
Contribution of an alternative 16S rRNA helix to biogenesis of the 30S subunit of the ribosome
- Department of Microbiology and Center for RNA Biology, The Ohio State University, Columbus, Ohio 43210, USA
- Corresponding author: fredrick.5{at}osu.edu
-
Handling editor: Marina Rodnina
Abstract
30S subunits become inactive upon exposure to low Mg2+ concentration, because of a reversible conformational change that entails nucleotides (nt) in the neck helix (h28) and 3′ tail of 16S rRNA. This active-to-inactive transition involves partial unwinding of h28 and repairing of nt 921–923 with nt 1532–1534, which requires flipping of the 3′ tail by ∼180°. Growing evidence suggests that immature 30S particles adopt the inactive conformation in the cell, and transition to the active state occurs at a late stage of maturation. Here, we target nucleotides that form the alternative helix (hALT) of the inactive state. Using an orthogonal ribosome system, we find that disruption of hALT decreases translation activity in the cell modestly, by approximately twofold, without compromising ribosome fidelity. Ribosomes carrying substitutions at positions 1532–1533 support the growth of Escherichia coli strain Δ7 prrn (which carries a single rRNA operon), albeit at rates 10%–20% slower than wild-type ribosomes. These mutant Δ7 prrn strains accumulate free 30S particles and precursor 17S rRNA, indicative of biogenesis defects. Analysis of purified control and mutant subunits suggests that hALT stabilizes the inactive state by 1.2 kcal/mol with little-to-no impact on the active state or the transition state of conversion.
Keywords
INTRODUCTION
The small (30S) subunit of the ribosome contains three main folding blocks. The 5′ domain of 16S rRNA (nt 1–559) and six ribosomal proteins (RPs) form the body; the central domain of 16S rRNA (nt 560–917) and six RPs form the platform; and the 3′ major domain of 16S rRNA (nt 918–1396) and eight RPs form the head. Each of these blocks can be independently assembled in vitro, resulting in ribonucleoprotein (RNP) particles with near-native rRNA structure and RP stoichiometry (Weitzmann et al. 1993; Samaha et al. 1994; Agalarov et al. 1998, 2000). The 3′ minor domain of 16S rRNA (nt 1397–1542), comprised of the penultimate and ultimate helices (h44–h45) and the 3′ tail, lies along the interface side of the subunit and makes extensive contacts with the body and platform blocks.
It is well known that the purified 30S subunit will transition to an inactive state when magnesium (or monovalent) cations are depleted (Zamir et al. 1969, 1971, 1974; Vogel et al. 1970; Ginzburg et al. 1973). Subunits in this state fail to bind the 50S subunit, tRNA, mRNA, and oligonucleotides complementary to the 3′ tail of 16S rRNA (Backendorf et al. 1981). Inactive subunits can be readily reactivated—for example, by incubating them at 42°C in the presence of 20 mM Mg2+. Chemical probing studies showed that the active-to-inactive transition involves a conformational change in the 16S rRNA, which exposes nucleotides of the “neck” helix (h28) and protects nucleotides of the 3′ tail (Hogan and Noller 1978; Backendorf et al. 1981; Chu et al. 1983; Chiaruttini et al. 1984; Moazed et al. 1986). Recent cryo-EM structures clarify the molecular basis of this conformation change (Fig. 1; Jahagirdar et al. 2020; Schedlbauer et al. 2021). The top of h44 undocks from the body, disrupting the decoding center, and the head tilts backward by 16° with respect to the body. Nucleotides 1398–1400 and 1504–1506 rearrange to form a short “linker” helix. Nucleotides 921–925 separate from nt 1391–1397, disrupting part of h28, and nt 921–923 re-pair with nt 1532–1534, forming an alternative rRNA helix, termed here hALT. To form hALT, the 3′ end of the 16S flips across the subunit and occupies the mRNA entry channel, occluding the P and A codons (Fig. 1; Jahagirdar et al. 2020; Schedlbauer et al. 2021). Recently, the same rRNA rearrangement was observed in Staphylococcus aureus (Garaeva et al. 2024), suggesting that this conformational switch is widespread in bacteria.
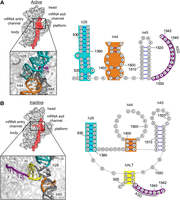
Structural basis of the active-to-inactive transition. Comparison of the 30S subunit in the active (A) and inactive (B) states, viewed from the 50S perspective. On the left are structures of the 30S where h28, h44, and h45 are colored red. Major regions of the subunit have been labeled. Panels show the relevant regions magnified with h28 (teal), h44 (orange), hALT (yellow), and the 3′ end of the 16S rRNA (purple). On the right are corresponding secondary structure diagrams with the same coloring scheme. (ASD) Anti-Shine–Dalgarno. This figure was made using PyMOL (PDB 6W6K, 6W77) and StructureEditor 1.0 (Mathews Lab).
An open question for many years was whether the inactive state holds any physiological relevance. In 2015, Weeks and coworkers performed chemical (SHAPE) probing of exponentially growing Escherichia coli cells and found that the free 30S particles largely adopt the inactive state. Most of these particles correspond to immature (pre-30S) subunits, suggesting that the inactive state normally contributes to 30S biogenesis (McGinnis and Weeks 2014; McGinnis et al. 2015). In the cell, ribosome assembly begins as soon as the nascent pre-rRNA emerges from RNA polymerase, and RPs bind rRNA in a hierarchical manner as the rRNA folds. The 30S body assembles first, followed by the platform and head. A number of assembly factors, including RNP-binding proteins, modification enzymes, and GTPases, facilitate the process and/or prevent immature particles from participating in translation. Interestingly, assembly factor RimP will induce the active-to-inactive transition in vitro with E. coli and S. aureus subunits in the presence of high (10 mM) Mg2+ (Schedlbauer et al. 2021; Garaeva et al. 2024). Moreover, it has been independently shown that ΔksgA cells accumulate pre-30S particles that adopt the inactive state (Sun et al. 2023). Collectively, these data point to a role for the inactive state in 30S assembly in the cell. Adoption of the inactive state may be the primary mechanism by which pre-30S particles are prevented from entering the translationally active pool.
In this work, we investigate the functional role of hALT of the inactive state. Using an orthogonal ribosome system, we find that disruptions of hALT cause small but significant decreases in translation activity. Mutant ribosomes unable to form hALT support the growth of strain Δ7 prrn (which carries a single rRNA operon), albeit at reduced rate, and these cells accumulate pre-30S intermediates. Biochemical analysis of these mutant ribosomes suggests that hALT stabilizes the inactive state by 1.2 kcal/mol, with no impact on the active state or transition state. This work helps clarify hALT's contribution to 30S biogenesis and dynamics.
RESULTS AND DISCUSSION
Effects of mutations at positions 1532–1534 of 16S rRNA on translation activity
One feature of the inactive state, hALT, consists of base pairs U921–A1534, G922–C1533, and A923–U1532. To test the functional importance of this helix in vivo, we used an orthogonal ribosome system to measure translation activity in cells (Fig. 2). In this system (Abdi and Fredrick 2005; Qin et al. 2007; Qin and Fredrick 2009; McClory et al. 2010, 2011; Warner et al. 2023), ribosomes made from plasmid-encoded 16S rRNA contain the alternative ASD sequence 5′-GGGAU-3′ and specifically translate reporter lacZ mRNA with the complementary SD sequence 5′-AUCCC-3′. Single, double, and triple mutations were introduced at positions 1532–1534 to disrupt hALT. Modest decreases in translation activity of approximately twofold or less were observed, even for the double and triple substitutions (Fig. 2A). Surprisingly, the triple mutation U1532A/C1533A/A1534U conferred one of the smaller defects (Fig. 2A). This could be explained by a register shift, for example with AAU (1532–1534) pairing to AUU (919–921). Overall, these data are consistent with a small role for hALT in 30S assembly or function.

Effect of nucleotide substitutions within h28 and hALT on translation activity in the cell. (A) Effects of mutations at positions 1532–1534 of the 16S rRNA, as indicated. A two-tailed t-test was used to evaluate differences from WT. Uncorrected P-values: (*) P < 0.05; (**) P < 0.005; (***) P < 0.005. (B) Effects of mutations at positions 922–923, 1393–1395, and 1532–1533, as indicated. Whether h28 and hALT contain a mismatch (red X) or are fully complementary (green ✓) is indicated for each construct. Data represent the mean ± SEM from four or more biological replicates. Raw data are given in Supplemental Table S1. (WT) Wild type, (VC) vector control.
Next, we tested whether these mutations altered translational fidelity, using various indicator strains (McClory et al. 2011; Warner et al. 2023). These strains contain mutations in lacZ that enable quantification of spurious initiation on AUC, readthrough of UGA, and spontaneous +1 frameshifting. Few significant effects were observed. Mutation U1532A increased spurious initiation and nonsense readthrough by approximately twofold (Supplemental Fig. S1A,B), whereas C1533G increased nonsense readthrough by approximately twofold (Supplemental Fig. S1A). Yet, surprisingly, double and triple substitutions (which include U1532A or C1533G) conferred no fidelity defects (Supplemental Fig. S1). These data suggest that hALT is dispensable for the formation of high-fidelity subunits.
Effects of mutations that disrupt and restore base-pairing in h28 and hALT
To further probe hALT function, we targeted G922 and A923, which contribute to either h28 or hALT. Substitution of G922 to A or C reduced translation activity by >200-fold, to background (vector control) levels (Fig. 2B). In both cases (1 and 2), a compensatory mutation that restores base-pairing in h28 also restored translation activity substantially, to levels of ∼20%–30% of the control (Fig. 2B). These data indicate that the 922–1395 bp of h28 is crucial, and G-C is better than A-U or C-G. Base pair G922–C1395 lies near the base of h28 and contacts A1398 via a Type I minor interaction, and one can imagine how disruption of these interactions might jeopardize 30S dynamics and function (Mohan et al. 2014). Substitution of A923 to G or U was much less deleterious, as translation activity was reduced by only two- to threefold, respectively (Fig. 2B, cases 3 and 4). Introducing compensatory mutations at position 1393 did little to improve translation activity further (Fig. 2B, cases 3 and 4). Thus, ribosomes tolerate a mismatch at 923–1393 or alternative base pairs, although the native A-U pair is optimal.
We also generated tandem mutations in the “top” strand of h28, G922A/A923G, and G922C/A923U (Fig. 2B, cases 5 and 6). As expected, each of these tandem substitutions eliminated translation activity. Compensatory tandem substitutions in the “bottom” strand of h28 were then introduced. Robust rescue of activity was seen for case 5, whereas significant but weaker rescue was seen in case 6 (Fig. 2B). These data underscore the degree to which base pair identity matters in this region of h28, which is universally conserved in bacteria (Noller et al. 2022).
Finally, we introduced substitutions at positions 1532–1533 designed to restore pairing in both h28 and hALT. In cases 1 and 5, there was some indication of functional rescue, consistent with a minor role for hALT in 30S biogenesis. Mutation C1533U seemed to improve activity in the context of case 1, and U1532C/C1533U seemed to improve activity in the context of case 5; although in neither case was the apparent increase deemed significant at the 95% confidence level. In the other four cases, no rescue was seen and the 3′ substitutions either decreased or had no effect on activity (Fig. 2B).
Mutations targeting hALT cause defects in 30S biogenesis
Tandem mutations U1532A/C1533G and U1532C/C1533U were each moved into Δ7 prrn, a strain which has no rRNA operons on the chromosome but carries one (rrnB) on a plasmid (Quan et al. 2015). Both mutant 16S alleles were able to support cell growth, albeit at rates 17% and 11% (respectively) lower than the control (Table 1).
Growth of control and mutant Δ7 prrn strains
Control and mutant strains were grown to the mid-log phase, cultures were rapidly cooled by pouring over crushed ice, and cell lysates were subjected to sucrose gradient sedimentation analysis. As reported previously (Warner et al. 2023), the control Δ7 prrn strain exhibited very low levels of polysomes compared to other, more natural, E. coli strains (Fig. 3A; Balakrishnan et al. 2014; Warner et al. 2023). The basis of this phenomenon remains unclear but is probably due to dysregulation of ribosome synthesis in this Δ7 prrn strain (Warner et al. 2023). Both mutant strains showed higher levels of 30S particles, compared to the control, and slightly lower levels of polysomes (Fig. 3A). Mutant U1532C/C1533U also exhibited a significantly reduced 50S peak (Fig. 3A). This may reflect a higher rate of 50S turnover, which for some reason depends on the particular 16S substitutions. Because polysome levels were so low in these strains, we repeated the experiment but added chloramphenicol (Cm; 100 µg/mL) 2 min before rapid cooling to help trap elongating ribosomes. Again, significantly higher levels of 30S particles were observed in the two mutant strains, and reduced 50S levels were seen for U1532C/C1533U (Fig. 3B). However, the apparent differences in polysome levels mentioned above (Fig. 3A) could not be confirmed in the presence of Cm (Fig. 3B). Nonetheless, the accumulation of 30S particles in the two mutant strains is consistent with defects in 30S biogenesis.
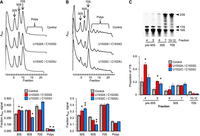
Evidence that hALT contributes to 30S biogenesis. (A,B) Polysome analysis of Δ7 prrn strains (as indicated) without (A) or with (B) chloramphenicol treatment before lysis. Shown are representative A254 traces of sucrose gradients, with peaks corresponding to subunits (30S, 50S), monosomes (70S), and polysomes (Polys) indicated. The bar graphs below show the quantification of 30S, 50S, 70S, and polysomes (as indicated) in control (gray) and mutant (red, U1532A/C1533G; blue, U1532C/C1533U) strains. Data represent the mean ± SEM from at least three biological replicates. A two-tailed t-test was used to evaluate differences from the WT. Uncorrected P-values: (*) P < 0.05; (**) P < 0.005. (C) Analysis of precursor 17S rRNA in the control and mutant strains. RNA was extracted from various fractions of sucrose gradients from experiments like those of A and analyzed by PAGE. A representative gel, analyzing RNA of the control strain, is shown, with specific fractions indicated. Arrowheads denote bands corresponding to precursor (17S) and mature (23S, 16S) rRNAs. For the bar graph below, 17S and 16S rRNA bands were quantified and used to calculate the proportion 17S (17S/[17S + 16S]) within each fraction from control (gray) and mutant (red, U1532A/C1533G; blue, U1532C/C1533U) cells. Data represents the mean ± SEM of three biological replicates. A two-tailed t-test was used to evaluate differences from the WT. Uncorrected P-value: (*) P < 0.05.
To further characterize these strains, fractions across the gradients were collected, and total RNA from fractions corresponding to pre-30S, 30S, and 70S regions of the gradient were analyzed by denaturing PAGE (Fig. 3C). Higher levels of precursor 17S rRNA were seen in pre-30S fractions for both mutant strains (Fig. 3C). These data provide strong evidence that disruption of hALT causes defects in 30S biogenesis.
Contribution of hALT to subunit dynamics in vitro
Wild-type and mutant (U1532A/C1533G) 30S subunits were purified from 70S ribosomes and analyzed with respect to conformational
state conversion. First, we measured the kinetics of inactivation by diluting active subunits into low Mg2+ and monitoring the loss of P-tRNA-binding activity as a function of time (Fig. 4A). Indistinguishable rates were observed for control and mutant subunits (0.57 ± 0.13 and 0.65 ± 0.10 min−1, respectively). This indicates that U1532A/C1533G has no impact on the free energy barrier (ΔG‡) for the active-to-inactive transition, calculated as 21.0 kcal/mol based on the equation 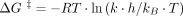 , where R is the gas constant, T is the temperature in Kelvin, k is the reaction rate, h is Planck's constant, and kB is Boltzmann's constant. Next, we measured the kinetics of activation. Inactive subunits (stored in low Mg2+) were supplemented with Mg2+ (10 mM) and shifted to 37°C at time t = 0, and the gain of P-tRNA-binding activity was monitored as a function of time (Fig. 4B). Control subunits were activated at a rate of 0.09 ± 0.01 min−1, whereas mutant subunits were activated six times faster, at a rate of 0.59 ± 0.05 min−1. The data indicate that ΔG‡ for the inactive-to-active transition is 1.2 kcal/mol smaller for the mutant (ΔG‡ = 21.0 kcal/mol) than for the WT (ΔG‡ = 22.2 kcal/mol). The simplest interpretation of these data is that U1532A/C1533G destabilizes the inactive state by 1.2
kcal/mol without influencing the transition state or active state (Fig. 4C). This interpretation is fully consistent with the structural data, as U1532A/C1533G is predicted to disrupt hALT of the
inactive state but no helix (or stabilizing features) of the active state.
, where R is the gas constant, T is the temperature in Kelvin, k is the reaction rate, h is Planck's constant, and kB is Boltzmann's constant. Next, we measured the kinetics of activation. Inactive subunits (stored in low Mg2+) were supplemented with Mg2+ (10 mM) and shifted to 37°C at time t = 0, and the gain of P-tRNA-binding activity was monitored as a function of time (Fig. 4B). Control subunits were activated at a rate of 0.09 ± 0.01 min−1, whereas mutant subunits were activated six times faster, at a rate of 0.59 ± 0.05 min−1. The data indicate that ΔG‡ for the inactive-to-active transition is 1.2 kcal/mol smaller for the mutant (ΔG‡ = 21.0 kcal/mol) than for the WT (ΔG‡ = 22.2 kcal/mol). The simplest interpretation of these data is that U1532A/C1533G destabilizes the inactive state by 1.2
kcal/mol without influencing the transition state or active state (Fig. 4C). This interpretation is fully consistent with the structural data, as U1532A/C1533G is predicted to disrupt hALT of the
inactive state but no helix (or stabilizing features) of the active state.
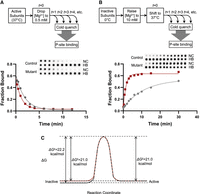
Disruption of hALT destabilizes the inactive state in vitro. (A,B) Comparing the rate of inactivation (A) or activation (B) of wild-type (gray circles) and mutant (red squares) 30S subunits. Reaction schemes (top) and representative primary data (inset) are shown. (NC) Nitrocellulose, (HB) Hybond-N+. Each plot shows fitted data for one replicate. Three independent replicates were performed, yielding the following rates (mean ± SEM): WT inactivation, 0.57 ± 0.13 min−1; mutant inactivation, 0.65 ± 0.10 min−1; WT activation, 0.09 ± 0.01 min−1; mutant activation, 0.59 ± 0.05 min−1. (C) Free energy diagram depicting inactive-to-active conversion for control (black) and mutant (red) subunits.
Conclusion
Here, we show that hALT stabilizes the inactive state and contributes to 30S biogenesis in the cell, consistent with current structural models (Jahagirdar et al. 2020; Schedlbauer et al. 2021; Sun et al. 2023). At the same time, our work reveals that hALT's role is quite small. Complete disruption of hALT decreases translation activity by approximately twofold, indicating no larger effect on the efficiency of 30S biogenesis. Ribosomes formed in the absence of hALT exhibit no fidelity defects, unlike ribosomes formed in the absence of various assembly factors, rRNA processing enzymes, or cis-acting RNA elements (Roy-Chaudhuri et al. 2008; Davies et al. 2010; Connolly and Culver 2013; Sharma and Anand 2019; Warner et al. 2023). Helix hALT stabilizes the inactive state, but by only 1.2 kcal/mol. Intuitively, one might expect these base pairs to be “worth” more, especially when they appear to redirect the 3′ tail of 16S rRNA by ∼180°. Yet, hALT is largely dispensable for 30S biogenesis and function. The observation that >10% of bacteria have one or more mismatches in hALT (Schedlbauer et al. 2021) is in line with our evidence that hALT plays a small and noncrucial role in 30S dynamics and biogenesis.
The fact that U1532A/C1533G subunits can be reversibly activated and inactivated, much like control subunits, shows that the inactive state does not strictly depend on hALT. Presumably, U1532A/C1533G subunits adopt an inactive state that closely resembles that of control subunits, without or with noncanonical interactions between nt 922–923 and 1532–1533. It has been shown that, in the presence of 10 mM Mg2+, assembly factor RimP promotes an inactive-like state in which h44 is displaced from the 30S body. In the cell, RimP and other assembly factors likely stabilize this inactive conformation in a mutual way, ensuring that the premature particles are excluded from the translationally active pool. Then, KsgA, RbfA, and RsgA promote h44 docking, h45 methylation, and 3′ tail repositioning to complete the assembly process, licensing the subunits for participation in translation. We envision that these events proceed similarly in wild-type and U1532A/C1533G cells, but the overall process is optimal when hALT can form.
MATERIALS AND METHODS
Orthogonal ribosome system
The translation activity of orthologous ribosomes was measured using lacZ as described previously (Abdi and Fredrick 2005; Qin et al. 2007; McClory et al. 2010; Warner et al. 2023). Plasmid pDQ207 contains the 16S rRNA gene, rrsB, with the alternative ASD sequence 5′-GGGAT-3′, under transcriptional control of the PBAD promoter. The marker mutation T1451A was engineered into pDQ207 to generate pBW022 (Warner et al. 2023). Mutations and deletions were constructed using QuikChange (Stratagene) or Phusion site-directed mutagenesis (New England Biolabs) (Supplemental Table S1).
Plasmid-borne 16S rRNA alleles (ASD 5′-GGGAT-3′) were expressed in indicator strains that harbor lacZ with the alternative SD sequence (5′-ATCCC-3′) on the chromosome. Indicator strain KLF2674, which has no mutations in the lacZ coding region, was used to measure overall translation activity (McClory et al. 2010). Isogenic strain KLF2672 has codon 1 of lacZ changed from ATG to ATC and was used to measure spurious initiation (Qin et al. 2007; Qin and Fredrick 2009; McClory et al. 2011). Isogenic strain KLF2723 has codon 585 of lacZ changed from TGG to TGA and was used to measure UGA readthrough (McClory et al. 2010, 2011). Isogenic strain KLF3361 has codons 250–252 of lacZ replaced with GGGTTTTAGC and was used to measure spontaneous +1 frameshifting (McClory et al. 2011). Cells from overnight cultures were diluted 300-fold into 3 mL fresh LB medium containing ampicillin (100 µg/mL), kanamycin (50 µg/mL), and l-arabinose (5 mM) and allowed to grow at 37°C for 4 h. Cells (1 mL) were pelleted and washed once with 1 mL Z-buffer (100 mM sodium phosphate at pH 7.0, 10 mM KCl, 10 mM MgSO4). Cells were permeabilized with B-Per (Thermo). Permeabilized cells (1 mL) were mixed with 200 μL of 1 mg/mL CRPG (Invitrogen) at time t = 0. Reactions were quenched with 500 μL of 1 M sodium carbonate, and the time was recorded. Cellular debris was pelleted, and the absorbance of the supernatant at 574 nm (A574), characteristic of the product, was determined. Specific activity was defined by the equation: 1 unit = 1000·(A574)/(OD600·v·t), where OD600 is the optical density of the cell suspension used, v is the volume of the cell suspension used (in milliliters), and t is time of incubation (in minutes) at room temperature. Rates of spurious initiation from AUC, readthrough of UGA, and +1 frameshifting were calculated as LacZ activity in the relevant indicator strain (KLF2672, KLF2723, or KLF3361, respectively) relative to that in the control strain (KLF2674).
Δ7 prrn strain construction and analysis
Mutations in the 3′ end region of rrsB were introduced into plasmid p278MS2 (Youngman et al. 2004) to generate pBW96 and pBW97 (Supplemental Table S1). These plasmids were transformed into SQZ10 (Quan et al. 2015), and transformants were plated on sucrose (5%) to select against the resident plasmid pHKrrnC-sacB (Qin et al. 2007), resulting in strains BRW176, BRW205, and BRW206 (Table 1). Strains were verified by plasmid purification and sequencing. For growth measurements, overnight cultures were diluted 300-fold into fresh LB media containing ampicillin (Amp; 100 µg/mL), and growth at 37°C was monitored by measuring OD600 as a function of time.
Polysome analysis of various Δ7 prrn strains was performed as described (Qin and Fredrick 2013). Strains were grown to the mid-log phase and poured over crushed ice to quickly cool the cells and stall elongating ribosome. Cells were pelleted, resuspended in Lysis Buffer (10 mM Tris-HCl pH at 8.0, 10 mM MgCl2, 10 mg/mL lysozyme), and subjected to three freeze/thaw cycles. Lysis was completed by the addition of sodium deoxycholate (0.3%, w/v) and clarified by centrifugation at 13,000 rpm for 10 min at 4°C. Clarified lysates were loaded onto 10%–40% sucrose gradients, subjected to ultracentrifugation at 35,000 rpm, 3 h, 4°C in a SW41 rotor (Beckman), and pumped using a syringe-pump (Brandel) with an in-line UV detector (UA-6, ISCO). Peaks of absorbance at 254 nm (A254) were integrated using the software Peak Chart (Brandel). Fractions (0.5 mL) were collected across the gradient. RNA from the pre-30S, 30S, and 70S regions of the gradient was extracted and analyzed by denaturing PAGE as described previously (Leong et al. 2013; Gibbs et al. 2017). Gels were strained with SYBR-Gold (Invitrogen) and scanned using a Typhoon 5 (Cytiva). Data were quantified using ImageQuant (Cytiva). For the chloramphenicol treatment experiment, chloramphenicol (100 μg/mL) was added to mid-log phase cultures 2 min before being poured over crushed ice. Samples were otherwise processed as described above.
Purification of 30S ribosomal subunits
Overnight cultures of strains BRW176 and BRW205 were diluted 500-fold into fresh LB media (1 L), grown to mid-logarithmic phase, and cooled on ice for 20 min. Cells were pelleted, washed, and lysed via French press. 30S subunits were then purified as previously detailed (Qin et al. 2007), except that the final storage buffer contained 50 mM Tris-HCl pH 7.1, 1 mM MgCl2,100 mM NH4Cl, and 6 mM βME.
Kinetics of activation
Inactive 30S subunits were diluted to 2 µM in a storage buffer. At time t = 0, the MgCl2 concentration was increased to 10 mM, and the reaction was incubated in a water bath at 37°C. At various time points, aliquots (2 µL) were removed, mixed with 8 µL of ice-cold buffer A (50 mM Tris-HCl pH 7.8, 10 mM MgCl2, 100 mM NH4Cl, 6 mM βME), and set on ice. P-tRNA-binding activity was then measured as detailed below.
Kinetics of inactivation
30S subunits (20 µM) were activated by incubation at 42°C for 20 min in buffer B (50 mM Tris-HCl pH 7.1, 10 mM MgCl2, 100 mM NH4Cl, 6 mM βME). At time t = 0, prewarmed buffer C (50 mM Tris-HCl pH 7.1, 100 mM NH4Cl, 6 mM βME) was added to dilute the subunits and Mg2+ to 1 µM and 0.5 mM, respectively. At various time points, aliquots (2 µL) were removed, mixed with 8 µL of ice-cold buffer D (50 mM Tris-HCl pH 7.8, 2 mM MgCl2, 100 mM NH4Cl, 6 mM βME), and set on ice to quench the reaction. P-tRNA binding activity was then measured as detailed below.
P-site tRNA binding
Natural E. coli tRNAPhe was radiolabeled using snake venom phosphodiesterase and ATP(CTP):tRNA nucleotidyltransferase as described (McGarry et al. 2005), except that the concentration of tRNA in the labeling reaction was 1 µM and the incubation time was 20 min. Binding of 3′-[32P]-tRNAPhe to the P site was measured using a double-filter method previously described (Fahlman and Uhlenbeck 2004; Qin et al. 2007). Each aliquot (10 µL) of 30S (1–2 µM) was incubated with poly(U) (0.5 µg) and 3′-[32P]-tRNAPhe (<0.02 µM) in 20 µL buffer A for 20 min on ice. Samples were diluted with 50 µL of ice-cold buffer A and immediately filtered through a prewet bilayer of nitrocellulose (Amersham Biosciences) and Hybond-N+ (GE Healthcare) membranes, using a 96-well dot-blot apparatus (Schleicher & Schuell). Membranes were then separated, dried, and exposed to a phosphoimager screen. Screens were scanned using a Typhoon 5 (Cytiva) and quantified with ImageQuant (Cytiva). The fraction of tRNA bound to nitrocellulose (F) was determined and plotted as a function of time, and data were fit to the equation F = F0 + (Fmax − F0)(1 − e−kx); where F0 = lowest fraction measured, Fmax = maximal fraction measured, k = rate, and x = time.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank A. Dunning and Q. Liu for providing initial data on G922A and C1395U, and J. Ortega and S. Connell for comments on the manuscript. This work was supported by a grant from the National Institutes of Health (GM072528 to K.F.) and by a fellowship from the Ohio State University Center of RNA Biology (to B.R.W.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079960.124.
-
Freely available online through the RNA Open Access option.
- Received January 22, 2024.
- Accepted March 23, 2024.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Benjamin R. Warner is the first author of this paper, “Contribution of an alternative 16S rRNA helix to biogenesis of the 30S subunit of the ribosome.” Benjamin is a graduate student at The Ohio State University in the Department of Microbiology. He studies the contribution of pre-rRNA structures in the biogenesis of the bacterial ribosome.
What are the major results described in your paper and how do they impact this branch of the field?
This work describes the contribution of an alternative helix (hALT) in vivo and in vitro. Disruption of the hALT results in 30S assembly defects and decreases translational activity, showing that this alternative helix has a small role in 30S assembly in cells. Also, hALT stabilizes the inactive state in vitro. Structural work has indicated that the inactive state, initially considered an in vitro phenomenon, is present in the cell and appears involved in assembly. This work provides experimental evidence that hALT, present in the inactive state, has a role in 30S assembly in the cell for the first time.
What led you to study RNA or this aspect of RNA science?
I have always been interested in cellular physiology, and one thing that is broadly overlooked is the role of RNA in physiology. The ribosome is present in all domains, with its core structure conserved. Yet, the process of biogenesis and assembly differs significantly from one organism to another. It fascinates me how organisms have constructed different mechanisms to overcome similar barriers in something as essential and fundamental as ribosome biogenesis.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
I found it surprising that 30S subunits lacking base-pairing potential to form hALT have no fidelity defects. Literature has shown that assembly defects typically result in fidelity defects. This probably shows that the quality control mechanisms send these misassembled subunits through the degradation pathway, resulting in high-fidelity translation still being achieved.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
I have always been interested in the sciences. However, my first research laboratory experience in Dr. Birgit Alber's laboratory greatly increased my interest in and love for science. Experiencing a laboratory setting in this fashion changed my view completely. I obtained a better understanding of the methodology rather than doing it once, as is typically done in teaching laboratories. Performing experiments and obtaining data on a firsthand basis also increased my interest in and appreciation for science.








