
Crystallographic and cryoEM analyses reveal SARS-CoV-2 SL5 is a mobile T-shaped four-way junction with deep pockets
- Laboratory of Nucleic Acids, National Heart, Lung and Blood Institute, Bethesda, Maryland 20892-8012, USA
- Corresponding authors: christopher.jones2{at}nih.gov, adrian.ferre{at}nih.gov
-
Handling editor: Eric Westhof
Abstract
Stem–loop 5 (SL5) is a structural element that is conserved across coronavirus genomic RNAs. It spans the start codon from which the long ORF1 is translated in full-length viral RNA. Phylogenetic conservation indicates that it is comprised of four paired elements, but the specific 3D arrangement of these helices has remained unknown. Now, we have solved the crystal structure of SL5 from SARS-CoV-2 at 3.3 Å resolution, finding that the RNA adopts a T-shaped four-way junction fold in which two coaxial stacks of two helices each pack orthogonally. This arrangement results in deep pockets at the helical junction, where cations bind. Except for limited interactions in this region, the structure is remarkable for the paucity of tertiary contacts. We confirmed the stability of this fold in solution by FRET and carried out single-particle cryogenic-sample electron microscopy (cryoEM). The resulting ∼5 Å resolution cryoEM map, and 3D variability analysis, suggest conformational flexibility at the junction. In vitro translation of structure-guided mutants demonstrated that SL5 inhibits protein synthesis. Thus, it is likely that SL5 recruits additional factors in vivo. This, and its characteristic clefts at the four-way junction, make SL5 an attractive target for the discovery of RNA-targeted antiviral small molecules.
Keywords
INTRODUCTION
During infection, SARS-CoV-2 translates polyprotein 1ab (pp1ab), which is subsequently proteolyzed into 16 nonstructural proteins (Nsp1–16). These ultimately take over the host cell (for review, see V'Kovski et al. 2021). The viral RNA recruits the translation machinery to synthesize proteins that dampen the host immune response, replicate the viral RNA, and ultimately selectively translate viral RNAs over host mRNAs. This multilayered control of viral protein synthesis is accomplished by viral translation regulatory proteins (Schubert et al. 2020; Thoms et al. 2020), such as Nsp1, and viral RNA structural elements, such as the frameshifting pseudoknot (Jones and Ferré-D'Amaré 2022). Consequently, translation early in infection produces proteins that limit host cell translation or facilitate viral RNA replication (V'Kovski et al. 2021). The roles that coronaviral RNA structure plays in tuning and timing selective translation of full-length and subgenomic RNAs remain to be fully elucidated.
Comprehensive investigations of the SARS-CoV-2 viral RNA as a whole have resulted in several models for its secondary structure (Manfredonia et al. 2020; Wacker et al. 2020; Lan et al. 2022). These models predict the 5′ end to fold into a series of stem–loops that span the 5′ untranslated region (UTR) and the start of ORF1. Consistently across these models, stem–loop 5 (SL5) is predicted (Lan et al. 2022). This ∼100 nt element is predicted to be a multistem motif with a duplex formed by the 5′ and 3′ ends (hereafter referred to as “SL5stem”), capped by a four-way junction that additionally includes stems SL5a, SL5b, and SL5c. Broader analyses of coronavirus genomes suggest that they all contain an SL5-like structure spanning the start codon. This high phylogenetic conservation has led to proposals that SL5 is involved in critical facets of viral biology. The repetitive loop sequences of SL5 have been suggested to be involved in viral RNA packaging (Chen et al. 2021). As SL5 is not present in subgenomic RNAs (V'Kovski et al. 2021), it functions only within full-length viral RNA, which is translated and packaged.
To provide a physical framework with which to elucidate the function of this RNA element conserved across coronavirus genomes, we have now determined the crystal structure of SL5 from SARS-CoV-2. Our structure confirms the presence of a four-way junction but reveals that the constituent helices adopt an atypical T-shaped arrangement. We extend insights from the SL5 crystal structure by performing single-particle cryogenic sample electron microscopy (cryoEM) analysis, which yielded evidence of conformational variability at the junction. To determine the functional significance of RNA features revealed by our analyses, we characterized the effect of structure-guided site-directed mutants in repressing translation in vitro. Our characterization of the SARS-CoV-2 SL5 element is the starting point for examining larger viral genomic RNA motifs and their interactions with binding partners.
RESULTS
Crystal structure of SARS-CoV-2 SL5
The structure of SL5 was determined de novo employing crystals of an in vitro transcribed 101 nt RNA construct. To facilitate crystallization, the distal loop of SL5a was mutated from UUUCGU (nt 200–205; SARS-CoV-2 NC_045512.2 reference genome numbering used throughout) to GAAA. The structure was solved by the single-wavelength anomalous dispersion (SAD) method using data from a CsCl-soaked crystal and refined at 3.3 Å resolution (Materials and Methods; Table 1; Supplemental Fig. S1A,B). Despite the intermediate resolution, the density-modified experimental SAD electron density maps were of high quality, allowing unambiguous tracing of the single RNA chain in the crystallographic asymmetric unit and revealing that the engineered SL5a distal loop forms crystal contacts with the SL5stem and A223 residue of symmetry-related molecules (Supplemental Fig. S1C,D).
Summary of crystallographic statistics
SL5 adopts an overall T-shaped fold that has the connectivity of cL-family (Laing and Schlick 2009) four-way junctions (Fig. 1) and comprises two perpendicular helical stacks. SL5a and SL5b stack coaxially on each other and point in opposite directions. Perpendicular to the SL5a/SL5b stack, SL5stem stacks coaxially on SL5c (Fig. 1B). Stem–loop SL5c terminates in a canonical GAAA tetraloop, which rather than participating in intramolecular tertiary contacts, is exposed to solution and forms crystal contacts with SL5a and A220 of symmetry-related molecules (Supplemental Fig. S1C,D). The perpendicular arrangement of the four helices produces two deep pockets on either side of the SL5c/SL5stem stack (Fig. 1B)—one formed by SL5c and SL5a and a second formed by SL5c, the base of SL5a, and a 5-nt bulge within SL5a from A220-U224 (Fig. 1B–D). As the SL5a bulge is looped out, stacking of SL5a and SL5b is mediated by a purine–purine sheared pair between A187 and G219 and by C186 (Fig. 1C). G174, which flips out of SL5stem, forms crystal contacts with C186, which is likely unpaired, given its high DMS reactivity (Lan et al. 2022). The residues A220, C222, and U224 stack on one another (Fig. 1D), and U221 forms a crystal contact by stacking on a symmetry mate. Thus, other than the helical stacking organizing the structure, there are no tertiary interactions of note (Fig. 1B).
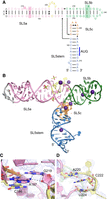
Structure of the SARS-CoV-2 stem–loop 5 (SL5) determined by X-ray crystallography. (A) Secondary structure of SL5, colored as stem SL5a (pink), SL5a bulge (yellow), stem SL5b (green), stem SL5c (orange), and SL5 stem (blue). Residues altered to facilitate crystallization are gray. Two notable base triples in SL5a and SL5b are boxed. Leontis–Westhof symbols (Leontis and Westhof 2001) denote base pairs. Arrows indicate chain connectivity. Blue bar denotes AUG start codon for ORF1 at nts 266–268. SARS-CoV-2 reference genome (NC_045512.2) numbering is used throughout. (B) Cartoon representation of the crystal structure (Cs+ derivative), colored as in A. Pink, purple, and lime spheres denote waters, Cs+ ions, and hydrated Mg2+ ions, respectively. (C) View of A187·G219 sheared pair above C186 and overlaid with the experimental (single-wavelength anomalous dispersion [SAD]) electron density map (blue mesh) for Data set 1, contoured at 2 σ. Putative hydrogen bonds are indicated by black dashes. (D) Stacking of SL5a bulge residues A220, C222, and U224 overlaid with the experimental electron density map (blue mesh) for Data set 1 contoured at 1.5 σ. For (C) and (D), ions were removed for clarity.
Seven RNA-bound Cs+ ions were identified through their anomalous dispersion in the data set from the CsCl-soaked crystal (Fig. 2). Two of these, hereafter Cs(A) and Cs(B), are bound at the four-way junction where the RNA chain changes direction (Fig. 2A–C) and likely screen charge to facilitate the acute turn of the phosphodiester backbone. Indeed, when mapped onto the molecular surface, the most negative regions of the electrostatic potential (calculated though the Poisson–Boltzmann equation) (Baker et al. 2001) are located at the four-way junction (Fig. 2A). Cs(A) is bound to the turn among all four stems at U182, C183, G252, and A253 (Fig. 2B). Cs(B) is bound between the turn between SL5b and SL5c to C230, U251, G252, A253, and C254 (Fig. 2C). The Cs+ ion binding-sites observed in our heavy atom derivative are likely to be occupied by K+ under physiological conditions.
Counterions localize to negatively charged pockets of the stem–loop 5 (SL5) four-way junction. (A) Electrostatic potential (red to white, −15 kBT/e− to 0 kBT/e−) for SL5 (Cs+ derivative) mapped onto its molecular surface. Two Cs+ ions and two hydrated Mg2+ ions are resolved in the structure. (B) Cs(A) site at the junction of the four helical elements. (C) Cs(B) site between SL5b and SL5c. (D) Mg(C) site between SL5b and the GAAA tetraloop of SL5c. (E) Mg(D) site between SL5a and SL5b. For (B)–(E), crystal structure, colored as in Figure 1, is shown overlaid with the anomalous difference Fourier synthesis (yellow mesh, contoured at 4 σ) or the |Fo|-|Fc| residual map (green mesh, contoured at 4 σ) prior to building non-Cs+ ions and waters. Putative hydrogen bonds between magnesium ion hydrate and the RNA are indicated by black dashes.
Positive features in residual |Fo|-|Fc| Fourier syntheses, for both the Cs+ derivative and the native data sets, suggested the presence of bound hydrated magnesium ions (Fig. 2D,E). Mg(C) sits in the deep cavity between SL5b and SL5c, and its inner-sphere water (aquo) ligands are in putative hydrogen-bonding distance with the nucleobase of G276 and the phosphate of A278 of the GAAA tetraloop of SL5c and the phosphate of G252 in SL5b (Fig. 2D). Previously, chemical shift perturbation nuclear magnetic resonance (NMR) analysis of an SL5 fragment in the presence and absence of Mg2+ demonstrated (Mertinkus et al. 2024) an ∼0.3 ppm change in G252, consistent with its interaction with Mg(C) observed in our crystal structures. The inner-sphere coordinated waters of Mg(D) are within putative hydrogen-bonding distance of the phosphates of C255 and G248 of SL5a and SL5b, respectively (Fig. 2E). Given the resolution limit of the diffraction data and the mean coordinate precision of ∼0.5–0.6 Å (Table 1), these interactions await confirmation at higher resolution. Overall, cation binding at negatively charged pockets in the four-way junction likely facilitates packing of the SL5a/SL5b and SL5c/SL5stem helical stacks into close proximity.
The U-rich bulge formed by U194, U211, and U212 appears to participate in noncanonical interactions, which we have modeled as a U195•G210 pair, U194•U212 pair, and C193•G213•U211 triple (Fig. 3A). These putative interactions are consistent with noncanonical U-imino resonances correlated (Wacker et al. 2020; Mertinkus et al. 2024) with C193•G213. Interestingly, the U195•G210 pair is not supported by an imino resonance (Mertinkus et al. 2024), which may be explained by the pronounced buckling of the pair as modeled here (Fig. 3A). In SL5b, the U234•G246•A235 triple (Fig. 3B) is consistent with nonstacked A235 chemical shifts (Mertinkus et al. 2022) and high DMS reactivity (Lan et al. 2022). The loop of SL5b, which possesses the wild-type (WT) UUUCGU sequence naturally present in SL5a and SL5b of some isolates, folds into a noncanonical loop that is partially disordered in our electron density maps, consistent with high DMS reactivities (Lan et al. 2022) of C203 and C241. The U238•G242 base pair in our crystallographic structure is likely not the only conformation of these nucleobases, as recent NMR characterization (Mertinkus et al. 2024) of the SL5a and SL5b loops found them to be dynamic.
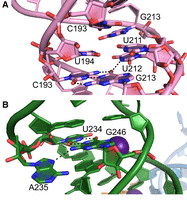
Base triples in the structure of SARS-CoV-2 stem–loop 5 (SL5). (A) SL5a base triple formed by C193·G213·U211 adjacent to the U194·U212 pair. (B) SL5b base triple formed by U234·G246·A235. Black dashes denote putative hydrogen bonds.
CryoEM and FRET analyses of SL5 conformation in solution
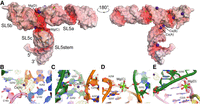
Other four-way junction RNAs have been found to populate multiple structures with different stacking arrangements (Hohng et al. 2007). To examine the overall structure of SARS-CoV-2 SL5 in solution and to gain insight into its conformational variability, we performed single-particle cryoEM analysis. Even though cryoEM reconstructions of RNAs of this size rarely attain better than 5 Å resolution, this is often sufficient for characterizing the overall shape of the RNAs (Kappel et al. 2020). Grids were prepared with RNA processed similarly to samples used for crystallization. Single-particle cryoEM data were analyzed without using the crystal structure, to avoid biasing particle picking (see Materials and Methods; Supplemental Fig. S2). This approach ultimately yielded a ∼4.5–5 Å resolution Coulomb potential map (Table 2) that is in good overall agreement with the T-shaped conformation present in our crystals (Fig. 4).
Single-particle cryogenic sample electron microscopy (cryoEM) analysis indicates a dynamic four-way junction and a SL5a swivel. (A) Coulomb potential map for stem–loop 5 (SL5) contoured at 10 σ in PyMOL (blue mesh) overlaid on the crystal structure (Cs+ derivative), colored as in Figure 1. (B) Orthogonal view.
Summary of cryoEM data collection statistics
At the current cryoEM map resolution, reliably observable features are limited to the shape and orientation of paired elements and gaps in Coulomb potential created by pockets at the four-way junction. The largest conformational difference between the crystal structure and the cryoEM reconstruction is that the helical axis of SL5a in the latter deviates ∼15° from that in the X-ray structure. CryoEM density for loop residues 220-4 is only partly resolved, suggesting that this portion of the RNA is dynamic and that the conformation present in crystals is one snapshot. Three-dimensional variability analysis (Punjani and Fleet 2021) of SL5 particles also indicated displacements at this region in which the SL5c loop and residues 220-4 appeared to move toward one another (Fig. 5).

Three-dimensional variability analysis of SARS-CoV-2 stem–loop 5 (SL5). The Coulomb potential maps are contoured at 0.25 map threshold in Chimera. The motion of interest is the changing density between the SL5c loop and SL5a at the four-way junction (arrows in bottom frame).
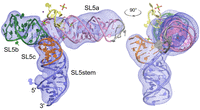
To further examine the conformation of SL5 in solution and specifically to distinguish H-shaped from T-shaped folds, Cy3 and Cy5 dyes were placed using two labeling strategies (Materials and Methods; Fig. 6A). For most plausible H-shaped folds (Supplemental Fig. S3), one high and one low EFRET value would be expected using this labeling strategy, depending on which two helices are brought proximal (i.e., 1-2, 1-3, or 1-4). Bulk fluorescence resonance energy transfer (FRET) was measured for each RNA in the presence and absence of Mg2+, using the Cy3-only–labeled specimen and Cy5-excited samples to calculate EFRET (see Materials and Methods; Supplemental Table S1). For doubly labeled RNAs, EFRET values were ∼0.33 (Fig. 6B), consistent with distances of ∼68 Å (Fig. 6C). In the presence of Mg2+, EFRET values increased to ∼0.38 for both RNAs, a difference that is not statistically significant. These results are consistent with the predicted distances for the FRET dyes based on the crystal structure and cryoEM reconstruction (Fig. 6A). As cyanine dyes can stack on the ends of helices and κ2 was not measured herein to account for the free rotation of dyes, these effects could confound our FRET measurements, which nonetheless are in excellent agreement with our crystal structure and cryoEM reconstruction.
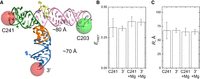
Fluorescence resonance energy transfer (FRET) analysis of stem–loop 5 (SL5) in solution. (A) Overview of labeling scheme. Cy3 was placed at C203, while Cy5 was at either C241 or the 3′ end. Distances predicted from the crystal structure are noted. (B) EFRET values measured for each RNA with Cy5 at C241 or the 3′ end in the presence and absence of Mg2+. (C) Corresponding distances (R) using an R0 of 60 Å for the Cy3–Cy5 FRET pair. Error bars indicate standard deviations (n = 6 independent measurements). The changes observed upon Mg2+ addition are not statistically significant.
Modulation of translation by structure-guided SL5 mutations
To evaluate the effects of SL5 structural features in translation, we used a reporter assay containing a firefly luciferase between the SARS-CoV-2 5′ and 3′ UTRs. Using this assay, we quantitated translation in rabbit reticulocyte lysate (RRL) from capped or uncapped mRNAs containing SL5 variants. The results were normalized to WT mRNA translation (Materials and Methods). As capped and uncapped mRNAs yielded similar results (Table 3), only results from uncapped mRNAs are discussed below, except for mutations adjacent to the start codon (i.e., G256U, A257U, A258U).
SL5 mRNA translation in vitro
Mutation of the start codon from AUG to GUG (Table 3, A266G) reduced translation by 65% relative to WT (0.35 ± 0.03, P = 5 × 10−8; t-test used throughout). All other mutations produced more modest effects on translation. Mutants made for crystallization (SL5aGAAA) were not significantly different from WT (0.97 ± 0.10, P = 0.47). Similar mutations converting SL5b into a GAAA tetraloop (Table 3, SL5bGAAA) increased translation by 12% (1.12 ± 0.09, P = 0.02). Mutation of the native SL5c GAAA tetraloop (G256U/A257U/A258U) did not result in activity significantly different from that of WT uncapped mRNA (0.94 ± 0.06, P = 0.06). However, the capped SL5c mutant was translated 45% less than WT (0.55 ± 0.09, P = 2 × 10−5), suggesting that the mutation could be affecting recruitment of a cellular factor involved in cap-dependent translation.
Variant mRNAs were also prepared to test the contributions to translation of looped-out residues, naturally occurring mutations, and stacking interactions at the four-way junction (Table 3). Deletion of G174 was not significantly different from WT (0.93 ± 0.10, P = 0.13). Deletion of looped-out residues U212 and 235A altered translation by ∼10%, changes which were statistically significant (1.12 ± 0.11, P = 0.04 and 0.92 ± 0.06, P = 0.02, respectively).
Absent from the parent strain, the mutation C241U occurred in viruses abundant early in the COVID-19 pandemic, and also in dominant circulating variants, while C203U occurs rarely. G210U circulates strictly in delta variants (Hadfield et al. 2018). Of the rare mutations that alter the A187•G219 pairing, G219U and G219C were not significantly different from WT (1.02 ± 0.04, P = 0.19 and 1.12 ± 0.17, P = 0.14, respectively). In contrast, A187C translation was reduced by 14% relative to WT (0.86 ± 0.08, P = 0.008). The adjacent C186A mutation did not significantly alter translation from WT (1.02 ± 0.11, P = 0.71).
For mRNAs bearing mutations that disrupted stacking interactions between SL5a and SL5b, translation was generally ∼20%–30% better than WT, showing an overall enhancement of translation concomitant with disruption of the four-way junction (Table 3). For example, the mutant G252A in SL5b was ∼30% better translated than WT (1.30 ± 0.11, P = 0.001), C228U was ∼20% better translated than WT (1.20 ± 0.04, P = 0.02), and the double compensatory mutant was not significantly different from WT (1.05 ± 0.15, P = 0.42). In SL5a, G227A was not significantly different from WT (1.17 ± 0.26, P = 0.08), C183U was ∼25% better translated than WT (1.25 ± 0.12, P = 0.002), and the double mutant was not significantly different from WT (1.10 ± 0.12, P = 0.10).
For mRNAs bearing disrupting mutations to stacking between SL5c and SL5stem, enhancement of translation was observed for SL5c (Table 3). Mutations to SL5stem, U182C and A263G, were not significantly different from WT (1.06 ± 0.13, P = 0.32 and 0.94 ± 0.16, P = 0.38, respectively). While SL5c mutations were better translated, as observed for U262G (1.24 ± 0.13, P = 0.004) and A253G (1.12 ± 0.10, P = 0.02), the double mutant bearing compensatory mutations was also significantly better than WT (1.09 ± 0.09, P = 0.04). The proximity of SL5c to the start codon (nt 266–268) raises the possibility for start codon context to play a role in these sequence features, as observed for SL5c loop mutants. Overall, our in vitro reporter assay shows that crystallographically observed structural features of SL5 have functional effects and that structural stability of this RNA element is in general inhibitory toward viral protein synthesis.
DISCUSSION
The crystal structure of the SL5 motif of SARS-CoV-2 demonstrates that this RNA element, which is conserved across coronavirus genomes, adopts a T-shaped four-way junction fold (Fig. 1). That this fold is stable and predominant in solution is further supported by our FRET experiments, which were designed to distinguish between possible alternative arrangements of the four constituent helices (Fig. 6; Supplemental Fig. S3). Our structure, including the noncanonical base pairs, triples, single-stranded nucleotides, and the SL5c GAAA tetraloop, is consistent with probing data (Manfredonia et al. 2020; Wacker et al. 2020; Lan et al. 2022; Mertinkus et al. 2022, 2024). GNRA tetraloops are known to facilitate long-range interactions (Nissen et al. 2001), and crystal contacts in our SL5 structure (Supplemental Fig. S1C,D) are consistent with this. While SL5c might have been expected to be involved in intramolecular (tertiary) contacts, our structure (Fig. 1) shows this not to be the case, and its high reactivity to DMS probing (Manfredonia et al. 2020; Lan et al. 2022) supports this. The junction residues A220-U224 (Fig. 1D), which form a bulge in the crystal structure, may transiently interact with the sugar edge of the GAAA tetraloop. Thus, while SL5c does not make interactions in the isolated RNA, the motif may do so with other binding partners through, for example, A-minor motifs. In this regard, it is notable that the 5′ UTR of SARS-CoV-2 has a weak internal ribosome entry site (Slobodin et al. 2022). This function of the RNA element may depend on SL5c interactions or its overall T-shaped structure. Thus, it is possible that the GAAA tetraloop of SL5 interacts with the ribosome through ES7 of 18S rRNA in a manner similar (Brown et al. 2022) to that of domain IIIe of the HCV IRES, or through another mode altogether.
By performing single-particle cryoEM reconstruction at ∼5 Å resolution, we show that the crystal structure adopts a similar overall conformation as in solution, noting some deviations. The Coulomb potential map suggested that the helical axis of SL5a can bend by as much 15° relative to its orientation in crystals (Fig. 4). Moreover, 3D variability analysis (Fig. 5) suggests that the core of the four-way junction can adopt a series of closely related structures. Recently, the results of cryoEM analyses at 5–10 Å resolution of SL5 orthologs from a number of related coronaviruses, as well as atomic force microscopy studies on the viral RNA motif, were reported (de Moura et al. 2024; Kretsch et al. 2024). Those studies also suggested flexibility at the SL5 four-way junction and indicated that, across phylogeny, the motif can adopt related but distinct conformations in isolation. The functional consequences of structural flexibility of coronaviral SL5 will be a future area of study.
Our analysis of in vitro translation of structure-guided mutants (Table 3) supports modest translational repression by the intact SL5 four-way junction and highlights the interaction between SL5a and SL5b. In vivo, it is possible that protein factors other than those present in reticulocyte lysate are involved. The modest translational effect of SL5 mirrors that recently observed (Mackeown et al. 2023) for stem–loops in the 5′ UTR of the model coronavirus OC43. The juxtaposition of two nearly equivalent U-rich loops at the ends of SL5a and SL5b imparts a partial pseudosymmetry to the RNA that could allow for its recognition by an oligomeric-binding partner capable of sensing the ∼9 nm of separation. Alternatively, the perpendicular arrangement of helical stacks in the center of the RNA may provide a unique phosphate-rich interface for protein recognition that could alter junction stability and bring SL5a and SL5b parallel to one another. The FRET assay (Fig. 6) developed here could be used to assay such large conformational changes in the presence of binding partners. The high information content (Warner et al. 2018) of the four-way junction and its deep electronegative pockets make this structural element attractive for RNA-targeted drug design aiming to dysregulate translation initiation or other functions of SL5. Given current progress in targeting SARS-CoV-2 RNA motifs (Zafferani et al. 2021), candidate small molecules could be structurally evaluated with SL5-derived RNAs rationally designed from this work.
MATERIALS AND METHODS
RNA preparation
DNAs (primers and plasmids, Supplemental Table S2) were purchased from Integrated DNA Technologies (IDT) or Eurofins Genomics, resuspended in diethylpyrocarbonate (DEPC)-treated water, and used without further purification. Mutations were introduced using site-directed mutagenesis kits from Ambion, and plasmid sequences were confirmed through Sanger sequencing at Eurofins Genomics or through whole plasmid sequencing at Plasmidsaurus. Prior to transcription template preparation, plasmids were transformed into GC10 competent cells (MilliporeSigma), which were plated on LB agar supplemented with 100 mg/L ampicillin and used to select single colonies for liquid LB cultures growth and plasmid midipreps (Qiagen). Plasmids were then used for preparation of DNA templates via PCR as described (Jones and Ferré-D'Amaré 2022). A plasmid encoding a luciferase flanked by the SARS-CoV-2 5′ and 3′ UTRs (pCov2-5UTR-Luc-3UTR) was used to prepare DNA templates for mRNAs for translation assays. RNAs for crystallization were transcribed from DNA templates in vitro using T7 RNA polymerase (5–10 mL transcription reactions incubated overnight at 37°C) and purified using denaturing polyacrylamide gel electrophoresis in 1× Tris-Borate-EDTA (TBE) buffer, followed by overnight electroelution at room temperature as described (Jones and Ferré-D'Amaré 2014). RNAs were concentrated in 10,000 Da MWCO Amicon centrifugal filters (Millipore), washed once with 0.5 M KCl, several times with DEPC-treated water, filtered through 0.1 µm centrifugal filters, and stored at −20°C until further use. For mRNA purification, 0.5 mL transcription mixtures were incubated for 4 h at 37°C, then supplemented with 10 mM CaCl2 and RQ1 RNase-free-DNase for 1 h at 37°C, prior to phenol:chloroform extraction (1:1 v/v pH 7.0 buffered phenol:chloroform), and ethanol precipitated overnight at −20°C. mRNA pellets were resuspended in 1 mL DEPC-treated water, concentrated in 100,000 MWCO centrifugal filters, and washed as above. Final mRNA purity was assessed using native electrophoresis through 1× TBE, 1% (w/v) agarose gels supplemented with 0.25 µg/mL ethidium bromide. For capped mRNAs, 0.1 mg mRNA was capped using Vaccinia virus capping enzyme (NEB) and cap 2′-O-methyltransferase enzyme (NEB) in 0.1 mL reactions for 1 h at 37°C, after which mRNAs were phenol:chloroform extracted, ethanol precipitated, and washed as above. Capped and uncapped mRNAs were diluted to 50 ng/µL stock solutions and stored at −20°C until further use.
Crystallization and X-ray data collection
SL5 RNAs were brought up at 6–9 g/L in a buffer containing 25 mM HEPES-KOH, pH 7.4, and 150 mM KCl. Then, RNAs were heated at 95°C for 2 min, placed on ice, and incubated at 37°C after adjusting MgCl2 to 10 mM. Crystallization through the vapor diffusion method (15-well EasyXtal trays, NeXtal) at 21°C was from 2 µL hanging drops prepared by mixing the RNA solution and a reservoir solution comprised of 1.2–1.4 M sodium citrate (MilliporeSigma) and 0.1 M HEPES-NaOH, pH 6.5–7.5 in ratios of 1:2, 1:1, or 2:1. Oblate ellipsoid-shaped crystals (Supplemental Fig. S1B) appeared after 2 weeks and grew to maximum dimensions of 300 × 300 × 200 µm3. Crystals were mechanically dislodged from the coverslip with a nylon loop prior to cryoprotection by addition of 1.4 M sodium citrate and 10% ethylene glycol to the mother liquor. For heavy-atom derivative preparation, crystals were transferred to cryoprotection solution in which 0.1 M HEPES-NaOH was substituted for 100 mM CsCl and incubated for 1 week. Data set 1 was collected from CsCl-treated crystals (Table 1). Crystals were also soaked similarly in a solution containing 10 mM cobalt hexammine and 40 mM spermidine for 1 week. Data set 2 was collected from such crystals (Table 1). For data collection, crystals were mounted on nylon loops, taking care to position the end of the crystal manipulated during coverslip dislodging at the loop tip, and flash frozen in liquid nitrogen. Diffraction data were collected in rotation mode at 100K at beamline 24-ID-C of the Advanced Photon Source, Argonne National Laboratory. Data were reduced with XDS (Kabsch 2010). Free-R flags from data set 1 were appended to data set 2 with resolution extension using CCP4 (Winn et al. 2011).
Structure determination and refinement
Six Cs+ sites were initially identified by Autosol in Phenix (Adams et al. 2010) in data set 1 (Table 1), yielding an initial mean figure of merit of 0.198. Despite the resolution limit, the experimental electron density maps were of excellent quality after density modification (Supplemental Fig. S1A) and allowed chain tracing in Coot (Emsley and Cowtan 2004), aided by RCrane (Keating and Pyle 2012). The nucleobases of weakly resolved residues in SL5b (U239, U240, and U243) were removed. Using secondary structure restraints, simulated annealing, conjugate gradient, and individual atomic B-factor refinement was performed, later with anomalous scattering factor refinement and TLS refinement, in Phenix. Aside from one additional Cs+ ion apparent from anomalous difference Fourier maps (its occupancy was set to 0.2), hydrated Mg2+ ions were placed in residual electron density features. For data set 2 (Table 1), the above structure was used as a search model for molecular replacement using Phaser (McCoy et al. 2007), yielding a single copy placed with an LLG of 2994 and TFZ of 49.6. No anomalous sites were apparent upon map inspection, and the strongest map features appeared to be due to other ions (and possibly a spermidine, which was not modeled). The model was refined as above, with simulated annealing during the first step, and without anomalous scattering factor refinement. Besides Cs+ ions, the structures are largely identical. Structural figures were prepared with PyMol (Schrödinger, LLC). Atomic coordinates and experimental structure factor amplitudes were deposited in the Protein Data Bank (Table 1). For calculating electrostatic potential using the Poisson–Boltzmann equation, the APBS web service (Baker et al. 2001; Jurrus et al. 2018) was used. The Cs+ derivative structure, with ions removed, was employed for the calculations, producing a map from −15 kBT/e− (red) to 0 kBT/e− (white) to +15 kBT/e− (blue) (the potential has no positive features).
CryoEM sample preparation, data collection, and data analysis
Grids purchased from Electron Microscopy Sciences were glow-discharged for 45 sec at 15 mAmp prior to use with a Pelco easiGlow (Ted Pella). Three microliters of 4 mg/mL RNA prepared as for crystallization were pipetted onto Quantifoil grids (R 1.2/1.3 Cu 300 mesh), which were plunge-frozen into liquid ethane using a Vitrobot (Thermo Fisher Scientific) at 4°C under 100% humidity, using a wait time of 15 sec, blotting force of 8, and blotting time of 5 sec. Prior to data collection, grids were screened for ice thickness and particle quality using a Glacios electron microscope with a Falcon 3 camera (Thermo Fisher Scientific). Movies were collected on a Krios microscope equipped with a Gatan BioQuantum K3 camera and imaging filter for over 3 days (Table 2). The data processing procedure is outlined in Supplemental Figure S2. Movies were initially analyzed in RELION 4.0 (Kimanius et al. 2021). After motion correction using MotionCor2 (Zheng et al. 2017) and contrast transfer function (CTF) correction using CTFFIND4 (Rohou and Grigorieff 2015), 9297 curated movies were taken for single-particle analysis. Topaz (Bepler et al. 2019) (trained on a previously collected Glacios data set) was employed for particle picking and identified 3.4 million (M) particles, which resulted in 1.23M particles after several rounds of 2D classification and 178 thousand (K) particles after further 3D classification using an ab initio model (two rounds) and 2D classification (one round) (Table 2; Supplemental Fig. S2). After Bayesian polishing and CTF refinement in RELION, as well as additional 2D classification to remove noisy particles (seven rounds), a final nonuniform refinement on 112K particles in cryoSPARC (Punjani et al. 2017) resulted in a map of ∼4.6 Å resolution, as judged by Fourier shell correlation of 0.143. The locally filtered map was created from this map (using a B-factor of 120). This map and the crystallographic model (Cs+ derivative) were aligned in Chimera (Fig. 3; Pettersen et al. 2004). No refinement of the model was performed against the cryoEM map. Three-dimensional variability analysis in cryoSPARC was conducted on a particle set of 128K particles prior to final rounds of 2D classification (Supplemental Fig. S2), using five frames and a 6 Å filter resolution. Uncurated micrographs and polished particle images were deposited (EMPIAR-12428).
In vitro translation assays
RRL was purchased from Green Hectares. Micrococcal nuclease-treated RRL was prepared as previously described (Feng and Shao 2018), aliquoted and frozen in liquid nitrogen, and stored at −80°C until further use. Aliquots were thawed and placed on ice until translation experiments were assembled by combining equal volumes of RRL and in vitro transcribed mRNAs (25 ng/µL final). Translation reaction volumes were 20 µL and incubated at 30°C for 90 min, prior to addition of 30 µL 1× lysis buffer from the Dual Luciferase Reporter Assay System (Promega). Technical 20 µL duplicates of this mixture were pipetted into white, µClear, flat-bottom, chimney 96 well microplates (Greiner). Upon addition of 50 µL of luciferase imaging buffer (LARII), per-well luminescence values were recording using a Tecan F200 plate reader. Duplicates from individual experiments were averaged and normalized using values obtained for WT mRNA luminescence within the same experiment (raw luminescence for uncapped and capped WT mRNA was 749,000 ± 90,000 units and 903,000 ± 98,000 units, respectively, n = 6 independent experiments). Raw luciferase values for capped mRNAs were 13% greater than uncapped mRNAs on average (P = 0.027). Means and standard deviations are reported for six independent experiments (Supplemental Table S1), and one- and two-tailed Student's t-tests were applied for significance.
Bulk FRET assay
Fluorescently labeled RNAs for FRET studies were produced by a three-piece ligation strategy using a 79-nt DNA splint (Supplemental Table S2, DNAsplint). In this strategy, a 40-nt Cy3-labeled 5′ segment spanning nts 171–210 was ligated to a 40-nt middle segment
spanning nts 211–250 and a 23-nt 3′ segment spanning nts 251–273 (Supplemental Table S2). A Cy3 dye was placed at a dT residue with a C6 linker at position C203 in all three RNAs, and the position of the Cy5 varied
in the other two segments. The middle segment was either unlabeled or Cy5-labeled using a C6 linker at position 241 in “Cy5
at C241” (Supplemental Table S1). The 3′ segment was either unlabeled or Cy5-labeled at the 3′ end in “Cy5 at 3′ end” (Supplemental Table S1). Ligated RNAs were gel purified using 8% denaturing polyacrylamide, electroeluted, and stored at −20°C until further use.
Twenty-five nanomolar ligated RNAs were refolded in the absence or presence of 10 mM MgCl2 prior to fluorescence measurements in black 384-well plates (Corning) using a CLARIOstar Plus (BMG Labtech). Spectra were
recorded by exciting at either 515 or 610 nm and recording emission from 550 to 740 nm or 615 to 740 nm, respectively, and
integrated in OriginPro 8.5 (OriginLab). EFRET was calculated as described (Clegg et al. 1992) using the extinction coefficients of Cy3 at 515 nm (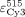 ) and Cy5 at 515 nm (
) and Cy5 at 515 nm (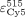 ) or 610 nm (
) or 610 nm (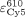 ), as well as the ratio r of the measured spectra:
), as well as the ratio r of the measured spectra:
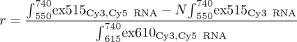
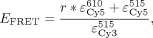 where integrals indicate the integrated fluorescence from spectra over given wavelengths, and the correction factor N is a ratio of the average intensity of the doubly labeled sample divided by the Cy3-only-labeled sample from 560 to 625 nm.
Six independent experiments were performed, and averages and standard deviations are reported in Supplemental Table S2. FRET distances were calculated from EFRET averages using an R0 of 60 Å.
where integrals indicate the integrated fluorescence from spectra over given wavelengths, and the correction factor N is a ratio of the average intensity of the doubly labeled sample divided by the Cy3-only-labeled sample from 560 to 625 nm.
Six independent experiments were performed, and averages and standard deviations are reported in Supplemental Table S2. FRET distances were calculated from EFRET averages using an R0 of 60 Å.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Drs. M. Banco, N. Demeshkina, Q. Elghondakly, L. Passalacqua, T. Dou, and J. Jiang for discussions. We thank Drs. J. Hogg and J. Chapman (National Heart, Lung and Blood Institute [NHLBI]) for use of the Tecan F200 plate reader. We thank Dr. R. Huang, A.J. Morton, and Z. Lang for help with cryoEM data collection, which used the National Cancer Institute/National Institutes of Health Intramural CryoEM Consortium Facility, and Dr. Ulrich Baxa for cryoEM screening, which used the National Institutes of Health (NIH) Multi-Institute Cryo-EM Facility. We thank Drs. S.-R. Shih, Y.-A. Kung, and K.-M. Lee for the plasmid pCov2-5UTR-Luc-3UTR. Diffraction experiments were conducted at the Northeastern Collaborative Access Team beamlines of the Advanced Photon Source (Argonne National Laboratory), a U.S. Department of Energy (DOE) Office of Science User Facility, and were funded by National Institute of General Medical Sciences of the National Institutes of Health (NIH) (P30 GM124165), NIH-ORIP HEI grant (S10OD021527), and contract DE-AC02-06CH11357. This work utilized the NIH Multi Institute CryoEM Facility. This work was supported by the NHLBI intramural program and the NIH Intramural Targeted Anti-COVID-19 (ITAC) Program of the National Institute of Allergy and Infectious Diseases. C.P.J. is the recipient of a K22 Career Transition Award from the NHLBI.
Author contributions: C.P.J. performed experiments and analyzed data, and C.P.J. and A.R.F.-D. wrote the paper.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080413.125.
- Received February 2, 2025.
- Accepted March 25, 2025.
This is a work of the US Government.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Christopher P. Jones is the first author of this paper, “Crystallographic and cryoEM analyses reveal SARS-CoV-2 SL5 is a mobile T-shaped four-way junction with deep pockets.” Chris is a postdoc in the lab of Adrian Ferre-D'Amare at the National Heart, Lung and Blood Institute. His recent research has focused on the roles of RNA structure in viruses.
What are the major results described in your paper and how do they impact this branch of the field?
Like many RNA viruses, SARS-CoV-2 uses highly conserved RNA motifs to regulate key steps in the viral life cycle. For example, in the 5′ untranslated region of the viral RNA, stem–loop 5 (SL5) overlaps with the start codon for ORF1, and the SL5 structure regulates translation of ORF1, the major SARS-CoV-2 polyprotein.
In this MS, X-ray crystallography and cryoEM are applied to solve structures of a portion of the SARS-CoV-2 viral RNA important for viral protein translation. With supporting experiments, the strengths of each approach reveal interactions within the viral RNA and suggest attractive pockets for small molecules.
What led you to study RNA or this aspect of RNA science?
From a cryoEM perspective, this ∼33-kDa RNA is small enough to be at the limit of current technology, meaning that resolution is limited. In contrast, the RNA is appropriate for crystallization, making it a suitable specimen for comparing and combining data from both techniques. In addition, this motif is derived from SARS-CoV-2, so choosing it for study sheds light on its viral function and potentially the development of RNA-targeted small molecules in the future.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
The most surprising results from structural studies were the lack of interactions within the RNA, a T-shaped four-way junction in which the helical elements point essentially in opposite directions. In particular, one helix contains a highly conserved GAAA tetraloop, a motif often involved in facilitating RNA–RNA interactions. Rather than forming such interactions, this GAAA tetraloop points into solution, raising the question of whether it is poised to contact another binding partner.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
My undergraduate research experience began in Ann Arbor in the summers. The goal was to test the oxidative effects of plant extracts on free radical production in the guts of caterpillars (which I raised, fed, and dissected). But for me, sitting in front of the EPR magnet was beyond cool. Since that time, training younger scientists in graduate school and at the NIH has been a joy. And of course, the science has been fun, too, especially when operating expensive, cutting-edge equipment.
If you were able to give one piece of advice to your younger self, what would that be?
I would take more breaks and do less bench work in favor of developing ideas and thinking deeply. I worry that this is getting harder and harder due to social media and attention deficit. I kept up with reading fiction and staying fit, but there were gaps (i.e., read and exercise more!).
Are there specific individuals or groups who have influenced your philosophy or approach to science?
The calm deliberative attitudes of my parents sent me down the math path, the largest influence on my approach to science. Growing up in a house of lawyers, surrounded by books, taught me to gather the facts and opinions and argue.
What are your subsequent near- or long-term career plans?
Due to the freeze on hiring at the NIH, I am likely out of a job by the time this is published. I may transition to industry or leave the field. It is impossible to make long-term career plans in science right now.








