed. Additionally, this study explores the consequences of these RNA exosome mutants for translation, including identifying some differences in translational defects between the rrp4-G226D and rrp40-W195R cap mutants and the rrp46-L191H core mutant. The results of this study underscore the significance of the link between impairment of RNA exosome function and defects in translation efficiency and fidelity, providing insights into the cellular defects that may contribute to distinct pathologies reported in exosomopathies.
The majority of studies that have explored the functional consequences of mutations underlying exosomopathies have focused on a single RNA exosome subunit (Morton et al. 2018; Fasken et al. 2020). Those studies that have compared across altered subunits have employed patient cells (Burns et al. 2018), which introduces inherent variability into these comparisons. The present study takes advantage of the evolutionary conservation of the RNA exosome complex to create a series of exosomopathy models that can be compared to one another to define similarities and differences in molecular phenotypes. These studies in budding yeast will not define cell-type specific RNA targets but can provide insight into the different classes of RNAs impacted.
We acknowledge that different RNA classes (e.g., tRNAs and snoRNAs compared to mRNAs) require different sequencing approaches to be comprehensively analyzed. We also note that some of the transcript differences detected will be indirect and reflect reduced growth rates and stress responses or different degrees of penetrance of the specific alleles, transcripts targeted for degradation or processing intermediates. Thus, whether the molecular consequences observed in RNA exosomopathy models are more systemic or are directly caused by changes in the integrity of the RNA exosome complex, remains to be fully established. Addressing these questions will require an in-depth biochemical analysis of each RNA exosome subunit variant to determine the impact of these changes on the RNA exosome complex and interactome. However, some predictions can be made based on the structures of the RNA exosome, considering the distinct consequences of each pathogenic RNA exosome subunit variant modeled in this study. The EXOSC2-G198D cap subunit variant is predicted to severely impact the structural organization of the cap (Di Donato et al. 2016; Sterrett et al. 2021). In contrast, the EXOSC3 cap subunit variants are predicted to impact interactions with interacting core subunits within the complex. The EXOSC3-D132 residue lies in a loop between strands in the S1 domain, and the EXOSC3-D132A variant would therefore likely be impaired in folding and interactions with neighboring subunits EXOSC5 and EXOSC9 (Fasken et al. 2017, 2020). Similarly, the EXOSC3-W238 residue is predicted to position other EXOSC3 residues to interact with neighboring EXOSC9 residues. Thus, the EXOSC3-W238R variant could weaken EXOSC3•EXOSC9 interactions (Fasken et al. 2017, 2020).
The EXOSC5 core subunit variants have been predicted to be unstable and impair complex interactions (Slavotinek et al. 2020). The EXOSC5-T114 residue interacts with A62 in the N-terminal region of the subunit and the EXOSC5-T114I variant would therefore likely be disrupted in this intra-subunit interaction (Slavotinek et al. 2020). The EXOSC5-M148 residue is at the interface with EXOSC3 and the EXOSC5-M148T variant would therefore likely cause impaired interactions between EXOSC5 and EXOSC3. The EXOSC5-L206 residue is buried in a hydrophobic pocket of the subunit. Therefore, the EXOSC5-L206H variant would likely cause destabilizing effects on the integrity of the subunit (Slavotinek et al. 2020). Lastly, the EXOSC9-L14 residue is located in the first alpha helix of EXOSC9 and EXOSC9-L14P variant would therefore likely cause disrupted interactions within the subunit (Liu et al. 2016). Thus, the pathogenic amino acid substitutions are predicted to have varied biochemical structural consequences. As such, each RNA exosomopathy protein variant may have differential impacts on the overall structure and function of the RNA exosome complex that require further investigation.
In growth assays, the rrp4-G226D mutant, modeling SHRF-linked EXOSC2-G198D, the rrp40-W195R mutant, modeling PCH-linked EXOSC3-W238R, and the rrp46-L191H mutant, modeling cerebellar atrophy-linked EXOSC5-L206H show the most impaired growth in budding yeast. These results are intriguing as the associated RNA exosomopathy disease pathologies linked to each mutation modeled in S. cerevisiae are diverse in the tissues impacted. Neither the EXOSC3-W238R mutation nor the EXOSC2-G198D mutation has been found in the homozygous state, suggesting these mutations may be lethal or highly deleterious in humans (Wan et al. 2012; Biancheri et al. 2013; Rudnik-Schoneborn et al. 2013; Eggens et al. 2014; Halevy et al. 2014; Di Donato et al. 2016). In contrast, the EXOSC5-L206H mutation, which has been found in the homozygous state, is associated with early infant mortality (Slavotinek et al. 2020). While the exosomopathy yeast mutant models all show defects, the modeled rrp4-G226D, rrp40-W195R, and rrp46-L191H mutants can support cell viability when expressed as the sole copy of the essential RRP gene, allowing us to analyze the functional consequences of these pathogenic amino acid changes in the budding yeast system.
Differential expression analysis reveals a significant decrease in transcripts that encode various heat shock protein (HSP) family members in the rrp4-G226D cells. The considerable reduction in HSP mRNAs could suggest that the rrp4-G226D cells have a compromised response to heat stress, thus explaining the significant growth defect observed at 37°C. Loss of Rrp6 also causes a decrease in levels of HSP transcripts (Wang et al. 2020); however, in a manner independent of the interaction between the exonuclease and the RNA exosome complex. Whether the loss of Rrp6 due to altered interaction with Rrp4 also contributes to a decrease in HSP mRNAs in the rrp4-G226D cells is not yet clear. The compromised response to stress observed in rrp4-G226D cells may also explain the increase in specific transcripts (e.g., DDR2 and PIR3 mRNAs), as expression of both these genes is activated in response to a variety of stress conditions (Gasch et al. 2001; Ribeiro et al. 2022). Furthermore, previous work has also linked RNA exosome activity to cellular integrity upon heat stress (Novačić et al. 2021). The changes in RNA exosome activity observed within all three rrp mutants may contribute to the slow growth observed upon heat stress. Additionally, the decrease in several biosynthetic transcripts observed in all three rrp mutants could further contribute to slow growth phenotypes. These changes to conserved metabolic and biosynthetic pathways observed in the model rrp mutants could also potentially contribute to disease pathologies in individuals with RNA exosomopathies. Among the increased transcripts detected in differential expression analysis of the three rrp mutant models, some had identifiable human homologs. GO analysis of the evolutionarily conserved transcripts revealed significant enrichment in synaptic vesicle priming and fusion biological processes. The link between these modeled RNA exosomopathy mutations and biological processes involved in synaptic vesicle fusion and trafficking may provide context for the numerous neurological defects common in individuals with mutations in EXOSC genes.
All three rrp mutant models analyzed in detail also show a significant decrease in the number of mRNAs. Many of these changes in mRNA levels could result from systemic effects, reflecting changes in cell signaling that can occur when the cell is under stress due to a dysfunctional RNA exosome. GO analysis of shared dysregulated transcripts reveals pathways related to ribosome biogenesis and translation are significantly enriched, pointing to potential changes in ribosome levels and/or function. In line with this observation, northern blots confirm a decrease in mature rRNA levels in rrp4-G226D, rrp40-W195R, and rrp46-L191H cells (Gillespie et al. 2017; Slavotinek et al. 2020). Depletion of individual RNA exosome subunits was previously shown to impact pre-rRNA cleavage, even at steps not directly involving the action of the RNA exosome, resulting in rRNA processing defects and reduction of mature rRNAs (Allmang et al. 2000). Indeed, besides involvement in 60S ribosome subunit maturation (Fernández-Pevida et al. 2015), the nuclear RNA exosome stably associates with late 90S pre-ribosomes during their transition to pre-40S and the RNA exosome is required for remodeling of 90S and maturation of pre-40S subunits (Lau et al. 2021). Our data corroborate the significant effect of these pathogenic RNA exosome mutants on ribosome biogenesis and further show that the effects of variants in specific subunits of the RNA exosome on translation goes beyond impacting solely the defined function of the RNA exosome in pre-rRNA processing. Some of these effects could be caused by accumulation or reduction of levels of transcripts that are important for ribosome assembly, including the snoRNAs and the specific mRNA transcripts coding for ribosomal proteins or ribosome assembly factors. Other effects could be direct or indirect due to reduced or incomplete processing of the precursor rRNAs. Impaired processing of rRNA could result in the assembly of dysfunctional ribosomes that are either quality controlled and discarded, causing reduced ribosome numbers, or escape into the translating pool of ribosomes.
Maturation of the 3′-end of the 5.8S rRNA from the 7S pre-rRNAs involves a series of cleavage events performed by different nucleases (Fernández-Pevida et al. 2015). In budding yeast, the trimming of 7S to 6S pre-rRNA happens in the nucleus, whereas the final processing of 6S pre-rRNA to 5.8S rRNA is cytoplasmic (Thomson and Tollervey 2010). However, immature 60S ribosome subunits have previously been reported to escape the nucleus and engage in translation in mutant yeast (Briggs et al. 1998; Rodríguez-Galán et al. 2015). In rrp6Δ cells, pre-60S ribosomes containing nuclear 5.8S rRNA and an additional 30 nucleotides of the ITS2 region can escape to the cytoplasm and enter the polysome pool (Briggs et al. 1998). Similarly, expression of a variant of the RNA exosome cofactor Mtr4 helicase was reported to result in the accumulation of nuclear pre-60S ribosomes containing 7S pre-rRNA in the cytoplasm (Rodríguez-Galán et al. 2015). These premature ribosomes were shown to engage in translation, suggesting 7S pre-rRNA processing defects do not prevent the export of 7S-containing pre-60S ribosomes (Rodríguez-Galán et al. 2015). In the context of 40S maturation, the trimming of the 3′-end of 18S rRNA from its precursor 20S pre-rRNA also occurs in the cytoplasm (Fatica et al. 2003, 2004; Lamanna and Karbstein 2009; Pertschy et al. 2009), and this process is strictly quality controlled to prevent the entry of premature 20S-containing 40S ribosomal subunits into the translating pool of ribosomes (Parker et al. 2019). However, cytoplasmic pre-40S ribosomes can escape into the translating pool in several mutant yeast mutants that fail the surveillance pathways during 40S assembly (Soudet et al. 2010; Strunk et al. 2012; García-Gómez et al. 2014; Parker et al. 2019; Parker and Karbstein 2023).
Our data indicate that the cap versus core RNA exosome subunit variants cause distinct translational defects, with cap variant Rrp4-G226D and Rrp40-W195R showing formation of halfmers and indicating a potential problem in 60S maturation or subunit joining, while the core Rrp46-L191H and Rrp41-L187P (Fasken et al. 2024) variants also indicating 40S biogenesis defect features. Our results suggest that a fraction of 7S pre-rRNA ribosomes in rrp4-G226D cells enter the translation pool, albeit not as efficiently as the 5.8S-containing ribosomes. Future mechanistic studies are needed to dissect how a shortage of ribosomes and the presence of 7S-containing ribosomes in the translation pool impact the cellular proteome in these RNA exosomopathy models. Overall, these studies demonstrate that variants in both the cap and core RNA exosome subunits could significantly change the translating ribosome pool and cellular proteome. However, the cap and core variants are likely to impact translation in distinct ways, possibly due to the different rRNA intermediates, contributing to unique molecular consequences and pathologies in each mutant model.
Our data also show that RNA exosomopathy variants result in translation fidelity defects. These defects could arise from the lower ribosome levels (Mills and Green 2017) or changes in transcripts related to ribosome biogenesis or translation, e.g., mRNAs encoding ribosome biogenesis and translation factors, tRNAs and/or snoRNAs. The decoding defect may be indicative of decreased rates of translation elongation in exosomopathy mutant cells, which likely provides more time for discrimination between aminoacyl tRNAs (Plant et al. 2007). Changes in tRNA abundance could also contribute to the decoding defect. Although we detected changes in transcripts that map to tRNAs, our sequencing approach was not ideally suited to measure tRNA abundance in an unbiased manner. Future studies are therefore required to reveal how decoding is altered in RNA exosomopathy mutant cells. Understanding whether RNA exosomopathy variants lead to changes in tRNA abundance is especially intriguing, as some RNA exosomopathies exhibit clinical features similar to those seen in patients with PCHs caused by mutations of the tRNA splicing endonuclease(TSEN) subunit genes (Hayne et al. 2023).
Of the two cap subunit variants modeled, the rrp4-G226D variant causes a severe growth defect at 37°C, whereas the rrp40-W195R variant causes a slight growth defect at 37°C. Thus, a global shortage of RNA exosome complexes cannot simply explain the severity of the molecular impact from each mutant or the overall growth fitness. Therefore, even though the simplest model to explain the severity of RNA exosomopathy mutants may be an RNA exosome concentration decrease model that would affect target transcript levels, the molecular pathology of RNA exosomopathy mutants appears far more complex and likely impacted by transcriptional, post-transcriptional, and translational mechanisms that require further investigation.
In summary, the data presented here provide novel insights into the molecular mechanisms of defects caused by disease-causing mutations in the RNA exosome genes modeled in yeast and reveal that different RNA exosomopathy mutations result in both unique and shared molecular changes across the variants. Our data suggest that while RNA exosomopathies result from a conglomeration of many direct and systemic molecular changes at the transcript level, translational defects could also contribute to the distinct cellular outcomes. Thus, even though distinct with respect to the molecular changes, RNA exosomopathies share phenotypic and mechanistic similarities among themselves and with ribosomopathies. Future work is required to reveal why, despite affecting molecular mechanisms that are seemingly universal to all cells, specific populations of neuronal cells are most affected by RNA exosomopathies. Likely, changes in the transcript levels, ribosome numbers, quality, and fidelity of translation in RNA exosomopathies alter the translation of specific mRNAs involved in neuronal differentiation and/or function to cause tissue-specific molecular defects. These studies in yeast models provide initial mechanistic insights and suggest models to explore in relevant cell types in the future.
MATERIALS AND METHODS
S. cerevisiae strains and plasmids
S. cerevisiae strains and plasmids used in this study are listed in Supplemental Table S2. Oligonucleotides used for plasmid construction are listed in Supplemental Table S3. The rrp4Δ (yAV1103), rrp40Δ (yAV1107), and rrp46Δ (yAV1105) strains used in this study were previously described (Schaeffer et al. 2009; Slavotinek et al. 2020). The rrp45Δ (yAV1410) strain used in this study was obtained by transforming a RRP45 URA3 plasmid into the heterozygous diploid RRP45/rrp45Δ strain from the knockout collection and sporulating the transformants to obtain a haploid rrp45Δ strain. The rrp6Δ strain (ACY1641) was obtained from Horizon Discovery. The wild-type RRP4 (pAC3656), RRP40 (pAC3652), and RRP46 (pAC3482) plasmids were constructed as previously described and each contains the ORF flanked by endogenous regulatory sequences (promoter, terminator, and 5′/3′ UTR) cloned into the pRS315 vector (ATTC #77144) (Sikorski and Hieter 1989), which harbors the LEU2 marker (Fasken et al. 2017; Slavotinek et al. 2020; Sterrett et al. 2021). The wild-type RRP45 (pAV975) plasmid contains the ORF flanked by endogenous regulatory sequences cloned into the pRS415 LEU2 vector. The rrp4-G226D (pAC3659), rrp40-S87A (pAC3654), rrp40-W195R (pAC3655), rrp46-Q86I (pAC3483), rrp46-L127T (pAC3534), and rrp46-L191H (pAC3484) mutant plasmids encoding the different RNA exosomopathy amino acid variants were generated by site-directed mutagenesis of the respective wild-type plasmids (pAC3656, pAC3652, pAC3482) using the QuikChange II Site-Directed Mutagenesis Kit (Agilent) and oligonucleotides containing the desired missense mutations as previously described (Fasken et al. 2017; Slavotinek et al. 2020; Sterrett et al. 2021). The rrp45-I15P (pAC3480) mutant plasmid was generated by site-directed mutagenesis of pAV975 using oligonucleotides AC8001 and AC8002 in this study. The wild-type RRP6 (pAC3752) plasmid was constructed by subcloning ApaI/SacI-digested RRP6 ORF with endogenous regulatory sequences from pAC2301 (RRP6 in pRS313; Fasken et al. 2015) into pRS315 cut with ApaI/SacI. The TEF1p-Cas9-CYC1t-SNR52p (pAC3846) pCas9 plasmid was constructed by PCR amplification of TEF1p with oligonucleotides AC8410 and AC8411 and Cas9-CYC1t with AC6802 and AC6803 using p414-TEF1p-Cas9-CYC1t plasmid template (Addgene #43802) (DiCarlo et al. 2013) and SNR52p with AC6804 and AC6805 using p426-SNR52p-gRNA.CAN1.Y-SUP4t plasmid template (Addgene #43803) (DiCarlo et al. 2013), followed by sequential cloning of SacI/SpeI-digested TEF1p, SpeI/KpnI-digested Cas9-CYC1t, and AgeI/KpnI-digested SNR52p into pRS316 plasmid digested with corresponding restriction enzymes. The TEF1p-Cas9-CYC1t-SNR52p-RRP4_668.gRNA-SUP4t (pAC3863), TEF1p-Cas9-CYC1t-SNR52p-RRP40_583.gRNA-SUP4t (pAC3861), and TEF1p-Cas9-CYC1t-SNR52p-RRP46_589.gRNA-SUP4t (pAC4342) pCas9 plasmids containing the gRNAs for targeting RRP4, RRP40, and RRP46, respectively, were constructed by PCR amplification of RRP4_668.gRNA with oligonucleotides AC8407 and AC6809, RRP40_583.gRNA with AC8402 and AC6809, and RRP46_589.gRNA with AC9888 and AC6809 using p426-SNR52p-gRNA.CAN1.Y-SUP4t plasmid template (Addgene #43803) and cloning of SphI/KpnI-digested gRNA products into pAC3846 digested with SphI/KpnI. All plasmids were fully sequenced.
Generation of S. cerevisiae mutant strains using CRISPR-Cas9 genome editing
The rrp4-G226D (ACY3110), rrp40-W195R (ACY3117), and rrp46-L191H (ACY3137) mutant strains were generated using CRISPR/Cas9 editing with a single pCas9-gRNA expression plasmid and double-stranded homology-directed repair (HDR) oligonucleotides in a wild-type BY4741 strain essentially as described before (DiCarlo et al. 2013). The single pCas9-gRNA plasmid on a pRS316 (URA3, CEN6) backbone is derived from p414-TEF1p-Cas9-CYC1t plasmid (Addgene #43802) and p426-SNR52p-gRNA.CAN1.Y-SUP4t plasmid (Addgene #43803). The constitutive expression of Cas9 is driven by the TEF1 promoter and the constitutive expression of the gRNA is driven by the SNR52 promoter. Specifically, 500 ng of pAC3846 (pCas9 without gRNA), pAC3863 (pCas9 + RRP4 gRNA) ± 1 nmol of double-stranded rrp4-G226D HDR oligonucleotide (AC8408/8409), pAC3861 (pCas9 + RRP40 gRNA) ± 1 nmol double-stranded rrp40-W195R HDR oligonucleotide (AC8404/8405), or pAC4342 (pCas9 + RRP46 gRNA) ± 1 nmol double-stranded rrp46-L191H HDR oligonucleotide (AC9889/9890) and 50 µg of salmon sperm DNA was transformed into wild-type BY4741 cells by standard lithium acetate (LiOAc) transformation protocol (Da et al. 2000). HDR oligonucleotides are listed in Supplemental Table S3. Cells were plated on SD -Ura media plates and incubated at 30°C for 2 days. Large colonies on plates with cells transformed pCas9-gRNA and HDR oligonucleotides were restreaked to new SD -Ura media plates and screened for the presence of rrp4-G226D, rrp40-W195R, and rrp46-L191H mutations via Sanger sequencing of genomic RRP4, RRP40, and RRP46 PCR products, respectively.
S. cerevisiae transformations and growth assays
All yeast transformations were performed using the standard LiOAc protocol (Da et al. 2000). Standard plasmid shuffle assays were performed to assess the in vivo function of the rrp variants as previously described (Fasken et al. 2017; Slavotinek et al. 2020; Sterrett et al. 2021). The rrpΔ cells (rrp4Δ [yAV1103], rrp40Δ [yAV1107], rrp45Δ [yAV1410], rrp46Δ [yAV1105]) transformed with the wild-type control LEU2 plasmid (RRP4 [pAC3656], RRP40 [pAC3652], RRP45 [pAV975], RRP46 [pAC3482]) or the mutant variant plasmid (rrp4-G226D [pAC3659], rrp40-S87A [pAC3654], rrp40-W195R [pAC3655], rrp45-I15P [pAC3480], rrp46-Q86I [pAC3483], rrp46-L127T [pAC3534], rrp46-L191H [pAC3484]) were streaked on 5-FOA SD -Leu media plates and incubated at 30°C for 2–3 days. Single colonies from the 5-FOA SD -Leu media plates were selected in quadruplicate and streaked onto selective SD -Leu media plates. The rrp6Δ cells (ACY1641) were transformed with empty LEU2 vector (pRS315) or wild-type LEU2 plasmid (RRP6 [pAC3752]) and selected on SD -Leu media plates. The rrpΔ cells containing only the wild-type RRP or mutant rrp LEU2 plasmid were used for the RNA-seq analysis and reporter assays. The CRISPR/Cas9-edited rrp4-G226D (ACY3110), rrp40-W195R (ACY3117), and rrp46-L191H (ACY3137) mutant cells were assessed for ts growth by a solid media growth assay, and rescue of the ts growth of the mutant cells by wild-type RRP4, RRP40, and RRP46 plasmids was confirmed. The rrp4-G226D, rrp40-W195R, and rrp46-L191H CRISPR mutant cells were used for the northern blots and the polysome profiling assay. Growth assays were performed on solid media and in liquid culture. The wild-type control and mutant model cells were grown to saturation at 30°C before the concentrations were adjusted to an A600 ∼0.5, and samples were serially diluted in 10-fold steps and spotted onto SD -Leu media plates. Plates were grown at 30°C and 37°C for 2–3 days. For growth in liquid culture, saturated overnight cultures grown at 30°C were diluted to an A600 ∼0.01 in SD -Leu in a 24-well plate, and growth at 37°C was monitored and recorded at OD600 in a BioTek Synergy MX microplate reader with Gen5 v2.04 software over 24 h. For each sample analyzed in growth assays, at least three independent biological replicates were used. In addition, for the liquid culture assays, technical triplicates for each biological sample were grown. Doubling times were calculated using GraphPad Prism version 9.3.1.
Sample collection for RNA-seq analysis
RNA-seq was performed on three independent biological replicates of rrpΔ cells containing the RRP wild-type control plasmids or the rrp variants as the sole copy of the RNA exosome gene. The rrp6Δ cells (ACY1641) contained either an RRP6 wild-type control plasmid (pAC3752) or an empty vector. Biological replicates of all samples were first screened by solid media growth assays prior to growth and collection for the RNA-seq experiment. For sample collection, cells were grown in SD -Leu media overnight at 30°C to saturation, diluted to A600 ∼0.2 in SD -Leu media, and shifted to 37°C for 5 h. Cells were washed, pelleted, and flash frozen, and cell pellets were sent to Zymo Research for total RNA preparation and RNA-seq analysis.
RNA-seq library preparation
RNA-seq library preparation was performed by Zymo Research. Total RNA-seq libraries were constructed from 300 ng of total RNA. To remove rRNA, a method previously described (Bogdanova et al. 2011) was followed with some modifications. Libraries were prepared using the Zymo-Seq RiboFree Total RNA Library Prep Kit (Cat # R3000). RNA-seq libraries were sequenced on an Illumina NovaSeq to a sequencing depth of at least 20 million read pairs (150 bp paired-end sequencing) per sample.
Sequence data alignments and differential expression analysis
NovaSeq paired-end 150-bp reads from total RNA-seq data files were first adaptor trimmed and then analyzed using the STAR program (version 2.6.1d) for alignment of short reads to S. cerevisiae reference genome. Transcript and gene expression estimates were measured using StringTie v2.1.7 (Pertea et al. 2015). The expression estimates FPKM were used with the Pheatmap R package v1.0.12 to generate heat maps (Kolde 2012). The raw reads per gene were counted using featureCounts v1.22.2 (Liao et al. 2013) to the S. cerevisiae S288C genome assembly R64-1-1 (Engel et al. 2014), annotated with CUTs and SUTs (Xu et al. 2009). Low feature counts (<10 reads total) were removed. Differential gene expression analysis on raw read counts was performed using the DESeq2 R package v1.38.1 (Love et al. 2014) to identify genes significantly changed (P-value <0.05, ≥1.5-fold change) in rrp mutant variant samples relative to RRP wild-type control samples. Shrinkage of effect size was performed on differential expression data for visualizations using the apeglm method (Zhu et al. 2019). Using DESeq2, PCA was performed, and MA plots were generated on raw read counts. Volcano plots of differential gene expression data were produced using EnhancedVolcano R package v1.16.0 (Blighe et al. 2019). UpSet plots were generated using UpSetR R package v1.4.0 (Conway et al. 2017), with transcripts identified through differential expression analysis in the mutant cells as significantly decreased by 1.5-fold or more (FC ≤ −1.5) and significantly increased by 1.5-fold or more (FC ≥ +1.5). UpSet intersections are shown in graphs as a matrix, with rows corresponding to the sets of samples (i.e., transcripts identified FC ≤ −1.5 or FC ≥ +1.5 within the three mutant models) and columns corresponding to the intersection between these sets. Pie charts and stacked bars of RNA class percentages in significantly altered genes were generated using GraphPad Prism version 9.3.1. Transcripts were sorted by class using the annotations available through the Saccharomyces cerevisiae Genome Database (SGD) (Cherry et al. 2012). GO analysis on significantly altered genes for the Biological Process category was performed using the YeastMine webserver. GO analysis on human homolog genes was performed using HumanMine. All GO analyses were performed by Holm–Bonferroni test correction.
Northern blot analysis of rRNAs
For analysis of ribosomal RNAs, yeast cells were grown to mid-log phase and RNA was extracted using the hot phenol method. Northern blotting was carried out essentially as previously described (Khoshnevis et al. 2019), using the probes listed in Supplemental Table S3.
Dual-luciferase reporter assays to monitor translation fidelity defects
For assaying translation fidelity defects, cells expressing wild-type or variant RNA exosome subunits and a dual-luciferase reporter plasmid were grown to mid-log phase in SD -Ura -Leu synthetic glucose liquid media. Cells were pelleted, washed, and stored at −80°C before analysis. Luciferase activities were measured using the Dual-Luciferase Reporter Assay kit (Promega). Thawed frozen cell pellets were resuspended in 1× passive lysis buffer and incubated for 10 min. LARII was mixed with lysate in clear bottom 96-well Microplates (Costar), and Firefly luciferase activity was measured. Stop and Glo solution was added, and Renilla luciferase activity was measured. Measurements were performed using a Synergy Microplate Reader (BioTek). For each biological replicate (single transformant), the Firefly luciferase signal was normalized to the Renilla luciferase signal. For each strain, Firefly/Renilla ratio was normalized to the average Firefly/Renilla ratio of replicates containing a control plasmid.
Sucrose density gradient analysis
To analyze polysomes by gradients, cells were grown to mid-log phase in YP-dextrose media at 30°C and 37°C and harvested after adding 0.1 mg/mL cycloheximide or no cycloheximide. Cells were washed and lysed in ice-cold gradient buffer (20 mM HEPES, pH 7.4, 5 mM MgCl2, 100 mM NaCl, and 2 mM DTT) supplemented with cOmplete Protease Inhibitor Cocktail (Roche) and 0.1 mg/mL cycloheximide or no cycloheximide. Cells were broken by cryogenic grinding and cell lysate was cleared by centrifugation at 10,000g for 10 min. The absorbance of cleared lysate was measured at UV260 and an equal amount of lysate was applied to 10%–50% sucrose gradients in the gradient buffer for all samples. To samples lacking cycloheximide, 2.5 mM puromycin was added to the cleared cell lysate, incubated on ice for 15 min, and then placed at 30°C or 37°C before loading onto the gradient. Gradients were centrifuged for 2 h at 40,000 RPM in a SW41Ti rotor and fractionated using a BioComp fraction collector.
DATA DEPOSITION
The raw RNA-seq data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO) under accession GSE246957.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank the Corbett and Ghalei laboratory members for critical discussions and input. We thank Dr. Benjamin Barwick for his contributions in assisting with the analysis of the CoMMpass data set. This work was supported by National Institutes of Health (NIH) awards R35GM138123 to H.G., R35GM141710 to A.v.H., and R01GM130147 to A.v.H. and A.H.C., as well as funds from a Synergy II Nexus Award provided by the Woodruff Health Sciences Center (WHSC), Emory School of Medicine, the Office of the Provost, and Emory College of Arts and Sciences (ECAS) to H.G. and A.H.C. M.C.S. was supported by an F31 fellowship (GM134649) from the National Institutes of Health. L.A.C. was supported by NIH T32GM149422 and F31GM157874.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080447.125.
- Received March 4, 2025.
- Accepted March 31, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Maria C. Sterrett is the first author of this paper, “Comparative analyses of disease-linked missense mutations in the RNA exosome modeled in budding yeast reveal distinct functional consequences in translation.” Maria received her PhD in Biochemistry, Cell and Developmental Biology from Emory University, working in Dr. Anita Corbett's laboratory. Utilizing budding yeast, she studied how human disease mutations impact RNA processing. She is currently an Institutional Research Career and Academic Development Award (IRACDA) postdoctoral scholar at Tufts University School of Medicine, working with Dr. Claire Moore. The focus of this paper comes from her doctoral work studying the molecular and functional consequences of pathogenic missense mutations in genes that encode different subunits of the molecular RNA processing machine, the RNA exosome.
What are the major results described in your paper and how do they impact this branch of the field?
In this paper, we assayed missense mutations in genes encoding subunits of the RNA exosome that are linked to a growing family of diseases, termed RNA exosomopathies. These have diverse disease pathologies, suggesting that these mutations could differentially impact RNA exosome function in vivo. To first investigate how RNA exosome function is differentially impacted, we performed a comparative RNA-seq analysis of several RNA exosomopathy mutations modeled in Saccharomyces cerevisiae, identifying broad transcriptomic changes as well as distinct consequences in translation and ribosomal RNA modification/processing pathways. Molecular studies revealed shared defects in ribosome biogenesis and global translation across these different RNA exosomopathy mutant models. However, using in vivo reporter assays and polysome profiling, we uncovered distinct defects in translation fidelity and efficiency between RNA exosomopathy mutant models. The work presented in this paper represents the first ever comparative analysis of several RNA exosomopathy mutant models, defining similarities and differences in molecular phenotypes of a collection of RNA exosome mutations. Additionally, our findings suggest that not only molecular changes at the transcript level, but also translational defects that impact protein homeostasis, may contribute to the distinct pathologies of RNA exosomopathies.
What led you to study RNA or this aspect of RNA science?
I was drawn to RNA biology, and particularly the RNA exosome field, because of collaboration, creativity and most of all, passionate and supportive mentorship. I always had an interest in science and discovery. As a younger student, I was fortunate to find mentors in high school and then college who bolstered my interest and encouraged me to continue following my passions in biology. My first research experience introduced me to the world of genetics and model systems biology. I became enamored with the tenacious and bold process of scientific research, as well as the versatility afforded by genetic model systems to not only answer complex questions but serve as a platform for trainees to engage in discovery. When I entered my graduate program, I knew I wanted to bring scientific research and learning to students as a career. I was immediately drawn to working with Dr. Anita Corbett, as she prioritizes mentorship, evidenced by her many accolades and awards, and supported my career goals. The Corbett lab uses genetic model systems, but the biochemical research questions regarding post-transcriptional regulation were new to me. I quickly fell for the creativity in the types of experiments and research questions you find in the RNA world and loved integrating cellular and molecular approaches to answer biochemical questions! I was also drawn to the collaborations particularly present in the RNA exosome field. My studies on the functional consequences of RNA exosomopathy mutations join a long-standing collaboration between the Corbett lab, the lab of Dr. Ambro van Hoof, and the lab of Dr. Derrick Morton. The research presented in this publication expanded the project to include the work and expertise of Dr. Homa Ghalei. I have learned from each of these mentors and seen the incredible value a team approach to science has not only in advancing a research question but in training younger scientists. The community of the RNA field, the creativity in the scientific questions, and the mentorship I have received as a trainee are ultimately what continue to motivate me as an RNA biologist!
If you were able to give one piece of advice to your younger self, what would that be?
The biggest piece of advice I would give my younger self is the same piece of advice I give any of my trainees now—find your people! Science research is fun and exciting, but it is also filled with frustrations, setbacks, and failure. While finding a research question that excites you is important, finding mentors and peers who champion your strengths, are excited by your passions, and bolster your confidence will ultimately sustain you as you pursue your goals.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My approach to research and science more broadly have been strongly influenced by my mentors and the scholarship communities I've been a part of during my training. As a graduate student at Emory University, I was a member of the NIH-funded Initiative to Maximize Student Diversity (IMSD), a mentoring system that supported students historically excluded from STEM and biomedical sciences by connecting researchers and trainees across levels of the academic system. I also served several diversity, equity, and inclusion initiatives during graduate school as well as outreach and mentorship opportunities. I am now part of the NIH funded IRACDA fellowship program that supplements my postdoctoral experience with training in mentorship, evidence-based pedagogy principles, and inclusive teaching practices. All these experiences taught me the value of a diverse community and the expansive power that inclusive science and training offers biomedical research. These lessons have made my goals as a scientific researcher inextricably linked with my goals as an educator: to foster creativity, tenacity, and boldness through impassioned scientific discovery and inclusive mentorship.
What are your subsequent near- or long-term career plans?
I am excitedly joining the Bowdoin College faculty as an Assistant Professor of Biochemistry this coming fall! I aim to bring my excitement for the RNA world to my future students, building a research program grounded in RNA biochemistry and biology. I will continue pursuing research questions about the consequences of disease-linked mutations on the function of the RNA exosome, utilizing the budding yeast system and integrating molecular techniques and bioinformatics analyses like those presented in this publication. I look forward to igniting my own students’ passions in scientific discovery, and for them to experience the collaboration, creativity and support I have felt through the broader RNA research community and RNA Society!








