
An MST-based assay reveals new binding preferences of IFIT1 for canonically and noncanonically capped RNAs
- Tomasz Spiewla1,2,
- Katarzyna Grab1,2,
- Anais Depaix3,
- Kamil Ziemkiewicz1,2,3,
- Marcin Warminski1,
- Jacek Jemielity3 and
- Joanna Kowalska1
- 1Division of Biophysics, Institute of Experimental Physics, Faculty of Physics, University of Warsaw, 02-093 Warsaw, Poland
- 2Doctoral School of Exact and Natural Sciences, University of Warsaw, 02-089 Warsaw, Poland
- 3Centre of New Technologies, University of Warsaw, 02-097 Warsaw, Poland
- Corresponding author: jkowalska{at}fuw.edu.pl
-
Handling editor: Eric Westhof
Abstract
IFITs (interferon-induced proteins with tetratricopeptide repeats) are components of the innate immune response that bind to viral and cellular RNA targets to inhibit translation and replication. The RNA target recognition is guided by molecular patterns, particularly at the RNA 5′ ends. IFIT1 preferably binds RNAs modified with the m7G cap-0 structure, while RNAs with cap-1 structure are recognized with lower affinity. Less is known about the propensity of IFIT1 to recognize noncanonical RNA 5′ ends, including hypermethylated and noncanonical RNA caps. Further insights into the structure-function relationship for IFIT1–RNA interactions are needed but require robust analytical methods. Here, we report a biophysical assay for quick, direct, in-solution affinity assessment of differently capped RNAs with IFIT1. The procedure, which relies on measuring microscale thermophoresis of fluorescently labeled protein as a function of increasing ligand concentration, is applicable to RNAs of various lengths and sequences without the need for their labeling or affinity tagging. Using the assay, we examined 13 canonically and noncanonically 5′-capped RNAs, revealing new binding preferences of IFIT1. The 5′ terminal m6A mark in the m7G cap had a protective function against IFIT1, which was additive with the effect observed for the 2′-O position (m6Am cap-1). In contrast, an increased affinity for IFIT1 was observed for several noncanonical caps, including trimethylguanosine, unmethylated (G), and flavin-adenine dinucleotide caps. The results suggest new potential cellular targets of IFIT1 and may contribute to broadening the knowledge of the innate immune response mechanisms and the more effective design of chemically modified mRNAs.
Keywords
INTRODUCTION
The innate immune response against foreign nucleic acids constitutes the initial cellular defense mechanism against bacteria and viruses (Kawasaki and Kawai 2019; Ghoreshi et al. 2022). To respond to viral infections, host cells need to identify the threat and activate efficient antiviral defense. This defense mechanism is set in motion in higher eukaryotes upon recognizing foreign pathogen-associated molecular patterns (PAMPs), such as unique viral RNA fragments, uncapped RNAs, and double-stranded RNAs. These viral PAMPs are identified by specific cellular pattern-recognition receptors (PRRs), such as Toll-like receptors (TLR3, TLR7, and TLR8) and RIG-I-like receptors (RLR: RIG-I and MDA-5), which are present both in endosomes and the cytoplasm, specifically recognizing foreign nucleic acids (Kumar et al. 2014; Dias et al. 2019; Rehwinkel and Gack 2020; Chen et al. 2021; Imaizumi et al. 2021). The interaction between these receptors and PAMPs triggers a signaling cascade resulting in the expression of type I antiviral interferons (IFN-α and IFN-β) (Franco et al. 2023). Secreted IFN-α and IFN-β then bind to type I IFN receptors (IFNARs), inducing the expression of numerous IFN-stimulated genes (ISGs) that encode proteins with regulatory and antiviral functions (Hyde et al. 2014; Pidugu et al. 2019a). Prominent members of this group are interferon-induced proteins with tetratricopeptide repeats (IFIT), which play pivotal roles in diverse biological mechanisms such as virus-induced translation inhibition, PAMP recognition, cell proliferation, replication, and signaling of double-stranded RNA (Fleith et al. 2018; Fitzgerald and Kagan 2020; Li and Wu 2021). IFIT proteins are immediate antiviral effectors during the innate immune response in humans and other mammals. While lacking enzymatic functions, they directly bind to non-self RNAs, preventing their translation and thereby inhibiting the production of viral proteins (Menachery et al. 2014; Hyde and Diamond 2015). IFIT1 can also selectively inhibit the translation of certain cellular mRNAs and modulate signaling pathways responsible for the production of immune mediators, further contributing to the antiviral response of the cell.
IFIT genes have been identified in numerous mammals, but the number and composition of the IFIT gene family can vary significantly from one species to another (Fensterl and Sen 2011). The human family consists of four canonical members, namely, IFIT1, IFIT2, IFIT3, and IFIT5, as well as uncharacterized IFIT1B and pseudogene IFIT1P1 (Fensterl and Sen 2015; Daugherty et al. 2016; Pidugu et al. 2019a). IFITs typically remain silent or are expressed at low levels under normal conditions. They are rapidly expressed upon viral infection, reaching concentrations 100–1000 times higher in the cytosol where they interact with foreign RNA (Fensterl and Sen 2015). IFITs consist of multiple repetitions of the tetratricopeptide repeat (TPR), which is a helix–turn–helix motif consisting of 34 amino acids. The TPR sequences are characterized by a degenerate pattern, with only nine residues at specific positions exhibiting limited conservation. Consequently, IFIT proteins display significant structural variability and different specificities toward RNA targets that are mostly guided by the structure of RNA 5′ terminus (Szretter et al. 2012). Both IFIT1 and IFIT5 directly bind to RNA–IFIT5 primarily recognizing uncapped RNA (i.e., 5′-triphosphorylated), while IFIT1 binds to RNAs carrying a cap-0 structure at the 5′ end (i.e., lacking the 2′-O methylation at the first transcribed nucleotide). The binding prevents the hijacking of the host's translational machinery by the virus and inhibits viral replication (John et al. 2017). The antiviral effect of IFIT1 is enhanced in the complex with IFIT3 (Abbas et al. 2013; Katibah et al. 2013; Johnson et al. 2018; Pidugu et al. 2019b). The RNA-binding site of IFIT1 is composed of a highly positively charged pocket with a distinguished 5′-end binding site, enabling the binding of various 5′-capped RNA ligands (Yang et al. 2012; Choi et al. 2018). While IFIT1 protein predominantly resides in the cytoplasm, ∼10%–15% of endogenous IFIT1 is present in the cell nucleus, with active shuttling of the protein occurring between the two compartments.
The IFIT1-capped RNA interaction has been already examined using various methods, such as primer extension inhibition (Fleith et al. 2018), electromobility shift assays (Habjan et al. 2013), surface plasmon resonance (Pichlmair et al. 2011), filter binding (Kumar et al. 2014), or biolayer interferometry (Miedziak et al. 2020). In all the studies reported so far, IFIT1 displayed a binding preference for cap-0 over cap-1 or uncapped RNAs (Abbas et al. 2017). These investigations consistently revealed a high affinity of IFIT1 for cap-0-RNA, with equilibrium dissociation constant (KD) values ranging from 23 to 175 nM, irrespective of the RNA's origin (viral or host) and sequence. Comparable studies have indicated that IFIT1 also binds to 5′-triphosphate and cap-1-RNAs, albeit with notably lower affinities (KD ∼250 nM to >1 μM and KD ∼450 to 710 nM, respectively) (Pichlmair et al. 2011; Kimura et al. 2013; Katibah et al. 2014; Miedziak et al. 2020). As concluded from primer extension assays, the critical elements for IFIT1 binding include the 5′ terminal mRNA cap residue and at least 4–5 nt downstream from the cap (Chung et al. 1994; Kumar et al. 2014). These additional nucleotides likely enhance the stability of IFIT1–RNA complex, blocking the recruitment of additional ligands to the binding pocket or eliminating nonspecific interactions by inducing conformational changes in the protein (Zhang et al. 2014). Significantly less is known about the affinity of IFIT1 for other cap structures, such as the noncanonical caps (NNCs) found on some endogenous, viral, and bacterial RNAs or chemically modified caps that are being developed for modification of in vitro transcribed mRNAs for therapeutic purposes (Warminski et al. 2023). We envisaged that a simple, easily accessible, material- and time-saving method for quantitative assessment of cap-IFIT would greatly expedite studies in this direction.
As such, we report here a simple, rapid, and cost-effective assay that enables the determination of the binding affinity of variously capped RNAs for IFIT1 based on the microscale thermophoresis (MST) phenomenon (Duhr and Braun 2006; Jerabek-Willemsen et al. 2014; Scheuermann et al. 2016; Tso et al. 2018). The assay enables precise measurement of KD values between fluorescently labeled IFIT1 and RNAs of various lengths. Utilizing this assay, we evaluated a library of 13 RNAs containing different 5′-terminal structures (Fig. 1). Our exploration included back-to-back comparison of all four 7-methylguanosine (m7G) cap-0 variants incorporating adenine, guanine, cytosine, and uridine as the 5′-terminal nucleobase, and some methylated derivatives commonly found in human mRNA, such as 2′-O-methyl and N6-methyl on adenosine as the 5′-terminal nucleoside. We also investigated several NNCs, including nicotinamide-adenine dinucleotide (NAD) found on some RNA across all domains of life, an unmethylated “G” cap that is associated with aberrant mRNA capping, hypermethylated trimethylguanosine (TMG) cap present in snRNAs but also found on some Rev/RRE-dependent HIV-1 RNAs (Chen et al. 2009; Yedavalli and Jeang 2010; Julius and Yuzenkova 2017), UDP-Glucose cap, which is a relatively abundant noncanonical RNA cap, and flavin-adenine dinucleotide cap (FAD), which has been recently identified as the major molecule modifying the 5′ end of HCV RNA (Fig. 1; Wang et al. 2019; Sherwood et al. 2023). Moreover, we also investigated if trinucleotide ligands can provide similar quantitative information regarding cap-IFIT interaction as the long-capped RNAs. The results revealed new 5′-terminal moieties that unexpectedly stabilize the interaction of capped RNA with human IFIT1, highlighting new potential cellular targets and underscoring the need for further investigation of the RNA cap-related aspects of the innate immune response.
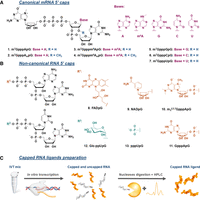
Structures of canonical and noncanonical RNA caps studied in this work. Structures of the (A) canonical and (B) noncanonical RNA 5′ cap analogs used for RNA preparation by IVT. (C) The workflow for RNA preparation and purification leading to highly homogenous 5′ capped RNA.
RESULTS AND DISCUSSION
An MST-based direct binding assay enables determining the affinity of IFIT1 for long-capped RNAs
To investigate the interaction of various 5′ RNA cap analogs and capped RNAs with the IFIT1 protein in a reproducible and time-effective manner, we developed an MST-based direct binding assay. MST is a biophysical method relying on the measurement of two factors: directed molecule movement in a temperature gradient (thermophoresis) and change in fluorescence intensity upon changing temperature (TRIC) (Magnez et al. 2022). The thermophoretic mobility of molecules is sensitive to small changes in the charge, size, and hydration shell caused by interactions with other compounds, making MST suitable for studying protein–ligand interactions across a wide range of ligand sizes. This, combined with a wide working range of measurable KD values, straightforward optimization, and low sample consumption, led us to choosing this method. In our setup, IFIT1 fluorescently labeled with a commercially available red dye via the protein's N-terminal His-tag was used to measure ligand concentration-dependent changes in the thermophoretic mobility of the protein. IFIT1 labeling was carried out using standard methods, and the labeled protein showed good stability in aqueous buffers as shown by DSF measurements (Supplemental Fig. S3). DSF was also used to verify how the addition of N-terminal His-tag affects protein stability and ligand recognition. The IFIT1 protein exhibited very similar stability both with and without the N-terminal His-tag. A similar increase in stability was observed for both proteins in the presence of cap-0-RNA, and was not observed in the presence of cap-1-RNA (Supplemental Fig. S4), indicating that the addition of N-terminal His-tag does not affect protein folding nor ligand-recognition capabilities significantly. To enable the synthesis of variously capped mRNAs, we generated a library of 13 different cap analogs, including cap-0, cap-1, and their further methylated analogs, as well as several noncanonical RNA 5′ ends (Fig. 1). The set contained both 5′ capped RNA that were previously evaluated against IFIT as well as the unstudied ones, which allowed us to compare our newly developed method with literature data and gain new insights into RNA 5′ ends recognition by IFIT1. The workflow of the assay is shown in Figure 2.

Workflow for the determination of KD values for the IFIT1-capped RNA70 interaction using an MST-based binding assay. Fluorescently labeled IFIT1 was mixed with RNA ligand at 16 different concentrations, equilibrated for 15 min, followed by the analysis by MST. The observed changes in fluorescence intensity were plotted as a function of ligand concentration and a theoretical binding curve was fitted to determine the dissociation constants (KD).
To find a suitable RNA model to compare the 5′ cap variants, we first explored four different RNA sequences as IFIT1 ligands, including two 35 nt RNAs differing in the secondary structure, a 70 nt fragment of human β-globin protein, and a 240 nt mRNA encoding V5x3 tag (Supplemental Table S1; Ranawakage et al. 2019). For each sequence variant, RNAs capped with m7GpppApG (cap-0) or m7GpppAmpG (cap-1) were synthesized by an in vitro transcription (IVT) reaction. To ensure a high purity of the analyzed RNA samples, uncapped RNA molecules were removed by enzymatic digestion, and the remaining material was purified by RP-HPLC. Since the preliminary analyses indicated significant differences in the affinity of RNA ligands for IFIT1 compared to the corresponding trinucleotide cap analogs, the ionic strength of the buffers for both types of ligands were differentiated to move the KD values toward the higher end of the range (Supplemental Fig. S3; Table 1). In each case, the MST analysis (Fig. 3; Tables 1 and 2) showed binding curves consistent with a 1:1 binding model (Fig. 3).
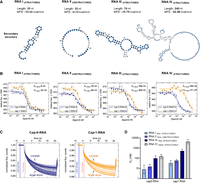
Determining KD values for four different sequence variants of cap-0 and cap-1 RNA using the MST method. (A) Predicted secondary structures of the analyzed RNA sequences (model Vienna RNAfold) and their MFE (minimum free energies). (B) The determined MST-binding curves from triplicate experiments (data points represent mean values ± SD). Binding curves are best fits for 1:1 binding model. (C) Representative MST traces for two of the curves shown in B (RNA III). Vertical marks (blue and red highlights) indicate cold and hot regions that were used for further analysis (Tjump). (D) Comparison of the determined KD values (mean values from triplicate experiments with confidence intervals).
Binding affinities of IFIT1 determined by MST for cap-0 and cap-1 structures depending on the RNA sequence
The cap-1:cap-0 KD values ratio for all tested sequence variants
The RNA I sequence forming a hairpin structure (Fig. 3A) bound to IFIT1 with KD values of 8 nM for cap-0 and 34 nM for cap-1. The RNA II (unstructured, 35 nt) that was designed to avoid formation of any secondary structures bound to IFIT1 with the slightly higher KD values: 15 nM for cap-0- and 60 nM for cap-1. For the third sequence variant, RNA III (structured, 70 nt), we observed notably weaker binding than for shorter RNAs, with KD values of 100 nM for cap-0 and 520 nM for cap-1. The RNA IV represented a model mRNA encoding a V5x3 tag (structured; 240 nt) and was used to verify if the method is suitable for measurements of functional mRNA molecules. For this RNA, even weaker binding was observed compared to shorter ligands, with KD values of 150 nM for cap-0- and 4.1 µM for cap-1. For shorter ligands, the affinity of IFIT1 for cap-0-RNAs was 4–5 higher than for RNAs with cap-1, which is consistent with previous results, and indirectly confirms that the differentiation of 5′-terminal structures by IFIT1 is RNA sequence independent. For the longer RNA ligand (RNA IV) the difference between cap-0 and cap-1 structures was more pronounced (∼30 fold), suggesting a potential participation of full-length mRNA body in ligand recognition.
The differences in the affinity exhibited by diverse RNA sequences may be attributed to electrostatic interactions between the RNA body and protein surface. RNA III was selected as the most suitable model for evaluating the influence of differently modified cap structures on RNA affinity for IFIT1 due to its reasonable length and KD values for both ligands well within the working range of the method.
Development of a direct binding assay for the evaluation of the IFIT1-capped RNA70 interactions
Next, we prepared a set of 11 additional variants of RNA III differing in the 5′ terminal modifications and determined their binding affinities for IFIT1 (Table 3). These variants included four additional cap-0 variants differing in the first transcribed nucleobase (G, C, U, m6A), cap-1 variant containing m6Am mark, and six noncanonical 5′ ends including G-cap, NAD, FAD, TMG, UDP-glucose, and UTP.
The KD values of binding between IFIT1 and 5′ RNA cap analogs or 5′ capped RNAs
The comparison of affinities for RNAs carrying different cap-0 variants (A, G, C, U, m6A) revealed a mild preference for A and U at the position of the first transcribed nucleotide (Table 3), albeit these differences cannot be considered statistically significant (note that throughout this discussion only values with nonoverlapping confidence intervals [P ≦ 0.32] will be considered as relevant differences). The methylation of cap-0 at the N6 position of adenosine (m7Gpppm6ApG-RNA) lowered the affinity of RNA for IFIT1, but not as much as methylation at the 2′-O position (the KD value of m7Gpppm6ApG-RNA was 2.3-fold higher than that of m7GpppApG-RNA but 2.2-fold lower than that for m7GpppAmpG-RNA). Notably, the effects of these two methylations were additive since the RNA probe carrying m7Gpppm6AmpG cap had ninefold lower affinity compared to cap-0 RNA ligand. This suggests that the N6-methylation of the 5′-terminal adenosine constitutes an additional layer of protection from RNA sequestering by IFIT1.
The investigated NNCs had a very variable effect on IFIT1–RNA interaction. The KD values determined for noncanonically capped RNAs showed broad affinity spectrum that extended over three orders of magnitude
(Table 3). FADpG-RNA bound to IFIT1 with affinity comparable to cap-0 RNA (KD 80 nM). GpppApG-RNA, an intermediate during cap biosynthesis (Shannon et al. 2022), was bound by IFIT1 even more avidly than cap-0-RNA (KD 11 nM). Surprisingly, the RNA containing hypermethylated m7G moiety, that is, the so-called trimethylguanosine cap (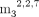 GpppApG-RNA), also had a very high affinity for IFIT1 (KD 23 nM). NADpG-RNA had a significantly lower affinity (KD 1.0 µM), and Glucose-ppUpG-RNA was a very weak binder (KD 8.2 µM), much weaker than the corresponding triphosphate RNA (KD 270 nM). Overall, these results may indicate that IFIT1 is capable of targeting more RNA 5′ terminal structural patterns
than previously anticipated, and that some noncanonical 5′ end modifications may protect RNA from binding by IFIT1.
GpppApG-RNA), also had a very high affinity for IFIT1 (KD 23 nM). NADpG-RNA had a significantly lower affinity (KD 1.0 µM), and Glucose-ppUpG-RNA was a very weak binder (KD 8.2 µM), much weaker than the corresponding triphosphate RNA (KD 270 nM). Overall, these results may indicate that IFIT1 is capable of targeting more RNA 5′ terminal structural patterns
than previously anticipated, and that some noncanonical 5′ end modifications may protect RNA from binding by IFIT1.
Finally, we also wanted to test if the use of short, trinucleotide cap analogs as IFIT1 ligands would provide meaningful data
on the relative affinities of different cap variants. This could significantly reduce the time and the total cost of the analysis.
The KD values determined for m7GpppApG (cap-0) and m7GpppAmpG (cap-1) suggested that IFIT is able to differentiate caps even more strongly if the RNA body is not present (Tables 1–3). However, a back-to-back comparison of all KD values determined for the capped RNA ligands and free caps (Table 3) suggests that the measurements for free cap analogs do not provide reliable quantitative data, albeit to some extent are
in qualitative agreement with the data obtained for RNA ligands. High binding affinity was observed for some particularly
strongly interacting RNA 5′ ends, even for “free” caps (m7GpppApG, 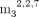 GpppApG). This may suggest that these structures specifically interact with IFIT1 and are particularly significant as its
cellular targets.
GpppApG). This may suggest that these structures specifically interact with IFIT1 and are particularly significant as its
cellular targets.
Conclusions
The continuous increase in the number of potential applications of RNA molecules highlights the need for the development of better methods to understand the structure–function relationships and biological effects for various RNA modifications. We have developed a robust and reproducible assay to evaluate the affinity of differently capped RNAs for IFIT1. The assay, which relies on measuring MST of fluorescently labeled protein in the presence of increasing ligand concentrations, enables more efficient analysis of IFIT1 RNA interactions compared to previously developed methods due to its low sample consumption, simple workflow, and easily quantifiable results. We have demonstrated that the assay is suitable for analysis of 35–240 nt long RNA ligands, including full-length mRNAs using HPLC-purified model RNAs. While for all studied RNA lengths, we saw differences between cap-0 and cap-1 RNAs, the absolute KD values strongly depended on RNA length, which underlined the importance of using high-quality, homogenous RNA samples for these types of measurements. We also showed that the analysis of small molecule cap analogs is technically possible using the MST assay, yet it does not deliver results that can be easily translated into properties of longer RNAs. Still, some qualitative trends in the structure–affinity relationship observed for RNA ligands are noticeable also for trinucleotide cap analogs, which suggests that the assay may be applicable for discovering high-affinity small molecule ligands of IFIT1 if desired.
We used the developed assay to back-to-back compare a set of 13 RNA ligands carrying different natural canonical and NNC structures. To our knowledge, this is the first such broad comparison of differently capped RNAs in the context of interactions with IFIT1, and many cap modification studies here have not been previously investigated (Table 3 and Figure 4). Starting with the reference molecules, the affinity for cap-0 was about five times higher than for the cap-1 molecule. All four tested cap-0 RNAs showed comparably strong binding to IFIT1 in the KD range of 100–300 nM. Comparing unmethylated m7GpppApG (cap-0) and m7Gpppm6ApG RNA ligands, we observed a tendency for decreased affinity with the methylated analog. However, given the overlapping confidence intervals for both ligands (Table 3), it is not possible to definitively conclude a significant decrease in affinity due to the m6A modification alone. The effect becomes more pronounced when combined with additional 2′-O-methylation, which suggests a general trend of weakening IFIT1 protein interaction with 5′ RNA structures that carry additional methylations on the first transcribed nucleotide. For the m7G modifications, both the deletion of methylation from m7G (GpppApG) and hypermethylation (TMGpG) resulted in increased affinity toward IFIT1. Although both these structures are formed during biosynthesis or maturation of some endogenous RNAs (Terns and Dahlberg 1994; Wurth et al. 2014), their exceptionally high affinity of IFIT1 for this hypermethylated structure implies that they may be another class of cellular IFIT1 targets.
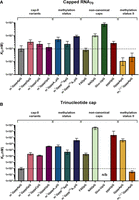
Comparison of KD values for different IFIT1 ligands. (A) Capped 70 nt RNAs. (B) Free cap structures. Data shown are mean values from minimum three replicates with confidence intervals. (n/b) No binding was observed at the studied concentration range.
The analysis of NNC structures revealed very high divergence in affinity. The FAD-capped RNA ligand strongly binds to IFIT1, while NAD-capped RNA binds more than 10-fold weaker, and the UDP-glucose-capped RNA is hardly recognized by IFIT1. The UDP-glucose moiety has a clearly “protective” effect against IFIT1, as it has an affinity lower than the corresponding uncapped RNA.
Interestingly, all structures that have been identified in this study as tightly binding to IFIT1 have been previously somehow linked to viral RNA. Since mRNA is capped in the nucleus, GpppApG-RNA is normally absent in the cytoplasm but is formed there during some viral RNA capping pathways. Besides its presence on endogenous RNAs, TMG cap has also been identified on some HIV-associated RNAs (Qiu et al. 2011; Boris-Lawrie et al. 2022). The 5′-terminal FAD cap has been found to modify ∼75% of HCV RNAs (Sherwood et al. 2023). The fact that our in vitro assay revealed comparably high or even slightly higher affinity of these three molecular patterns to cap-0 RNA may suggest that they are also targeted by IFIT1. The biological relevance (if it exists) of noncanonical modifications that had significant protective effects against IFIT1, with UDP-glucose being the most striking example, needs to be verified, but it may also guide future design of noncoding RNAs with therapeutic function.
The use of mRNA in therapies offers numerous advantages over conventional methods, as evidenced by the rapid development of mRNA technology in recent years (Xu et al. 2020; Huang et al. 2022; Yao et al. 2024). However, the administration of exogenous mRNA to patients carries the risk of triggering an innate immune response against foreign RNA (Schlee and Hartmann 2016). The innate immune response executed by IFIT1 can efficiently and selectively influence the translation by shutting down selected viral and cellular mRNAs and inhibiting the amplification of foreign genetic material. However, these same mechanisms may reduce the efficacy of mRNA-based therapeutics (Andrejeva et al. 2013). The influencing factor in the occurrence of such adverse effects is the recognition of therapeutic mRNA molecules by IFIT1 (Abbas et al. 2017; Pingale et al. 2019; Russ et al. 2022). The incorporation of cap-1 in therapeutic mRNAs is anticipated to mitigate the risk of an adverse immune response while concurrently reducing susceptibility to translational inhibition mediated by IFIT1 (Pinto et al. 2015; Fleith et al. 2018; Johnson et al. 2018). Recent discoveries increasingly underscore the importance of chemical modifications of RNA in modulating the innate immune response (Tong et al. 2022). The use of improved mRNA molecules in therapy has the potential to enhance the translation of the target protein, expediting therapeutic effects (Baptista et al. 2021). We hypothesize that the design of chemically modified mRNA cap structures that can evade sequestering IFIT1 may additionally benefit the therapeutic mRNA field. We hope that the assay reported will become an essential tool for verifying this hypothesis as well.
MATERIALS AND METHODS
RNA cap analogs
Canonical 5′-cap analogs (m7GpppApG, m7GpppAmpG, m7GpppGpG, m7GpppCpG, m7GpppUpG, m7Gpppm6ApG, m7Gpppm6AmpG) were synthesized as previously described (Sikorski et al. 2020). Noncanonical 5′-cap analogs (NADpG, FADpG, Glucose-ppUpG) and uncapped pppUpG were synthesized as previously described
(Depaix et al. 2022). 5′-cap analogs with modification in m7G structure (GpppApG and 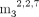 GpppApG) were synthesized as previously described (Grab et al. 2024).
GpppApG) were synthesized as previously described (Grab et al. 2024).
Synthesis of capped RNA
The capped RNA ligands used in the study were synthesized by in vitro transcription (IVT) on the DNA template containing the
T7 promoter Φ6.5 (TAATACGACTCACTATAGGG). A typical transcription reaction (100 µL) was conducted for 1 h at 37°C and contained
the concentrations of nucleotides (NTPs, Thermo Fisher Scientific) and cap analogs as specified below. Transcription conditions
for each class of cap analogs were optimized to ensure high incorporation of the analog at the RNA 5′ end (capping efficiency).
For m7GpppApG, m7GpppAmpG, m7Gpppm6ApG, and m7Gpppm6AmpG, the transcription mixture contained 5 mM concentrations of UTP/ATP/CTP, 4 mM GTP, and 10 mM trinucleotide cap analog.
For m7GpppGpG, m7GpppCpG and m7GpppUpG, the transcription mixture contained 4 mM UTP/ATP/CTP, 2 mM GTP, and 8 mM trinucleotide cap analog. For all other
analogs (NADpG, FADpG, Glc-ppUpG, 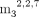 GpppApG, GpppApG and pppUpG), the transcription mixtures contained 4 mM UTP/ATP/CTP, 2 mM GTP and 12 mM trinucleotide/dinucleotide
analog. The reaction mixtures were supplied with RNA polymerase buffer (Thermo Fisher Scientific), 20 mM MgCl2, 1U/µL RiboLock RNase Inhibitor (Thermo Fisher Scientific), 0.002 U/µL inorganic pyrophosphatase (Thermo Fisher Scientific),
T7 RNA polymerase (20 U/µL, Thermo Fisher Scientific), and 40 ng/µL annealed oligonucleotides (GenoMed) as a DNA template.
Four different sequences were used for RNA sequence optimization (Supplemental Table S1). After 1 h incubation of IVT mix, 7 U of DNase I (Thermo Fisher Scientific) was added, followed by incubation for another
30 min at 37°C. To stop the reaction, 10 µL of 500 mM aqueous Na2EDTA solution (equimolar amount of EDTA respective to Mg2+) was added. The crude RNA was preliminarily purified using Monarch RNA Clean-up kit (New England Biolabs), according to the
manufacturer's protocol. Then a two-step enzymatic digestion using 5′ polyphosphatase (Lucigen) and Xrn-1 (New England Biolabs)
enzymes was applied to all RNAs except pppUpG-RNA to remove uncapped RNA. The content (in µg) of uncapped RNA was estimated
based on the amount of RNA material and the capping efficiency of the individual analogs. In the first step, the reaction
with 5′ polyphosphatase (100 µL) was carried out for 1 h at 37°C and contained: digested RNA, 1 U/µL RiboLock RNase Inhibitor
(Thermo Fisher Scientific), 5′ polyphosphatase buffer (Lucigen), and 5′ polyphosphatase (Lucigen), using 1 µL of enzyme per
5 µg of uncapped RNA. RNA samples were again purified with Monarch RNA Clean-up (New England Biolabs). In the second step,
the reaction with Xrn-1 (200 µL) was carried out for 2 h at 37°C and contained digested RNA, 1 U/µL RiboLock RNase Inhibitor
(Thermo Fisher Scientific), Xrn-1 buffer (New England Biolabs) and Xrn-1 (New England Biolabs), using 1 µL of enzyme per 1
µg of uncapped RNA. The samples were then purified again with Monarch RNA Clean-Up (New England Biolabs). Finally, all RNA
samples were purified using HPLC and Phenomenex Clarity 3 µM Oligo-RP column 150 × 4.6 mm (using method: A: 100 mM TEAAc,
B: 200 mM TEAAc/ACN; 10%–35% [v/v] of eluent B in 32 min), precipitated as sodium salts (3 M NaOAc pH 5.2, isopropanol) and
dissolved in water. The micromolar concentrations of the RNA samples were determined using NanoDrop (Thermo Fisher Scientific).
GpppApG, GpppApG and pppUpG), the transcription mixtures contained 4 mM UTP/ATP/CTP, 2 mM GTP and 12 mM trinucleotide/dinucleotide
analog. The reaction mixtures were supplied with RNA polymerase buffer (Thermo Fisher Scientific), 20 mM MgCl2, 1U/µL RiboLock RNase Inhibitor (Thermo Fisher Scientific), 0.002 U/µL inorganic pyrophosphatase (Thermo Fisher Scientific),
T7 RNA polymerase (20 U/µL, Thermo Fisher Scientific), and 40 ng/µL annealed oligonucleotides (GenoMed) as a DNA template.
Four different sequences were used for RNA sequence optimization (Supplemental Table S1). After 1 h incubation of IVT mix, 7 U of DNase I (Thermo Fisher Scientific) was added, followed by incubation for another
30 min at 37°C. To stop the reaction, 10 µL of 500 mM aqueous Na2EDTA solution (equimolar amount of EDTA respective to Mg2+) was added. The crude RNA was preliminarily purified using Monarch RNA Clean-up kit (New England Biolabs), according to the
manufacturer's protocol. Then a two-step enzymatic digestion using 5′ polyphosphatase (Lucigen) and Xrn-1 (New England Biolabs)
enzymes was applied to all RNAs except pppUpG-RNA to remove uncapped RNA. The content (in µg) of uncapped RNA was estimated
based on the amount of RNA material and the capping efficiency of the individual analogs. In the first step, the reaction
with 5′ polyphosphatase (100 µL) was carried out for 1 h at 37°C and contained: digested RNA, 1 U/µL RiboLock RNase Inhibitor
(Thermo Fisher Scientific), 5′ polyphosphatase buffer (Lucigen), and 5′ polyphosphatase (Lucigen), using 1 µL of enzyme per
5 µg of uncapped RNA. RNA samples were again purified with Monarch RNA Clean-up (New England Biolabs). In the second step,
the reaction with Xrn-1 (200 µL) was carried out for 2 h at 37°C and contained digested RNA, 1 U/µL RiboLock RNase Inhibitor
(Thermo Fisher Scientific), Xrn-1 buffer (New England Biolabs) and Xrn-1 (New England Biolabs), using 1 µL of enzyme per 1
µg of uncapped RNA. The samples were then purified again with Monarch RNA Clean-Up (New England Biolabs). Finally, all RNA
samples were purified using HPLC and Phenomenex Clarity 3 µM Oligo-RP column 150 × 4.6 mm (using method: A: 100 mM TEAAc,
B: 200 mM TEAAc/ACN; 10%–35% [v/v] of eluent B in 32 min), precipitated as sodium salts (3 M NaOAc pH 5.2, isopropanol) and
dissolved in water. The micromolar concentrations of the RNA samples were determined using NanoDrop (Thermo Fisher Scientific).
Expression and purification of IFIT1
Human interferon-induced protein with tetratricopeptide repeats 1 (IFIT1) gene (Gene ID: 3434) was obtained from Addgene in the plasmid vector pET28a_IFIT1, dedicated to protein expression. The hIFIT1 protein (∼55 kDa) with 6× histidine at the N terminus (His-Tag) was overexpressed in the BL21 (DE3) RIL Escherichia coli (Invitrogene) procaryotic expression system in LB medium supplemented with kanamycin (30 mg/mL). Cells were grown to the optical density OD600∼0.7 at 37°C. Then, the temperature was adjusted to 18°C and expression was induced with 0.5 mM isopropyl b-D-1-thiogalactopyranoside (IPTG), and the cells were further cultured for 16 h. Bacterial cultures were harvested by centrifugation and lysed in a buffer containing 20 mM HEPES (pH 7.5), 250 mM NaCl, 20 mM imidazole, 5 mM β-Mercaptoethanol (2-ME), 5% glycerol, 1 mg/mL of lysozyme and mixture of protease inhibitors (Aprotinin, Pepstatin, PMSF, Leupeptin). The lysate was sonicated (15 min, amplitude 50%, 15 sec on/off) and centrifuged. The supernatant was then purified by immobilized metal affinity chromatography (IMAC). The protein solution was loaded on a 2 × 5 mL HisTrap FF column (Cytiva) previously equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 250 mM NaCl, 20 mM imidazole, 5 mM β-Mercaptoethanol (2-ME), and 10% (v/v) glycerol. The His6-IFIT1 protein was washed with a buffer containing 1 M NaCl and eluted with buffer: 20 mM HEPES (pH 7.5), 250 mM NaCl, 300 mM imidazole, 5 mM β-Mercaptoethanol, and 10% glycerol. Affinity chromatography on a heparin column was then used to remove the remaining endogenous nucleic acids and some protein contaminants from the preparation (after IMAC). The protein fractions collected were diluted with 20 mM HEPES (pH 7.5), 1 mM DTT to a final salt concentration of ∼100 mM NaCl and loaded onto a 5 mL HiTrap Heparin HP column (Cytiva) previously equilibrated with a buffer containing 20 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM DTT (phase A). The method was carried out with a constant increasing proportion of phase B at 1%/min from 0 to 100% (v/v); phase B: 20 mM HEPES, 1000 mM NaCl, 1 mM DTT. The final step in protein purification was gel filtration. The eluted fractions were loaded onto a 75 pg HiLoad 26/600 gel filtration column (Cytiva). Samples containing IFIT1 were centrifuged (10 min, 10,000 rcf, 4°C) to remove any aggregates, concentrated to a final concentration of 12 μM, flash frozen, and stored at −80°C in a buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM DTT, 10% (v/v) glycerol. The nucleotide sequence of His6-IFIT1 and MS analysis are included in Supplemental Figure S1.
Protein labeling
The stock solution of His6-IFIT1 (12 µM) was centrifuged (10 min, 10,000 rcf, 4°C) and diluted to 200 nM in MST-buffer containing 20 mM HEPES (pH 7.5), NaCl, 0.075% Pluronic F-127. The buffer (buffer L1) contained 100 mM NaCl for cap analog ligands, whereas for capped RNA ligands—200 mM NaCl (buffer L2). The RED-tris-NTA dye (NT-647, Nano Temper Technologies) was diluted in the same buffer to 100 nM. Protein and dye were mixed in a 1:1 volume ratio and incubated for 30 min at room temperature. Before the MST measurements, every sample of the labeled protein was centrifuged to remove potential aggregates (10 min, 10,000 rcf, 4°C). A dye affinity assay was performed using RED-tris-NTA dye to assess its binding affinity to IFIT1 protein. The dye was titrated with IFIT1 protein following the manufacturer's recommendations (NanoTemper).
Optimization of conditions
Several optimization measurements were performed for labeled IFIT1 and ligands. Three types of buffers were used for this purpose: MST-buffer, 1× PBS, and 20 mM HEPES. To avoid aggregation and suppress unspecific protein adsorption onto capillary walls, we tested two different detergents: Tween-20 and Pluronic-F127 at concentrations of 0.01–0.1% (v/v). The use of BSA (0.1 mg/mL) instead of detergent was also explored. The effect of ionic strength was tested at three concentrations of NaCl (100 mM, 200 mM, and 300 mM) in three types of buffers above. Furthermore, three different pH buffers were also tested: (pH 7, pH 7.5, and pH 8). Finally, 20 mM HEPES pH 7.5 buffer supplemented with 0.075% Pluronic F-127 and 100 mM or 200 mM NaCl (for free cap analogs and RNA ligands, respectively) was chosen. Under these conditions, the determined dissociation constant (KD) for the protein–dye interaction was 13 nM, indicating a suitably high affinity of the N-terminal His6-IFIT1 protein for the fluorescent dye (Supplemental Fig. S2A). We also confirmed lack of interactions between the free dye and ligand RNA (Fig. 2B) The stability of the protein in measuring buffers analyzed by nanoDSF (Supplemental Figs. S3 and S4) revealed that the labeled protein was stable; no aggregation, no adsorption, no photobleaching, nor photoenhancement were observed (Supplemental Figs. S3–S5).
Microscale thermophoresis
For all thermophoretic measurements, MST Premium coated capillaries (Nano Temper Technologies) were used. The final concentration of His-tag-labeled IFIT1 was set to 50 nM. Sixteen point half-log serial dilutions of ligands at different concentration ranges were prepared. The concentration ranges were adjusted for each analog to cover a minimum 80% of the binding curve. The final samples were prepared by mixing an equal volume (10 µL) of 100 nM His-tag labeled IFIT1 with the ligand at a specified concentration. For RNA cap analogs, measurements were performed in buffer L1 (20 mM HEPES [pH 7.5], 100 mM NaCl, 0.075% Pluronic F-127) at ligand concentrations typically ranging from 10 mM to 305 nM. For capped RNA, measurements were performed in buffer L2 (20 mM HEPES [pH 7.5], 200 mM NaCl, 0.075% Pluronic F-127) and with ligand concentrations ranging from 20 µM to 610 pM.
Measurements of IFIT1–ligand interaction were performed on a Nano Temper Monolith NT.115 instrument. Before MST measurement, samples were equilibrated for 15 min at room temperature in darkness, then loaded into capillaries, and inserted into the data collection instrument (with a temperature set at 25°C). The measurement of each ligand was performed in triplicate. The final RED-tris-NTA dye concentration of 50 nM yielded the fluorescence intensity of labeled IFIT1 around 600 counts at a LED power of 40%. The samples were measured at medium MST power with a pre-MST period of 5 sec, a laser-on time period of 30 sec, and a laser-off time period of 5 sec.
Data analysis
The data (time-traces) were loaded into PALMIST v1.5.8 software and evaluated using a simple 1:1 binding model. For every measurement, the relative fluorescence value was determined by a rapid change in normalized fluorescence in phase II of thermophoresis caused by the temperature dependence of fluorescence (T-jump preset). Triplicates for given measurement points were averaged, but a weighted fitting was not used. The fluorescence values were normalized, and a logarithmic concentration scale was applied. The plots were rendered using the GUSSI v1.4.2 program.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the National Science Centre, Poland (NCN) (UMO-2018/31/B/ST5/03821 to J.K.).
Author contributions: T.S. and J.K. designed the study. J.J. and J.K. supervised the study. T.S. performed experiments. A.D., K.G., K.Z., and M.W. provided resources. T.S. and J.K. wrote the first draft of the manuscript. The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080089.124.
- Received May 10, 2024.
- Accepted October 22, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








