
The influence of downstream structured elements within mRNA on the dynamics of intersubunit rotation in ribosomes
- Bassem Shebl1,
- Anna Pavlova2,
- Preston Kellenberger1,
- Dongmei Yu4,
- Drew E. Menke1,
- James C. Gumbart3 and
- Peter V. Cornish1
- 1Department of Biochemistry, University of Missouri, Columbia, Missouri 65211, USA
- 2School of Physics, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
- 3School of Physics and School of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, Georgia 30332, USA
- 4Departments of Chemical and Biomedical Engineering, University of Missouri, Columbia, Missouri 65211, USA
- Corresponding author: cornishp{at}missouri.edu
-
Handling editor: Marina Rodnina
Abstract
Proper codon/anticodon pairing within the ribosome necessitates linearity of the transcript. Any structures formed within a messenger RNA (mRNA) must be unwound before the respective codon is interpreted. Linearity, however, is not always the norm; some intricate structures within mRNA are able to exert unique ribosome/mRNA interactions to regulate translation. Intrinsic kinetic and thermal stability in many of these structures are efficient in slowing translation causing pausing of the ribosome. Altered translation kinetics arising from atypical interactions have been shown to affect intersubunit rotation. Here, we employ single-molecule Förster resonance energy transfer (smFRET) to observe changes in intersubunit rotation of the ribosome as it approaches downstream structured nucleic acid. The emergence of the hyperrotated state is critically dependent on the distance between downstream structure and the ribosome, suggesting interactions with the helicase center are allosterically coupled to intersubunit rotation. Further, molecular dynamics (MD) simulations were performed to determine ribosomal protein/mRNA interactions that may play a pivotal role in helicase activity and ultimately unwinding of downstream structure.
Keywords
INTRODUCTION
The ribosome is a ubiquitous macromolecular complex that translates the information encoded in the messenger RNA (mRNA) into functional proteins. Despite the high processivity of the ribosome, protein synthesis is not a continuous process. Intermittent pausing of the ribosome slows down or even completely halts translation and is an example of discontinuity (Guisez et al. 1993; Buchan and Stansfield 2007; Zhang et al. 2009). Ribosomal pausing has been functionally linked to cotranslational protein folding (Watts et al. 2009), membrane localization (Young and Andrews 1996), modulating protein expression levels (Nackley et al. 2006), and programmed ribosomal frameshifting (PRF) (Jacks et al. 1988; Tsuchihashi 1991). A common feature in these is the utilization of a secondary RNA structure within the mRNA coding region to pause the moving ribosome, permitting cells to regulate protein production. Thus, folded secondary structures within the mRNA coding region present a thermodynamic and kinetic barrier to translation and must be disrupted for translation to proceed.
The average diameter of the mRNA entrance tunnel (∼15 Å) is smaller than the diameter of a double-stranded helix (∼20 Å) (Takyar et al. 2005). Thus, for translation to proceed forward, any nucleic acid structure requires unfolding. Unwinding nucleic acid duplexes is typically carried out via dedicated helicases (Delagoutte and von Hippel 2003). In the bacterial ribosome, however, the required helicase activity is intrinsic to the macromolecular complex itself (Takyar et al. 2005). Findings by Takyar et al. (2005) predicted the position of the helicase center of the ribosome to be within the downstream mRNA entrance tunnel, ∼11 nt away from the P-site. The predicted distance of 11 nt, interestingly, corresponds to the optimal distance for RNA pseudoknots to be capable of inducing −1 PRF, along with the upstream slippery site in a few studies (Brierley and Pennell 2001; Caliskan et al. 2014), although some have found as little as 6 nt to be optimal, as for dnaX (Larsen et al. 1997). Ribosomal proteins S3, S4, and S5 encircle and interact with the incoming mRNA at the 30S entrance tunnel (Yusupova et al. 2001; Zimmermann et al. 2016). The three proteins surrounding the incoming mRNA resemble the well-known sliding clamps of DNA and RNA polymerases, suggestive of a similar function to S3, S4, and S5 (Kuriyan and O'Donnell 1993). However, no known helicase motifs have been identified in these proteins. Furthermore, despite the ribosome itself being a GTPase, these proteins demonstrate no direct use of ATP or GTP (Takyar et al. 2005). The highly dynamic nature of the ribosomal structure, along with the lack of evidence to any external energy sources required for the unwinding activity, suggests the involvement of conformational changes of the two subunits in engaging and unwinding downstream structures facing the ribosome (Frank and Agrawal 2000; Namy et al. 2006).
Structural and biophysical studies have shown that tRNA and mRNA translocation are accompanied by large-scale conformational changes (Takyar et al. 2005; Cornish et al. 2008; Qu et al. 2011; Guo and Noller 2012; Chen et al. 2014, 2015; Qin et al. 2014). The most notable change is the thermally driven counterclockwise rotation of the small 30S subunit relative to the vertical axis of the large 50S subunit (Frank and Agrawal 2000; Valle et al. 2003; Cornish et al. 2008). Thus, subunit ratcheting renders the ribosome between two states: the rotated and nonrotated states. Accompanied by the 30S subunit rotation is a process known as head swiveling. This head domain exhibits 5° rotation about an axis within itself (Ratje et al. 2010). The region between the head and the body of the 30S subunit forms the mRNA entrance and exit tunnels on opposite sides of the subunit. The two tunnels were found to alternatively expand and contract with subunit rotation, presumably allowing the mRNA to advance through the ribosome and maintain a grip on the reading frame (Frank and Agrawal 2000). The mRNA entrance tunnel with its circular clamp-like entrance has been proposed to host the intrinsic helicase center of the ribosome. Two distinctive mechanisms have been suggested to achieve this goal (Qu et al. 2011). First, the ribosome can bind and stabilize the open conformation of the duplex during helical fraying. In the second mechanism, the ribosome can mechanically pull apart the two strands through an interplay between the various conformational changes that accompany translation (Ratje et al. 2010; Guo and Noller 2012; Qin et al. 2014). Thus, it is possible that an intricate balance of conformational changes regulates which mechanism is used in the process of unwinding.
Various approaches have been employed to relate structural changes to functional activity during translation, varying from cryo-electron microscopy (cryo-EM) and X-ray crystallography to fluorescence imaging. Using single-molecule Förster resonance energy transfer (smFRET), we have shown previously that ribosomal subunits spontaneously fluctuate between two conformations, corresponding to the rotated and nonrotated states, when translating an unstructured mRNA (Cornish et al. 2008). The presence of structured mRNA within the coding region was shown to induce further rotation of the small subunit when the structure is placed at the mRNA entrance tunnel. This creates a third conformation, the hyperrotated state, alongside the two identified states: the rotated and nonrotated states (Fig. 1; Qin et al. 2014). The presence of the hyperrotated state is independent of the identity and amino-acylation status of the tRNA occupying the P site. Chen et al. (2014, 2015) showed a similar observation of a noncanonical rotated state, characterized by a long pause, coupled to programmed frameshifting and bypassing of noncoding regions. The structural constraints imposed by the dimensions of the mRNA entrance tunnel facilitate the presence of an optimal position for an RNA structure, allowing the ribosome to sense the structure and respond in the form of unwinding. As mentioned above, investigating the highly concerted movement of the ribosomal subunits could shed light on the unwinding mechanism of the helicase center of the ribosome. Given the uniqueness of the ribosomal helicase activity and the lack of an observed direct energy source for the process, subunit rotation seems highly likely to be integral in the sensory mechanism by which ribosomes engage structured mRNA, and unwind it. Thus, a further quantitative analysis of the influence of structured mRNA sequences on the conformational states present and the population distribution is required.

Schematic depicting the two ribosomal subunits (50S—yellow; 30S—blue) in various rotational states: nonrotated, rotated, and hyperrotated. Approximate locations of Cy3 (green sphere) attached to ribosomal protein L9 and Cy5 (red sphere) attached to ribosomal protein S6 are shown.
Here, we use an smFRET approach to track the conformational changes of intersubunit rotation during translation. Consistent with our previous work, we show that hyperrotation is formed in the presence of secondary structures within the coding region of the mRNA at a close proximity to the ribosome (Qin et al. 2014). Furthermore, using a DNA–RNA hybrid duplex, we track the formation of the hyperrotated state and illustrate the importance of the relative spatial position of a structured element on the conformational dynamics of the ribosome. We show how the presence of such a structure alters the equilibrium of the existing states toward the newly formed state: the hyperrotated state. Alongside subunit rotation, “How does the ribosome respond near the mRNA entrance tunnel to incoming structures?” is a standing question. Using molecular dynamics (MD) simulations, we investigated the local structural rearrangements of the clamp-like structure encircling the mRNA entrance tunnel when the ribosome encounters a structured nucleic acid at various distances. Taken together, smFRET and MD provide valuable and complementary insights into the mechanism by which the ribosome can sense incoming structured elements and how it can transduce that across the entire structure through major structural rearrangements with functional implications on translation.
RESULTS
S6 (Cy5)/L9 (Cy3) ribosomes report on intersubunit rotation
For this study, ribosomal proteins S6 and L9 were individually labeled with Cy5 and Cy3, respectively, and reconstituted in vitro into ΔS6/ΔL9 ribosomes purified by Ni-NTA via a 6× His tag on the L7/L12 stalk, as described in Materials and Methods (Ermolenko et al. 2007; Cornish et al. 2008; Qin et al. 2014; Sharma et al. 2016). The dual-labeled ribosomal constructs were assembled with an unstructured control mRNA employed in previous studies (Takyar et al. 2005; Ermolenko et al. 2007; Cornish et al. 2008), m291, and immobilized to the passivated surface of a quartz slide via a biotin-neutravidin linkage. Time trajectories of the individual molecules showed fluctuations between two conformations/FRET states: the nonrotated (∼0.58) and rotated (∼0.38) states, as described previously (Cornish et al. 2008). The two conformations coexist in equilibrium at a ratio highly dependent on the occupancy of the A-site tRNA and the acylation status of the P-site tRNA. Ribosomes assembled with a single deacylated tRNAfMet in the P site bias the conformational equilibrium toward the rotated state (Supplemental Fig. S1). Using deacylated tRNAfMet provides the highest rotational freedom for the ribosome among different tested conditions, as reflected in the number of dynamic molecules (Cornish et al. 2008). In agreement with previously reported data on m291, we showed that the presence of a deacyated tRNAfMet in the P site, regardless of the absence or presence of a A-site tRNA, shifted the existing equilibrium toward the rotated conformation (∼72% and 68%, respectively) (Cornish et al. 2008; Sharma et al. 2016).
Visualization of the hyperrotated state
Previously, we reported an additional conformational state of the 50S and 30S ribosomal subunits while interacting with the dnaX hairpin (Fig. 1; Qin et al. 2014). The dnaX hairpin (Fig. 2C) has been well characterized and used for studying ribosome dynamics whether in isolation (Qin et al. 2014) or in the context of a full frameshifting signal (Chen et al. 2014; Kim et al. 2014; Yan et al. 2015). We observed that the placement of a structured RNA sequence within the mRNA and in close proximity to the mRNA entrance tunnel of the ribosome induces a new population with a significant reduction in FRET (0.22) as compared to the rotated (0.38) and nonrotated (0.58) states (Supplemental Fig. S2). Characterization of the hyperrotated conformation showed that it has a substantial influence on the equilibrium among the different conformational states of the ribosome. Furthermore, ribosomes showed less fluctuation events in comparison to the control; indicative of a decrease in molecular dynamicity (see “The kinetics of intersubunit rotation in the presence of a folded structure” section).

(A) Ribosomes were immobilized by the hybridization of the 3′ end of the mRNA (m291 or dnaX) to a complementary biotinylated DNA oligo bound to the slide surface via a neutravidin-biotin-PEG linkage. (B) The sequence of m291 (mRNA) and oligonucleotides used. The nucleotide register is shown starting at the first nucleotide in the P site as +1. Oligonucleotides are labeled as +10 up to +16 in single nucleotide increments. Shine–Dalgarno (SD) sequence and codons occupying the P and A sites are highlighted. (C) The sequence of the GC-rich dnaX hairpin.
His-tagged, tight coupled ribosomes show more activity (increased/comparable levels of P-site tRNA binding, PTC activity, and A-site tRNA binding) compared to the ribosomes employed in the past study (Shebl et al. 2016); thus, we are reproducing these experiments to provide consistency throughout the results in the present study. We assembled the His-tagged S6 (Cy5)/L9 (Cy3) 70S ribosomes with the dnaX hairpin and tRNAfMet in the P site. The hairpin of the dnaX gene was placed at the mRNA entrance tunnel using a 6 nt linker (Larsen et al. 1997). Consistent with previous results, the dnaX hairpin induced a significant portion of the population of ribosomes into hyperrotation (∼39%, previously 68%) and rotation (∼56%, previously 20%) (Fig. 3H; Table 1) coupled with suppression of the nonrotated conformation (∼6%, previously 20%), which shows a significant change from observed values for the linear control sequence, m291, assembled under the same conditions (0% hyperrotated, previously 0%; 72% rotated, previously 71%; and 28% nonrotated, previously 29%) (Cornish et al. 2008; Qin et al. 2014; Shebl et al. 2016). Furthermore, the percentage of fluctuating/dynamic molecules, or molecules that switch between the nonrotated and rotated conformations, decreased to ∼10%, an ∼80% reduction as compared to the linear control (∼52%). Thus, constructs capable of inducing hyperrotation seem to reduce molecular dynamicity (see “The kinetics of intersubunit rotation in the presence of a folded structure” section).
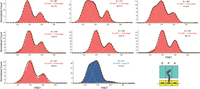
Normalized histograms complied from hundreds of traces showing distributions of FRET values for S6-Cy5/L9-Cy3 ribosomes containing a deacylated tRNAfMet in the P site and nucleic acid duplexes. N, number of molecules used to compile each histogram; yellow lines represent individual Gaussian fits centered at ∼0.26 (hyperrotated state), 0.38 (rotated state), and 0.58 (nonrotated state) FRET efficiency; black lines represent the sum of two (A, D–G) or three (B, C, H) Gaussians; % represents the percentage of fluctuating molecules for a given ribosomal complex.
Statistical analysis for all tested ribosomal complexes
Pretranslocation complexes (PREs) represent an intermediate step in translation where the A-site tRNA contains an elongated peptide chain while the P-site tRNA is deacylated. In our experiments, PREs were formed by adding N-Ac-Phe-tRNAPhe nonenzymatically to the A site with tRNAfMet in the P site while bound to dnaX. The addition of an A-site tRNA had a modest effect on the conformational equilibrium and the percentage of dynamic molecules (Table 1). Nevertheless, the hyperrotated state was significantly present (∼25%), with a concomitant increase in the nonrotated state population (Fig. 4H; Table 1). Additionally, our data show a slightly higher population of the rotated state as compared to previous results (Qin et al. 2014) (∼58%, previously 40%). Likewise, the total percentage of dynamic molecules showed a significant drop as compared to the linear control but was comparable to the singly bound ribosomal complexes (∼10%). Our observations confirm the presence of a significant conformational change upon the presence of a structured hairpin in close proximity to the entrance tunnel. The data are comparable to previously published work and to the control, m291 (Cornish et al. 2008; Qin et al. 2014).
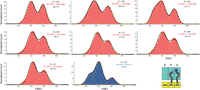
Normalized histograms compiled from hundreds of traces showing distributions of Förster resonance energy transfer (FRET) values for S6-Cy5/L9-Cy3 ribosomes containing a deacylated tRNAfMet in the P site, N-Ac-Phe-tRNAPhe in the A site, and nucleic acid duplexes. N, number of molecules used to compile each histogram; yellow lines represent individual Gaussian fits centered at ∼0.26 (hyperrotated state), 0.38 (rotated state), and 0.58 (nonrotated state) FRET efficiency; black lines represent the sum of two (A, D–G) or three (B, C, H) Gaussians; % represents the percentage of fluctuating molecules for a given ribosomal complex.
Duplex walking and the induction of the hyperrotated state
We used a duplex walking assay to monitor the induction of the hyperrotated state. A DNA–RNA helix was formed downstream from the ribosome-binding region by hybridizing a complementary DNA oligomer to the mRNA (Fig. 2). Shown previously in vitro, the helicase activity of the bacterial ribosome does not discriminate against RNA–RNA versus DNA–RNA duplexes, acting on both with a comparable unwinding efficiency (Takyar et al. 2005). Seven DNA oligomers were designed to be of the same length, 20 mers, and have comparable stability (Table 2). DNA-absent constructs have been performed in separate experimentation (Shebl et al. 2016). The first DNA–RNA duplex was formed 10 nt away from the +1 nt in the P site. The added oligonucleotide was prehybridized to the mRNA during assembly and added in excess to the imaging buffer (1 µM, 1000× the concentration of the mRNA) (see Materials and Methods). Additional complexes were formed by walking different DNA–RNA duplexes 1 nt at a time, in +1 increments, up to the +16 position (Fig. 2B; Table 2). The chosen region, +10 to +16, covers the accessible region of the mRNA entrance tunnel based on X-ray crystallography data, and spans the length of the tunnel to the outside rim (Culver 2001; Jenner et al. 2010).
List of oligonucleotides used and their Gibbs free energy (kcal mol−1)
Following the same experimental design as for the dnaX hairpin, the first ribosomal complex was assembled with a single deacylated tRNAfMet in the P site, and m291 preannealed to a complementary DNA oligo spanning the +10 to +29 region (Fig. 2B). Binding of a single deacylated tRNA in the P site drove the rotational equilibrium to favor the rotated conformation (∼70%) as per the linear unhybridized control (Fig. 3A–H; Table 1). As shown previously, the assembled complexes show spontaneous fluctuations between the rotated and nonrotated conformations (∼52%) (Cornish et al. 2008). Only when the DNA–RNA duplex was placed 11 or 12 nt away from the P site, were we able to induce hyperrotation (Fig. 3B,C). When the +11 oligo was bound to the ribosomal complex, we observed a significant redistribution of the rotational equilibrium of the ribosome, where ∼43% of the ribosomal constructs were observed in the low FRET, hyperrotated state versus ∼38% for the rotated state and ∼19% for the nonrotated state (Table 1). The appearance of the hyperrotated state was concurrent with a drop in the percentage of fluctuating/dynamic ribosomes to ∼28%. Shifting the duplex downstream by a single nucleotide (+12 oligo) resulted in a reduction of the hyperrotated population to ∼26% with a concomitant increase in the populations of the rotated (∼51%) and nonrotated (∼23%) conformations. The reduction in the hyperrotated population was reflected in an increase in the percentage of fluctuating/dynamic molecules to 41.8%. Beyond the +12 position, +13 to +16, except for few molecules sampling the hyperrotated state, the low FRET, hyperrotated population was suppressed to background levels and the return of the rotated and nonrotated conformations to their normal distributions (Fig. 5). In the absence of the hyperrotated state, the ribosomal constructs again displayed normal levels of fluctuation between the rotated and nonrotated states (∼52%), indicating DNA beyond the +12 position has little to no influence on intersubunit rotation (Fig. 5).
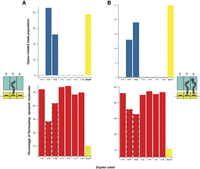
The negative correlation between the prevalence of the hyperrotated population and the percentage of dynamic molecules. (A) The left panel shows data from the singly occupied P site. (B) The right panel shows data for a pretranslocation complex. Red bars represent the percentage of fluctuating molecules. Blue bars represent the population of the hyperrotated state. Highlighted data: dnaX is bolded and shown in yellow; +11 and +12 are bolded and highlighted with a dotted margin.
Next, we wanted to investigate the influence of the A-site occupancy on intersubunit rotation in the presence of a structured element. Generally, the addition of the A-site tRNA shows a modest effect on the equilibrium distributions of the rotated versus nonrotated states, with a slight increase of the nonrotated conformation and a slight decrease in the percentage of fluctuating/dynamic molecules (∼45%) (Fig. 4A–H; Table 1; Cornish et al. 2008). To investigate any influences, a PRE was assembled by adding an N-Ac-Phe-tRNAPhe to the A site. As shown above for single tRNA complexes, hyperrotation was induced only in the presence of a DNA–RNA duplex placed 11–12 nt away from the P site (Fig. 2B). Surprisingly, the introduction of the A-site tRNA resulted in a dramatic reduction in the observed hyperrotated population in the presence of the +11 oligonucleotide (∼13%) as compared to the single P-site tRNA construct (∼43%) (Table 1). However, on the addition of the +12 oligonucleotide, we only observed a modest decrease (∼19%) in the hyperrotated population as compared to its single tRNA counterpart (26%). As shown for the single tRNA data, the introduction of DNA oligonucleotides at the +13 position and beyond, up to +16, did not affect the equilibrium distribution between the rotated and nonrotated conformations, and we observed a suppression of the hyperrotated state. These results show that a DNA–RNA duplex, spatially positioned at a specific distance from the entrance tunnel (11–12 nt away), induces the hyperrotated state formation regardless of the occupancy of the A site.
The kinetics of intersubunit rotation in the presence of a folded structure
FRET distribution histograms are useful for elucidating thermodynamic equilibrium distributions. To extract kinetic information pertaining to the dynamic heterogeneity of fluctuating ribosomes, smFRET traces showing fluctuations were analyzed using hidden Markov modeling (HMM), extracting idealized FRET trajectories (Supplemental Fig. S3A). Next, individual data sets were categorized into three populations, hyperrotated, rotated, and nonrotated, based on a thresholding algorithm and cut-off limits (see Materials and Methods). Dwell times, time spent within a specific state before transitioning to a different state, were calculated. The first and last dwell times of each trace were discarded to minimize the bias, since the origin and/or the termination transitions were undetectable, respectively. Next, calculated dwell times were binned and plotted as a cumulative probability distribution. The data were then fitted to a single exponential. Kinetic rates were extracted from the fitting parameters (Supplemental Fig. S3B,C). Reduced χ2 values were found to be nearly identical when fitting to a biexponential.
Due to the limited observation window and the static nature of the hyperrotated state, molecules transitioning from the rotated state into the hyperrotated state showed only infrequent transitions, and thus were removed from our analysis. However, we did note the presence of these events in the collected traces (a total of 330 of 5827 and 36 dwell times for all the ribosomal complexes with the P or the P- and A-site tRNA, respectively). The remaining fluctuating traces showed clear transitions between two distinct conformations: rotated and nonrotated states. Regardless of the presence or absence of nucleic acid duplexes downstream from the ribosome binding site, the measured kinetics of intersubunit rotation between the rotated and nonrotated states is unaffected (less than a factor of 2). The mean forward rate (nonrotated→rotated) is 0.79 ± 0.06 sec−1 and the mean reverse rate (rotated→nonrotated) is 0.41 ± 0.1 sec−1 at room temperature, under used in vitro conditions (Table 3). As compared to previously reported rates (NR→R is 0.27 sec−1 and R→NR is 0.19 sec−1), transition rates between the rotated and nonrotated conformations are higher, which could be attributed to the higher activity of the ribosomes employed (Cornish et al. 2008). Further investigation is required to detect forward and reverse transitions between the rotated and hyperrotated conformations.
Kinetic rates measured for transitions between the nonrotated conformation and the rotated conformation
Molecular dynamics simulations show a change in ribosomal structure encountering structured mRNA
MD simulations were used to investigate how the proximity of structured mRNA to the entrance tunnel can affect the ribosomal structure and what could be the possible points of interaction between an incoming structured nucleic acid sequence and the mRNA entrance tunnel. Based on the structure 5AFI (Fischer et al. 2015), we created three model systems of the Escherichia coli ribosome that were similar to systems studied by Qin et al. (2014) (see Materials and Methods in Supplemental Material for system preparation and MD protocol). All systems contained the whole ribosome, including tRNAs at P and A positions, as well as the mRNA. Starting from +1 position to the final base at the position +30, the sequence of the mRNA in our model was identical to that in m291 (Fig. 6A). The three systems differed by the position of the complementary DNA 15 mers, which were added to the mRNA strand at either +11, +13, or +15 position (Supplemental Fig. S3). The systems are referred to as either +11, +13, or +15 system, and three 100-nsec-long simulations were performed for each system.
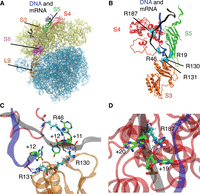
(A) The ribosome model used in MD simulations with water and ions omitted for clarity. The 50S subunit is shown in light blue, and the 30S subunit is shown in yellow. The ribosomal proteins S3, S4, and S5 are colored orange, red, and green, respectively. The mRNA is colored black, while the DNA 15 mer is colored blue. Proteins S6 and L9 used in the FRET experiments are shown in magenta and brown, respectively. (B) The position of arginines in the ribosomal proteins S3, S4, and S5 that were found to maintain hydrogen bonds with either DNA or mRNA residues 50–70 in more than 50% of the trajectory frames for the +11 construct. The arginines are shown in cyan, and DNA and mRNA are shown in blue and black, respectively. Proteins S3, S4, and S5 are colored as in A. (C) Selected snapshot from our simulations illustrates hydrogen bonding of protein residues R130 and R131 from S3 and R46 from S4 with the DNA–mRNA strand. The backbone colors for DNA, mRNA, S3, and S4 are the same as in A. The protein and nucleic residues directly involved in hydrogen bonding are shown in licorice representation. For easier visualization, the carbon atoms are colored cyan for arginine residues and green for nucleic acid residues. Hydrogen atoms are shown in white and are omitted for nucleic acid residues for clarity. For all residues, oxygen atoms are colored red, and nitrogen atoms are blue. Hydrogen bonds are visualized as dotted blue lines. (D) Selected snapshot from our simulations illustrates hydrogen bonding between R187 from S4 and mRNA residues +19 and +20 in the +11 system. The colors and representations are the same as in C.
All MD simulations showed high flexibility for the mRNA residues +10 to +30 and the complementary DNA strand. The DNA and mRNA strands remained in contact with each other for all three systems in all three simulations. However, the helical structure of the DNA–mRNA construct was not maintained by the end of any of the simulations of the +13 and +11 systems; in contrast, it was maintained in two of three of the +15 system simulations (Supplemental Fig. S3), suggesting a higher structural stability of the latter. Although the DNA strand remained in the vicinity of S4 and S5 proteins in all systems, only the +11 system allows for interaction between the DNA strand and S3 (Supplemental Fig. S3).
Analysis of hydrogen bonding during the simulations found numerous interactions between the DNA–mRNA strand and S3, S4, and S5 in the +11 system (Fig. 6B; Supplemental Tables S1 and S2) that were maintained in more than 50% of the trajectory frames. Specifically, residues R130 from S3, R46 and R187 from S4, and R19 from S5 formed hydrogen bonds with mRNA residues +10 to +30 in the +11 system. R131 from S3 was the only hydrogen bond between the DNA strand and the ribosome. A significant reduction in hydrogen bonding between DNA–mRNA construct and S3, S4, and S5 proteins was found for +13 and +15 systems (Supplemental Table S1). Hydrogen bonding with R130 and R131 from S3 and R187 from S4 was only transiently present in +13 and +15 systems (<35% of the frames), with R131 interacting with mRNA instead of DNA. Bonding with R46 from S4 maintains similar levels in +11 and +13 systems and is reduced in the +15 system. Finally, hydrogen bonding with R19 from S5 was absent in the +13 system and present at a similar rate in the +15 as in the +11. Previous work by Takyar et al. (2005) found that triple alanine mutation of R131, R132, and K135 in S3 and double mutation of R44 and R47 (our R46*) in S4 showed decreased helicase activity, whereas double mutation of R19 and R28 in S5 did not alter helicase activity. Our analysis suggests that residues R130 and R131 from S3, and R187 and R46 from S4 are likely to be important for helicase activity, based on the persistence of these interactions in the +11 system and their significant reduction or complete absence in the less active +13 and +15 systems. The observed interactions with R19 from S5 appear less impactful because they are still present in the inactive +15 system to a similar degree as in the active +11 system. These four residues primarily interact with the DNA–mRNA backbone at the following positions: R130 with DNA +11, R131 with mRNA +11, R46 with mRNA +12, and R187 with mRNA +19 and +20 (Fig. 6C,D).
Additionally, analyzing changes in the structures of S3, S4, and S5, we noted significant changes in the fluctuations of S4 protein loop containing residues 25–35 in the +11 system (Supplemental Fig. S5B), suggesting that this loop could also be important for helicase activity. We also investigated structural changes in S6 and L9 proteins, which are labeled in the FRET experiments. The two proteins remained in the vicinity of each other in all of the +15 and +13 simulations; however, a large separation of these proteins was observed for one of the +11 simulations (Supplemental Fig. S6). The distance between the FRET-labeled residues is not increased in this structural change, and significantly longer simulation times may be needed to observe the residue separation found in the hyperrotated state. This simulation is also the only one of the three +11 runs that has high maintenance of hydrogen bonds (>80%) with four residues from ribosomal proteins (R130 and R131 from S3, R187 and R46 from S4) (Supplemental Table S2).
DISCUSSION
Ribosomes undergo extensive conformational rearrangements during translation; the most notable of which is intersubunit rotation. Understanding these conformational changes accompanying translation and how the ribosome interacts with different elements, such as folded coding regions, provides a valuable insight into translation regulation mechanisms (Chen et al. 2014, 2015; Musalgaonkar et al. 2014; Qin et al. 2014). Structured nucleic acids introduce a high energy kinetic barrier to the ribosome, thus pausing translation; a process exploited in cotranslational folding, colocalization, and recoding mechanisms.
Inspired from work by the Noller laboratory, a DNA–RNA duplex was placed at variable distances from the ribosomal P site to determine the positional effect of the duplex on ribosomal conformation. We observed that structural elements close to the mRNA entrance tunnel drive the subunits into a hyperrotated state, consistent with previous observation.18 The results show a stable low FRET state (∼0.22), as compared to the well-characterized rotated (∼0.38) and nonrotated states (∼0.58). The determinant factor for the observed FRET states was decidedly the distance of the nucleic acid duplex to the ribosome. Under our experimental conditions, 7–8 nt spacers from the P site were required to induce hyperrotation. Such spacers place the nucleic acid duplex at the +11 to +12 position from the P site, a region close to the proposed helicase center of the ribosome. The +11 to +12 alignment is in agreement with the required spacer length for a folded mRNA structure to efficiently induce frameshifting (Lin et al. 2012). A 2017 report from Agirrezabala and colleagues presents a structure of the 30S subunit assuming rolling (5–10 Å) upon encountering structured mRNA +12 from the P site (Agirrezabala et al. 2017). It would be interesting to verify if our 0.22 FRET state arises from subunit rolling alone, or if it occurs subsequently as suggested by the previous study (Agirrezabala et al. 2017). A total of +13, +14, +15, and +16 spacers were ineffective in inducing a significant percentage of ribosomes in the hyperrotated conformation. In addition, we observed a significant reduction in the hyperrotated population for the +12 position but not for the +11 position (Fig. 5). It is possible that this decline in the hyperrotated population is due to the occupancy of the A site. Transitioning from initiation to elongation is accompanied by a conformational change of the shoulder of the 30S subunit which lays at an interface of the 30S and 50S subunits and near the mRNA entry tunnel. This domain closure leads to the contraction of the downstream tunnel and thus constricts its diameter by ∼1–3 Å (Jenner et al. 2010). Although it remains to be determined, the tunnel narrowing accompanying the addition of N-Ac-Phe-tRNAPhe to the A site may limit the accessibility of the oligo +12 from the P site.
Folded nucleic acid structures impose a kinetic and thermodynamic challenge to the ribosome. This challenge becomes particularly intriguing when ribosomes encounter stable downstream structured nucleic acid causing interaction of mRNA entrance tunnel proteins with the transcript (Namy et al. 2006). Recent smFRET studies have provided insight into the conformational dynamics of ribosomes during translation (Namy et al. 2006; Cornish et al. 2008; Wen et al. 2008; Chen et al. 2014; Kim et al. 2014). For instance, ribosomes translating frameshifting signals with a downstream hairpin exhibit significant pausing events. These pauses have been linked to a noncanonical rotated conformation (Chen et al. 2014). Additionally, it has been demonstrated by Wen et al. (2008) through optical tweezers that the rate-limiting step in structural interactions is not translocation, but rather pausing in a likely attempt to unwind mRNA interactions. Increasing the pulling force on either end of the mRNA lowered the amount of pausing observed (Qu et al. 2011). In our investigation, we found that the hyperrotated conformation represents a static state, while dynamic equilibrium exists between the rotated and nonrotated conformations. The kinetics of this preexisting dynamic equilibrium remain unaffected even in constructs capable of inducing hyperrotation. However, due to infrequent traces capturing the transition from the rotated to the hyperrotated state, this shift occurs over a longer interval than the dynamicity expressed between the rotated and nonrotated states. We propose that this metastable local minimum in the ribosome's energy landscape corresponds to the long-paused state, which has been previously observed by Puglisi and colleagues and referred to as a long-paused noncanonical rotated state (Chen et al. 2014, 2015). Unlike the studies conducted by Puglisi and colleagues, which study active translation, we sought to precisely examine the effects of intersubunit rotation by utilizing stationary ribosomal complexes.
At equilibrium, and in the absence of translation factors, bacterial ribosomes rotate freely between the rotated and nonrotated states. Maintaining the delicate balance at equilibrium is crucial for translational fidelity. Furthermore, it has been demonstrated by Horan and Noller (2007) that translation is impossible when intersubunit rotation is restricted by the presence of a disulfide bond formed between small subunit protein S6 and large subunit protein L2. Binding of several translation factors and antibiotics (viomycin, sparsomycin, lincomycin, clindamycin, and chloramphenicol) at the A site preferentially stabilizes one conformation or the other (Cornish et al. 2008; Ermolenko et al. 2013; Sharma et al. 2016). Previously and within our current studies, the dynamic equilibrium between the rotated and nonrotated state in ribosomes encountering stable hairpins in close proximity of the mRNA entrance tunnel becomes disrupted, shifting a population into the hyperrotated conformation (Qin et al. 2014). Seemingly, the close presence of structured elements at the mRNA entrance tunnel is sensed and transduced across the ribosomal network propagating a series of conformational changes resulting in hyperrotation. Perturbations to the rotational freedom could disrupt the resulting allosteric communication network throughout the ribosome, affecting the binding affinity of translation factors (Horan and Noller 2007; Musalgaonkar et al. 2014; Sulima et al. 2014). The consequences of perturbation in rotational freedom may impact the fidelity of translation and frame-maintenance, a property that could be exploited in recoding mechanisms such as PRF. In other words, stably folded structures elevate the thermodynamic barrier such that a pool of molecules is shifted to the hyperrotated conformation. Due to this thermodynamic barrier, we wanted to determine what interactions the ribosome would have with the nucleic acid structure.
Our simulation data showed structural differences in +11, +13, and +15 systems and interactional difference between the oligos and the ribosome, in agreement with previous results of Qin et al. (2014). Notably, helical structure of the DNA–mRNA construct was only maintained in the +15 system, suggesting some degree of unwinding in the +13 and +11 systems. We also found that the DNA was sufficiently close enough to interact with the S3 protein at the +11 position but not at the +13 and +15 positions, suggesting the importance of this protein for helicase activity. Specific interactions were observed between the mRNA–DNA construct and positively charged residues of the ribosomal proteins, which were most prominent in the +11 system (Supplemental Table S1). We propose that the following residues could play a role in mRNA unwinding, allosteric communication, and subsequent hyperrotation: R130, R131 from S3, and R46, R187 from S4. The S4 loop containing residues 25–35 could also play a role in the unwinding process, based on movement of these residues in the +11 system (Supplemental Fig. S5). Consistent with previous results from Takyar et al., the importance of residues R130, R131 of S3 and R46 of S4 in ribosomal helicase activity have been shown experimentally (Delagoutte and von Hippel 2003). However, the effect of S4 R187 has yet to be demonstrated. Thus, residues R130, R131 from S3, and R46, R187 from S4, then may serve as integral parts of the helicase center. Further experimental validation is needed to confirm the role of these residues in helicase activity. While the studies carried out by Takyar and colleagues utilized mutations of S3 and S4, many other mutations occur on proteins in the helicase center resulting in the phenotype of ribosomal ambiguity (Takyar et al. 2005). Ribosomal ambiguity mutations (ram) result in high rates of inaccuracies in the ribosome's ability to incorporate the correct amino acid. Several mutations in S4 and S5, including D49Y and E68D in S4, have been shown to exhibit ribosomal ambiguity, and thus may have a larger capacity to enable hyperrotation due to difficulties in unwinding downstream structures, something which would be interesting to experimentally verify (Agarwal et al. 2015). In our simulations, we do not observe any significant interactions between the mRNA/DNA construct and known ram mutations, suggesting that these mutations are more likely to alter S4 and S5 structures and the interface between the two proteins. These structural changes could in turn diminish the helicase functionality of the ribosome.
Recently, a cryo-EM structure was released from Bao et al. (2020) depicting the HIV frameshifting sequence hairpin bound to the A site of the 70S ribosome. In addition to this structure, S6/L9 labeled ribosomes were used to determine intersubunit rotation dependent of A site tRNA occupancy in both dnaX and HIV frameshifting sequences. While no hyperrotation is reported, a few key experimental factors can explain this observed absence. As shown in our duplex-walking experiments, the contingency of the hyperrotated state is dependent upon the distance of the nucleic acid duplex to the ribosome. Additionally, analysis of these data is challenging due to the hyperrotated state exhibiting low FRET close to that of noise. Our results show that hyperrotation occurs within the confines of an interaction between the helicase center of the ribosome and a frameshifting hairpin. As the A site is reported to be occupied by the HIV hairpin in ∼64% of molecules, the possibility of observing hyperrotation would greatly decrease. Indeed, it is observed that the HIV hairpin exhibits lower A site tRNA binding than that of the dnaX hairpin, a result which could arise from the competitive A site binding due to hairpin occupancy. Although a few structures of frameshifting hairpins bound to the A site exist (Yusupova et al. 2001; Agirrezabala et al. 2017), it remains unclear how the hairpin would move from outside of the ribosome to inside of the ribosomal A site during translation, and furthermore, how such a hairpin could move back outside the ribosome.
The hyperrotated metastable state presumably lies within a deep energy well on the ribosome's energy landscape with a high energy barrier (>kBT), in contrast to the lower energy barrier allowing the dynamic interconversion between the rotated and nonrotated states (∼kBT) (Munro et al. 2009). Thus, molecules trapped in this state are eliminated from the total pool of molecules in dynamic equilibrium as evidenced by a reduction in the percentage of dynamic/fluctuating molecules. We found a significant reduction of dynamic molecules when the self-complementary dnaX hairpin was utilized rather than a nucleic acid duplex with two fraying, and less stable, ends. Thus, structures that are more difficult to unwind, like dnaX hairpin, could possibly restructure the energy landscape, directing more molecules into the hyperrotated state. As we show, even single-nucleotide displacements (+11 and +12 vs. +13 and beyond) may significantly restructure the rugged energy landscape to divert ribosomes into different local pockets as evidenced by the appearance/disappearance of the hyperrotated state. Similarly, EF-G-bound GDPNP and the antibiotic viomycin, respectively, are capable of structurally diverting the vast majority of ribosomes into the rotated conformation and raising the surrounding energy barrier (>kBT) enough to suppress the thermally driven intersubunit rotation (Cornish et al. 2008). Identifying ribosome–mRNA interactions on residues of S3 and S4 through MD simulations leads us to believe that the interplay of mRNA-entry proteins with nucleic acid structure may have a significant impact on hyperrotation.
Despite the mounting knowledge of the intrinsic helicase activity of the ribosome and the conformational changes accompanying translation, many of the mechanistic details are missing. The intricacies of local structural rearrangement around the mRNA entrance tunnel are essential in how ribosomes sense and unwind structured mRNA elements. How, then, would the ribosome transduce such signals allosterically throughout the macromolecular complex? Further investigations are required to address the remaining challenges and questions involving the impact of the stability of structured nucleic acids on the conformational dynamics of the ribosome.
MATERIALS AND METHODS
Ribosome preparation
We introduced specific cysteine mutations via site-directed mutagenesis to recombinant ribosomal proteins S6 (D41C) and L9 (N11C). Next, we conjugated fluorescent reporters Cy3 and Cy5 maleimide to S6 (D41C–Cy5) and L9 (N11C–Cy3). Fluorescently labeled proteins were then reconstituted into ΔS6/ΔL9 dual knockout His-tagged 70S E. coli ribosomes (Shebl et al. 2016). Testing the biochemical activity of the generated ribosomes using filter-binding assays and puromycin-reactivity assays showed that the partially reconstituted ribosomes have comparable tRNA binding and EF-G translocation efficiency to wild-type ribosomes (Shebl et al. 2014, 2016).
Preparation of charged tRNA and mRNA
m291 and dnaX were transcribed using T7 RNA polymerase and purified on a denaturing polyacrylamide gel (Milligan and Uhlenbeck 1989; Fredrick and Noller 2002; Qin et al. 2014). m291 is a derivative of T4 bacteriophage gene 32 (Takyar et al. 2005). dnaX hairpin is a derivative of the dnaX gene encoding DNA polymerase in E. coli and, along with a slippery site and a SD sequence upstream, is involved in –1 programmed frameshifting (Tsuchihashi 1991). tRNAfMet and tRNAPhe were purchased from MP Biomedicals and tRNA probes, respectively. N-Ac-Phe-tRNAPhe were aminoacylated using DEAE-purified S100 enzymes (Dubnoff and Maitra 1971; Moazed and Noller 1989; Walker and Fredrick 2008). Aminoacylated tRNAs were purified from the charging reaction and the extent of aminoacylation was verified by acid gel electrophoresis. DNA oligonucleotide variants were ordered from Integrated DNA Technologies (IDT) (Table 3).
m291 sequence:
-
5′GUAAAGUGUCAUAGCACCAACUGUUAAUUAAAUUAAAUUAAAAAGGAAAUAAAAAUGUUUGUAUACAAAUCUACUGCUGAACUCGCUGCACAAAUGGCUAAA CUGAAUGGCAAUAAAGGUUUUUCUUCUGAAGAUAAAG 3′
dnaX sequence:
-
5′UAAGGAAAUAAAAAUGUUUAGUGAACCGGCAGCCGCUACCCGCGCGCGGCCGGUGAGGUUUUUCUUCUGAAGAUAAAG 3′
Preparation of ribosomal complexes for smFRET
All ribosomal complexes were assembled in polyamine buffer (20 mM Hepes, pH 7.5, 6 mM MgCl2, 150 mM NH4Cl, 6 mM ßME, 2 mM spermidine, and 0.1 mM spermine) (Cornish et al. 2008). m291 was preannealed to the desired complementary DNA oligonucleotide and the biotin-labeled DNA oligonucleotide (5′ biotin – CTTTATCTTCAGAAGAAAAACC-3′, IDT) at 65°C for 5 min and then cooled on ice for 10 min. To initiate ribosomes with a P-site tRNA only, 1 μM S6/L9-labeled ribosomes were incubated with 2 μM of the preannealed m291/DNA duplex and 2 μM tRNAfMet at 37°C for 20 min. Pre-translocation complexes were prepared in a similar manner, except including incubation of the A site tRNA, N-Ac-Phe-tRNAPhe at 37°C for an additional 20 min. dnaX-containing samples were prepared following the same protocol with the elimination of the duplex forming DNA oligonucleotide in the preincubation step.
Quartz slides were precleaned and passivated with a mixture of m-PEG-succinimidyl valerate (MW 5000) and Biotin-PEG-succinimidyl valerate (MW 5000) (Laysan Bio Inc.) and pretreated with neutravidin (0.2 mg/mL) (Pierce) (Roy et al. 2008; Joo and Ha 2012; Qin et al. 2014). Ribosomal complexes were diluted to a final concentration of 1 nM and immobilized on a quartz slide (Fig. 2A). To minimize photobleaching, all samples were imaged in polyamine buffer supplemented with an oxygen-scavenging system; 0.8 mg/mL glucose oxidase (Sigma), 0.625% β-d-glucose, 0.02 mg/mL catalase (Roche), and 1.5 mM Trolox (Sigma) (Cornish et al. 2008; Roy et al. 2008). Duplex-forming DNA oligonucleotides complementary to the desired region were added to the imaging buffer to a final concentration of 1 μM.
Acquisition and analysis of smFRET data
All of the experiments described were performed at room temperature, in accordance with our previous work (Qin et al. 2014; Shebl et al. 2016). A 532 nm laser (Spectra-Physics) was used to excite the donor dye (Cy3) via prism-based total internal reflection microscopy (TIRFM) (Roy et al. 2008; Joo and Ha 2012). The resulting fluorescence emission was split into two pathways (Cy3 and Cy5 emission) with a 630dcxr dichroic mirror (Chroma). The emission signal was collected via an Andor iXonEM + 897 Electron Multiplying Charge Coupled Device (EMCCD) camera. Movies were recorded using Single (custom software generously provided by Taekjib Ha) at an acquisition rate of 100 msec. A calibration image was acquired using crimson fluorescent FluoSpheres (Invitrogen). The movies were processed to extract donor and acceptor intensities using custom IDL scripts. Next, data were sorted and analyzed using custom MATLAB scripts (MathWorks).
The acquired traces were sorted and validated based on predefined criteria: only traces showing single step photobleaching for both dyes, Cy3 and Cy5, were considered; the trace had a minimum of 10 data points (1 sec). Traces showing positive correlation between the intensities of the donor and acceptor dyes, intensities <100 arbitrary units (au) or FRET states less than a set threshold (0.15) were manually excluded. Each trace was normalized to its temporal length to ensure equal contribution to the final histogram (Elvekrog and Gonzalez 2013; Qin et al. 2014). Data points were smoothed with a five-point window average to construct the FRET histograms; all other points remain unsmoothed. A comparable number of molecules were selected to contribute to the histogram. Peaks within the normalized histograms were identified automatically via the peak finder function of IGOR Pro (WaveMetrics) and fitted to either two or three Gaussian distributions (Qin et al. 2014). The viability of the fit was partially assessed from the consistency of the width of the individual peaks.
For rate determination, a combined approach was used. Discrete FRET states were identified within traces using HMM (vbFRET) (Bronson et al. 2009). Next, dwell times of the different states were categorized into three bins (hyperrotated, rotated, and nonrotated) based on a thresholding algorithm (custom Matlab script) (Blanco and Walter 2010). Individual dwell times were then calculated and plotted as a cumulative probability distribution that was then fit to a single exponential curve using IGOR Pro (WaveMetrics).
Molecular dynamics simulations
To prepare the models of the ribosome, we started with the structure of the E. coli ribosome (PDB code 5AFI) that contains three tRNAs at the A/T, P, and E positions (Fischer et al. 2015). The tRNA in the E site was deleted to match the ribosomes used experimentally. Because the mRNA strand in this structure was very short, we also used the PDB structure 4V4Y that contains a full mRNA strand in the Thermus thermophilus ribosome (Yusupova et al. 2006). We aligned the 16S segments in 5AFI and 4V4Y, which have very similar structures, to obtain the correct positions of the mRNA strand and the SD region of the 16S for our model. The 16S rRNA and the mRNA in 5AFI were replaced with the ones from 4V4Y after alignment to 16S. The replaced structures were minimized in the E. coli ribosome to eliminate any steric clashes. We used the cIonize plugin (Stone et al. 2007) in VMD to neutralize the system by adding Mg2+ ions with completed octahedral solvation shells at points of electrostatic potential minima. The last base in the mRNA strand was at the +30 position; between positions +1 and +30, the bases were mutated to the ones in the 291mRNA. We added the DNA 15 mers by first using the Nucleic Acid Builder (Macke and Case 1997) to generate the structures of 291mRNA starting from the +13 position and the complementary DNA 15 mers at either +11, +13, or +15 position. The three generated structures were equilibrated in water for 10 nsec, after which they were covalently linked to the mRNA in our E. coli ribosome system at the corresponding position, resulting in the +11, +13, and +15 systems. Bulk water was added with the VMD Solvate plugin, after which chloride and potassium ions were added at a concentration of 0.1 mol/L with the Autoionize plugin.
All molecular dynamics (MD) simulations were performed with NAMD (Phillips et al. 2005). The CHARMM36 protein (Best et al. 2012) and nucleic acid (Hart et al. 2012) force fields were used for the ribosome, whereas the TIP3P model was used for water (Jorgensen et al. 1983). We used the particle-mesh Ewald method to calculate long-range electrostatics (Darden et al. 1993). A cutoff of 12 Å was used for the van der Waals interactions and a potential switching function was applied from 10 to 12 Å to ensure a smooth decay to zero. All covalent hydrogen bonds were kept rigid, which allowed us to integrate the equations of motion with a 2 fsec time step. The temperature and pressure were kept constant at the biologically relevant values of 310 K and 1 atm with a Langevin thermostat and piston, respectively. For hydrogen bond analysis, bonds were defined as present if the acceptor-donor distance was 3.5 Å or less and the bond angle was between 145° and 180°.
The constructed systems were minimized in two steps. In the first step, only water and ions were minimized, followed by minimization of all atoms in system in the second step. The equilibration was done in three 2-nsec-long steps. In the first step, water and ions were equilibrated, after which mRNA bases in positions +1 to +3 and the complementary bases in the P-site tRNA were equilibrated, followed by equilibration of all side chains with only the backbone restrained in the final equilibration step. After equilibration, all systems were sampled for 100 nsec in triplicate.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
P.V.C. acknowledges support from the National Science Foundation (NSF; MCB-2122902) and Mizzou Forward Undergraduate Research Training Grant, University of Missouri Agricultural Institute. J.C.G. acknowledges support from the National Institutes of Health (R01-AI148740). For simulations, we used the Hive cluster, which is supported by the NSF (MRI-1828187) and is managed by the Partnership for an Advanced Computing Environment at Georgia Tech.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080291.124.
- Received October 17, 2024.
- Accepted March 31, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








