
Translation regulation by a guanidine-II riboswitch is highly tunable in sensitivity, dynamic range, and apparent cooperativity
- 1Institute of Biochemical Design and Discovery, Yale University, West Haven, Connecticut 06516, USA
- 2Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut 06510, USA
- 3Department of Chemistry, Yale University, New Haven, Connecticut 06511, USA
- Corresponding author: scott.strobel{at}yale.edu
Abstract
Riboswitches function as important translational regulators in bacteria. Comprehensive mutational analysis of transcriptional riboswitches has been used to probe the energetic intricacies of interplay between the aptamer and expression platform, but translational riboswitches have been inaccessible to massively parallel techniques. The guanidine-II (gdm-II) riboswitch is an exclusively translational class. We have integrated RelE cleavage with next-generation sequencing to quantify ligand-dependent changes in translation initiation for all single and double mutations of the Pseudomonas aeruginosa gdm-II riboswitch, a total of more than 23,000 variants. This extensive mutational analysis is consistent with the prominent features of the bioinformatic consensus. These data indicate, unexpectedly, that direct sequestration of the Shine–Dalgarno sequence is dispensable for riboswitch function. Additionally, this comprehensive data set reveals important positions not identified in previous computational and crystallographic studies. Mutations in the variable linker region stabilize alternate conformations. The double mutant data reveal the functional importance of the previously modeled P0b helix formed by the 5′ and 3′ tails that serves as the basis for translational control. Additional mutations to GU wobble base pairs in both P1 and P2 reveal how the apparent cooperativity of the system involves an intricate network of communication between the two binding sites. This comprehensive examination of a translational riboswitch's expression platform illuminates how the riboswitch is precisely tuned and tunable with regard to ligand sensitivity, the amplitude of expression between ON and OFF states, and the cooperativity of ligand binding.
Keywords
INTRODUCTION
Riboswitches, RNA motifs found predominantly in the 5′ untranslated regions of bacterial mRNAs, control gene expression largely via two mechanisms. Transcriptional riboswitches use terminator/antiterminator systems to modulate premature transcription termination. Translational riboswitches control access to the Shine–Dalgarno (SD) sequence to temper the mRNA loading onto the ribosome (Breaker 2018). Extensive structural biology studies have established how these RNA motifs recognize their small molecule effectors (Garst et al. 2011; Peselis and Serganov 2014); however, the intricacies of how such binding is translated into changes in gene expression remain underexplored.
In the two decades of riboswitch studies to date, our grasp of transcriptional riboswitch function has outstripped that of other riboswitch regulatory mechanisms. Given the facility of transcriptional assays, the functional and folding landscapes of various transcriptional riboswitches have been explored (Wickiser et al. 2005; Quarta et al. 2012; Watters et al. 2016; Steinert et al. 2017; Scull et al. 2021). These studies have revealed a crucial interplay between cotranscriptional folding and ligand binding in some systems, indicating kinetic regimes of riboswitch gene regulation (i.e., the K1/2 of the system exceeds the KD of the aptamer) in those instances.
The details of translational riboswitch regulatory requirements are sparse by comparison. Translational riboswitches are currently modeled with OFF states where the SD sequence is sequestered and with ON states where the SD sequence is free for association with the 30S ribosomal subunit. While some studies have proposed kinetic models for translational riboswitch function (Guedich et al. 2016), other translational riboswitches appear to operate in a thermodynamic regime (Rieder et al. 2007), relying on conformational dynamics over the lifetime of the transcript, rather than during the short window of nascent transcription. Recent work has revealed the importance of ribosomal protein S1 in unwinding secondary structure around the ribosome binding site in an adenine riboswitch (de Jesus et al. 2021, 2022), but more work is needed to study the energetic requirements of these translational regulators. Given that some classes of riboswitches comprise only translational representatives, probing the functional landscape of these elements is imperative not only for the fundamental understanding of these regulatory motifs but also for any future engineering efforts.
The guanidine-II (gdm-II) riboswitch is an exclusively translational class found predominantly upstream of multidrug resistance transporters (Sherlock et al. 2017) and is one of four guanidine-responsive riboswitch families that have been identified in bacteria (Nelson et al. 2017; Sherlock and Breaker 2017; Sherlock et al. 2017; Lenkeit et al. 2020; Salvail et al. 2020). Bioinformatically identified as two similar hairpins (P1 and P2) capped by identical ACGR tetraloops and connected by a variable length linker, the gdm-II riboswitch class binds two guanidinium molecules cooperatively, one in each loop. A Hill coefficient greater than 1 suggests positive cooperativity between multiple binding sites, and in-line probing data for the Gloeobacter violaceus gdm-II riboswitch fit with a Hill coefficient of 1.4 (Sherlock et al. 2017). The bound structure has been solved by X-ray crystallography (Huang et al. 2017; Reiss and Strobel 2017) revealing a kissing loop interaction between the two hairpins, with a guanidine molecule bound in each hairpin. These structures, however, comprise only the hairpin dimers—the linker and flanking regions containing the SD sequence and anti-Shine–Dalgarno (aSD) were excluded from structural studies. These dynamic regions are predicted to be integral to the switching behavior of these motifs.
For the Pseudomonas aeruginosa gdm-II riboswitch, the aSD element is held apart from the ribosome binding site when the P1 and P2 helices dimerize in the bound state (Fig. 1A,B). No structural information is available for the full-length riboswitch in the OFF state. The OFF state is modeled to include an additional third helix, P0, comprising the hybridization of the 5′ tail with the 3′ tail (P0b) and the SD with the aSD (P0a) (Fig. 1A). This P0 helix is proposed to inhibit the association of the ribosome in the OFF state, while dimerization of the P1 and P2 hairpins in the ON state pulls the P0 apart so that the ribosome can bind the SD sequence. While the crystal structures have provided crucial information about ligand binding and recognition, analysis of the full sequence of the riboswitch is needed to understand how this RNA element drives ligand-dependent expression modulation.

(A) Secondary structure of the P. aeruginosa gdm-II riboswitch. (B) Helical dimerization of the P. aeruginosa gdm-II riboswitch.
Recent studies have explored regulatory RNA function through high-throughput methods (Buenrostro et al. 2014; Torgerson et al. 2018; Strobel et al. 2019; Niederer et al. 2022). We recently reported the use of RelE as an efficient method to monitor ligand-dependent changes in ribosomal initiation (Focht and Strobel 2022). As a ribosome-dependent endonuclease, RelE exclusively cleaves ribosome-bound transcripts (Pedersen et al. 2003); therefore, changes in cleavage reflect changes in translation initiation. As a result, we have been able to quantify ligand-dependent changes in riboswitch-regulated ribosomal initiation. Here, we have integrated RelE cleavage with next-generation sequencing to examine the ligand responsiveness of more than 23,000 variants of the P. aeruginosa gdm-II riboswitch. Quantitative single and double mutant functional data revealed a delicate expression platform and key positions that tune the switch's sensitivity, dynamic range, and apparent cooperativity.
RESULTS
Developing a sequencing-based assay for translational riboswitch function
We performed a comprehensive mutational analysis of the P. aeruginosa gdm-II riboswitch using a sequencing-based translation initiation assay. We have previously reported the use of RelE cleavage to quantify changes in ribosome initiation (Focht and Strobel 2022). Since RelE cleavage produces a measurable length difference between ribosome bound and unbound RNAs, we integrated RelE cleavage with the existing high-throughput pipeline (Fig. 2A) for transcriptional riboswitches, which leverages the intrinsic length difference between full-length and terminated transcripts to report functional changes (Torgerson et al. 2018). We generated a mutant RNA library of the Pae gdm-II riboswitch by in vitro transcription. Positions 1–72 were doped to maximize for double mutations. Translation initiation complexes were formed at various guanidine concentrations, and the RNAs bound to the ribosome were cleaved in the A site by RelE. RNAs were sequenced, and the number of full-length and cleaved reads were calculated at each ligand concentration for all 23,220 single and double mutants to generate quantitative ligand–response curves for over 97% of these variants. This made it possible to assess the sensitivity and dynamic range of each sequence.
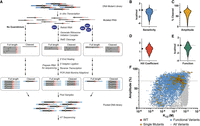
(A) Schematic of the high-throughput mutational analysis of RNA translation initiation. (B) Distribution of functional K1/2 values as ΔΔGSensitivity (n = 7222). (C) Distribution of functional amplitude values (n = 7222). (D) Distribution of single mutant Hill coefficient values (n = 127). (E) Distribution of function parameter values (n = 7222). Wild-type value for all parameters is indicated on the y-axis. (F) Plot of K1/2 versus amplitude for all functional variants (dark blue), all functional single mutants (yellow), and the wild type (red). Dotted lines indicate the intersection of wild-type values.
Previous analysis indicated this riboswitch is cooperative (Sherlock et al. 2017). The percent cleaved at each concentration was fit to the Hill equation (Equation 1). The fit produces an apparent K1/2 that reflects the sensitivity of the riboswitch (Supplemental Fig. 1A), an amplitude that reflects the range of the response (Supplemental Fig. 1B), and a Hill coefficient that indicates the degree of cooperativity between the binding sites. The trends in the fit data are recapitulated among replicates (Supplemental Figs. 2, 3, 5). Single mutant heat maps of the fit data are available in Supplemental Figure 7.
This data set reveals that variants within a very limited sequence space can cover almost the entire functional range. Sensitivities span three orders of magnitude, and amplitudes range from 0% to 97% (Fig. 2B,C). Significant variation is seen in the Hill coefficient, which is extremely sensitive to the quality of the fit, but the distribution is still centered close to the wild-type value (Fig. 2D). Notably, the single and double mutants populate almost every combination of K1/2 and amplitude values (Fig. 2F), indicating the tunability of the riboswitch within close sequence space. The maximum K1/2 value that can be measured is in the mid-millimolar range due to the concentration limits of the assay. The riboswitch can improve its sensitivity by close to a 100-fold with a single point mutation (shown in yellow if Fig. 2F). While the upper right quadrant is not densely populated, as expected, we still observe thousands of variants with K1/2 and amplitude values tighter and greater than wild type.
Our data set contains almost every possible combination of K1/2 and amplitude within the range of values, but not every combination represents a functional switch. We developed a parameter that combines the apparent K1/2 and amplitude to describe the functionality of each variant (Materials and Methods). Integrating these two values into a single metric of riboswitch function facilitated the analysis of the tens of thousands of variants in this data set. The riboswitch function depends not only on the concentration of ligand it senses but also on the dynamic range of the expression response. A riboswitch that responds to extremely low concentrations of ligand but barely alters translation initiation is not a functional switch. On the other hand, a riboswitch with a large change in translation initiation that only responds at physiologically irrelevant concentrations of ligand is similarly not functional. Thus, a riboswitch functional parameter requires the integration of the K1/2 and the amplitude. To do this, we transformed the apparent K1/2 into a free energy (ΔGsensitivity) and the amplitude into a pseudoenergy (ΔGamplitude). We compared each variant value with the wild type to create ΔΔG values. The ΔΔGsensitivity and the ΔΔGamplitude are similar in magnitude (Supplemental Fig. 4) and were weighted equally. The function parameter (F) is thus centered around a wild-type value of zero. Variants that function better than wild type (tighter K1/2, larger amplitude) have F less than zero, and variants that function worse than wild type (weaker K1/2, smaller amplitude) have F greater than zero (Supplemental Fig. 1C).
The distribution of F is skewed toward values less than zero since the most broken mutants are excluded due to poor fits (Fig. 2E). Imposing a function cutoff, we separated functional switches (dark blue in Fig. 2F) from the entire pool (gray in Fig. 2F). The functional switches maintain the range of amplitude and K1/2 values, and the functional space achievable by single mutants is similar to that of the double mutants. Thus, the functional landscape of this sequence is broad and rugged, with steep peaks and deep valleys within reach of a single mutation. The double mutant data allow us to probe that landscape further by illuminating interactions within the riboswitch.
Double mutant covariation indicates structural features
The functionality of each double mutant variant reveals covariation in the P1, P2, and P0b helices. We calculated the difference between the observed and expected function of each double mutant (Materials and Methods), where the expected value is the sum of the function parameters for the relevant single mutations. We plotted these differences as a heat map to identify possible interactions in the riboswitch (Fig. 3A). Negative values indicate favorable interactions between two mutations in the ON state, and positive values indicate interactions unfavorable to the ON state.
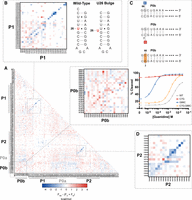
Double mutant interactions reveal structural features. (A) Epistasis heat map of double mutant function parameters. Negative values reveal P1 and P2 helices, while positive values identify P0b. (B) Inset of P1 covariation with parallel diagonal indicating the U26 bulge shown in the accompanying secondary structure. (C) Inset of P0b covariation and secondary structure of key single mutations to a P0 base pair and the double mutant restoration. (D) Inset of P2 covariation.
As arranged in this heat map, strong diagonals indicate covariation within structured regions of the riboswitch. Since the P1 and P2 helices are both present in the ON state, covariation presents as diagonals of negative values. A single dark blue diagonal between G44–G49:C54–C59 highlights the P2 helix. The P1 helical trend is more complex (Fig. 3B). Instead of a single diagonal, P1 exhibits a set of two parallel diagonals between G10–G13:U27–C30. This parallel diagonal suggests an alternative pairing within the helix, both of which appear to be functional. The data are consistent with a P1 helix with a bulge at U26 and alternative pairing of the stem (Fig. 3B). Replacement of the G–U wobble between G10 and U29 with a G–C pair between G10 and C30 may energetically compensate for the bulged nucleotide. Evidence of mutation-induced structural rearrangements has previously been seen in the mutational analysis of the glmS ribozyme (Andreasson et al. 2020), but these data suggest that bulging U26 may occur as a natural alternative conformation in the wild-type switch.
Covariation also identifies the riboswitch's expression platform. We had previously modeled the P0a and P0b helices based upon the complementarity of the putative aSD element in the linker region with the SD sequence and confirmed that mutations to this region are functionally relevant (Focht and Strobel 2022). However, the extent of this helix and its role as the riboswitch's expression platform had not been measured. As modeled, the P0a helix sequesters the majority of the SD sequence with an aSD element. The P0b helix hybridizes the region between the SD and start codon with the tail 5′ to P1. Dimerization of P1 and P2 via a kissing loop interaction in the ON state is proposed to open the P0a and P0b helices by physically separating the strands in space (Fig. 1A). Interestingly, we observe no evidence for functional covariation in the P0a region. Single and double mutations in this region are also extremely well-tolerated (Supplemental Fig. 9A,B). Double mutations to the SD sequence itself are largely functional, indicating that ribosome binding occurs efficiently in this in vitro context even given poor pairing between the riboswitch and the 16S rRNA. Thus, sequestration of the majority of the SD sequence is dispensable for riboswitch function.
The expression platform of the Pae gdm-II riboswitch appears to lie in the P0b helix. Covariation in P0b presents as a red diagonal of positive values, indicating interactions unfavorable to the formation of the ON state. This helix is at least 7 bp and harbors the only positions with variants that break the riboswitch ON (Fig. 3C). Mutations to G65 and G66 appear to produce drastic improvements in riboswitch function. Thus, covariation is observed not as rescue among broken variants, but as regression back toward wild-type function through the reinforcement of P0b with the complementary mutation. For example, the G66C variant (Fig. 3C) responds with an average apparent K1/2 ∼ 65 times tighter than wild type. Across the base pair, the C7G variant (Fig. 3C) is broken ON. The covarying double mutation C7G;G66C (Fig. 3C) decreases sensitivity back toward the wild-type value (Fig. 3C).
Interestingly, the increase in sensitivity does not originate simply from the weakening of the P0 helix. Breaking the helix with mutations to C7 and C8 does not produce the dramatic increases in sensitivity seen when breaking the helix with mutations to G65 and G66, the other side of the base pair. Modest increases in functionality are seen with C to U transitions, but transversions completely break the switch ON. Since the 3′ side of the helix will interact with the ribosome during initiation, asymmetry across base pairs in the P0b helix is reasonable. Mutations to G65 and G66 weaken P0b but also weaken the riboswitch's interaction with the 16s rRNA (Fig. 4A). These two competing factors result in a riboswitch that is easier to turn ON but is not constitutively ON due to its less effective SD (Fig. 4B). Mutations to C7 and C8, on the other hand, weaken P0b but maintain strong 16s rRNA pairing. These mutations therefore produce a constitutively ON riboswitch (Fig. 4B).

P0b mutational asymmetry. (A) Secondary structure of the Pae gdm-II riboswitch pairing with its internal aSD compared with pairing with the Escherichia coli 16S rRNA aSD. (B) Representative response curves for transversions to C7, C8, G65, and G66
Comprehensive mutational analysis is consistent with bioinformatic consensus
We used these double mutant data to compare prior phylogenetic conservation with the biochemical consensus sequence. As previously reported, bioinformatic conservation of the gdm-II riboswitch class is largely restricted to the two hairpins, P1 and P2 (Fig. 5A). While the identity and length of the stems of P1 and P2 are not highly conserved, they display a high degree of covariation. The L1 and L2 loops are highly conserved as ACGR tetraloops. The linker region is variable in both length and sequence, and neither the 5′ nor 3′ tail is conserved.
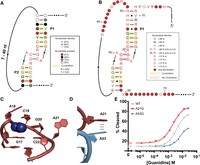
(A) Bioinformatic consensus of the gdm-II riboswitch class. (B) Biochemical consensus of the P. aeruginosa gdm-II riboswitch. (C) Conserved P1 binding pocket nucleotides. (D) Representation of the stacking interaction between A21 from P1 and A53 from P2. (E) Response profiles of two linker nucleotide variants with marked functional deficiencies.
We developed a biochemical consensus diagram of the Pae gdm-II riboswitch based upon the functional data (Fig. 5B). We screened each single and double mutant based on the function parameter (F) and calculated the conservation of each nucleotide at each position (Materials and Methods). The biochemical conservation of the Pae gdm-II riboswitch recapitulates many features of the bioinformatic consensus. Because bioinformatic conservation reflects features that are maintained across diverse sequences and diverse species, we expect the most highly conserved positions to be immutable in the Pae gdm-II sequence as well. In the biochemical consensus, covariation is observed for as many as four base pairs in both P1 and P2. The closing base pair of both helices is strongly conserved without covariation, consistent with the structural observation that the closing G–C pairs in both helices engage in π–cation interactions with the ligands (Fig. 5C).
We also observe strict conservation of the ACGR tetraloops as observed in the bioinformatic consensus. These nucleotides in both loops form the two guanidine binding pockets, and structural studies have shown their direct contact with the ligand (Fig. 5C). The 3′ nucleotide of each loop (A21 and A53 in Pae gdm-II) is bioinformatically conserved as a purine, and we observe this in the biochemical consensus as well. These nucleotides project away from the binding pocket and stack on top of one another in the kissing loop dimerization that occurs upon ligand binding (Fig. 5D). Since the transition from A to G maintains stacking ability, conservation as a purine is reasonable. Previous work with the E. coli gdm-II riboswitch reported an asymmetry in binding between the loops (Schamber et al. 2022). Mutational analysis indicated a preference for A at the fourth position in both loops, despite having a G endogenously in P2. We also noticed a functional asymmetry between the loops in the Pae gdm-II riboswitch, but while the A to G transition in P2 (A53G) is less functional, the same transition in P1 (A21G) is slightly more functional (Fig. 5E). Thus, while the bioinformatic consensus indicates an equal tolerance of both A and G at these positions, the individual sequence context of each riboswitch creates a functional preference.
The biochemical consensus has also identified positions that do not appear in the bioinformatic consensus sequence. While mutations to the variable linker region between P1 and P2 have largely no effect on riboswitch function, the U33G or U36C mutation each break the switch into an OFF state (Fig. 6, Supplemental Fig. 10). This linker has no published conservation with respect to nucleotide identity or length. While unexpected, the deleterious effects of these mutations can be explained by the stabilization of alternate secondary structures favoring the OFF state that are specific to this sequence background. We hypothesize that the U33G mutation dramatically stabilizes an alternate fold that disrupts the P1 binding site (Supplemental Fig. 10). While the SD is not directly sequestered in this competing conformation, the ribosome still struggles to initiate on this sequence at any concentration of guanidine. This mutation is not flagged as detrimental in prior bioinformatic analyses since the alternative conformation is unique to this particular sequence.
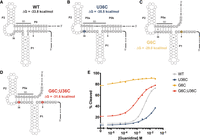
Modeling the role of U36C in riboswitch function. (A) The WT MFE secondary structure of the Pae gdm-II riboswitch. Ribosome binding site shown in navy. (B) The U36C MFE secondary structure. U36C mutation is shown in yellow. (C) The G6C MFE secondary structure. G6C mutation shown in red. (D) The G6C;U36C MFE secondary structure. G6C and U36C mutations are shown as light blue. (E) Representative response profiles of the WT, G6C, U36C, and G6C;U36C Pae gdm-II riboswitches.
The U36C mutation reveals the delicate energetics of this riboswitch's expression platform. While U36 is part of the linker region that is not bioinformatically conserved, it must be anything but a C in this sequence context. Interestingly, we noticed an interaction between U36C and G6: Any mutation to G6 recovered function. We interrogated the connection between these two positions using secondary structure modeling. The minimum free energy (MFE) structure of the wild-type sequence includes the previously modeled P0a and P0b helices (Fig. 6A). P0a sequesters the SD with an aSD element, and P0b comprises the 5′ and 3′ tails. The U36C mutation produces an even better aSD element, increasing the number of base pairs in P0a to 5 in the MFE structure (Fig. 6B). We hypothesize that this OFF state stabilization largely breaks the riboswitch OFF (Fig. 6E). Mutation of G6 (e.g., G6C), on the other hand, completely opens up the P0b helix in the MFE, expanding P0a to 7 bp (Fig. 6C). Destabilization of the OFF state may be the reason this variant is broken constitutively ON (Fig. 6C). The G6C;U36C double mutant, however, recovers function with minimal destabilization of the OFF state (Fig. 6D,E). P0b remains open in this double mutant, but replacing the GU wobble in the 7-bp P0a with a GC pair in the MFE structure may stabilize the OFF state enough to recover ligand responsiveness. Thus, we propose that the U36C point mutation dysregulates riboswitch function in this fixed sequence background by populating an alternate conformation that requires more energy for switching than supplied by ligand binding. Mutations elsewhere in the sequence may energetically compensate and restore the ligand-dependent response.
Wobble base pairs affect apparent sensitivity and cooperativity
The wild-type gdm-II riboswitch exhibits cooperative behavior, with a Hill coefficient greater than 1. While the Hill coefficient produced from fitting this global data set with the Hill equation is extremely sensitive to the quality of the fit, some variants display significant differences in the apparent cooperativity of their guanidine response profiles. The wild-type Pae gdm-II riboswitch cooperatively responds to guanidine with a Hill coefficient of 1.4 ± 0.2, which agrees with the Hill coefficient previously reported for the G. violaceus gdm-II riboswitch (Sherlock et al. 2017). The sensitivity of the Hill coefficient is such that we could only reliably calculate Hill coefficients for the single mutants in this data set.
Among the positions with changes in apparent cooperativity, the GU wobble base pairs in P1 and P2 appeared to tune both the sensitivity and the apparent cooperativity of the riboswitch. These base pairs covary in the bioinformatic consensus but are not strictly conserved as GU wobbles. Transitions to canonical G–C base pairs in either P1 or P2 (Fig. 7A) produce riboswitches that maintain wild-type sensitivity with an average K1/2 of 1.96 mM ± 0.06 mM (Fig. 7B). These mutants display heightened positive cooperativity, however, with an average Hill coefficient of 1.8 ± 0.2 (Fig. 7C). This value is in the fourth quartile of the distribution of Hill coefficient values among functional variants. Transitions to canonical A–U base pairs in either P1 or P2 (Fig. 7A), however, produce riboswitches with 50-fold tighter K1/2 values on average but with an apparent loss of cooperativity (Fig. 7B,C). The average Hill coefficient for these A–U base pairs is decreased to 0.98 ±0.2, which is in the first quartile. The tightening of sensitivity cannot, therefore, be attributed to the stabilization of P1 or P2 since the transition to G–C base pairs maintains wild-type cooperativity and sensitivity.

(A) Secondary structure highlighting the two GU wobbles in the P1 and P2 stems. (B) Representative SMARTI response profiles of the four single mutations restoring Watson–Crick pairing in the P1 and P2 wobbles. (C) Gel-based RelE cleavage curves for the G13A (n = 3), U26C (n = 2), G13A;G46A (n = 3), and U26C;U57C (n = 3) mutants.
To further explore the effects of these mutations on cooperativity, we used a gel-based readout of RelE cleavage to test individual mutant sequences (Fig. 7C). In this assay, the G13A mutant again shows an order of magnitude increase in sensitivity and a modest decrease in apparent cooperativity with a Hill coefficient of 1.3 ± 0.2 (n = 3). The U26C mutant showed a tighter K1/2 but an increased Hill coefficient of 1.8 ± 0.1 (n = 2). The G13A;G46A and U26C;U57C double mutants behave the same as the single G–U wobble mutants, with Hill coefficients of 1.4 ± 0.2 and 1.9 ± 0.2, respectively. These G–U wobble positions are clearly integral to the communication between the two guanidine binding sites, but how mutations at these sites impact this energetic network is not immediately apparent.
DISCUSSION
The expansion of the RelE cleavage assay to a sequencing-based readout establishes an efficient and quantitative approach for the comprehensive mutational analysis of translational riboswitches. We generated quantitative functional data of ligand responsiveness for over 23,000 single and double mutants of the 72-nt P. aeruginosa gdm-II riboswitch. Using these functional data, we determined the biochemical conservation of each position in this sequence. Conservation of the binding pockets is consistent with previous bioinformatic analyses, and we hypothesize that variants of the biochemically conserved positions not observed in prior computational and structural studies may stabilize functionally incompetent alternative structures in this fixed sequence background. While there is no evidence for pairing between the SD and aSD in the P0a helix, these data have confirmed the functional relevance of the putative P0b helix and identified key nucleotides that tune the riboswitch's sensitivity and apparent cooperativity.
When riboswitches respond to ligands with poorly understood metabolisms, studying these regulatory systems may also further our understanding of biology. The Pae gdm-II riboswitch cooperatively senses two molecules of the small toxic metabolite guanidine. The riboswitch responds with a weak apparent K1/2 in the low millimolar range. Little is known about guanidine metabolism, although a guanidine hydrolase was recently identified in cyanobacteria (Funck et al. 2022). The KM of this enzyme is similarly in the low millimolar range, which, in conjunction with the wild-type K1/2 value reported for the Pae gdm-II riboswitch, implies that significant accumulation of guanidine is required to trigger a cellular response. The riboswitch can improve its sensitivity by two orders of magnitude with a single mutation; therefore, we suggest that the riboswitch has been finely tuned to respond to guanidine at a physiologically relevant concentration.
For engineering purposes, mutational data factors dramatically into construct development. The single and double mutant fits in this data set indicate that a wide range of amplitudes and sensitivities are achievable with conservative sequence changes. This comprehensive functional survey of single and double mutations provides an atlas of potential guanidine biosensors covering an extensive range of possible response profiles. If a biosensor responding to guanidinium in the mid-micromolar range were needed, for instance, a single P0b point mutant may be used to tighten the sensitivity of this riboswitch.
Riboswitches operate within two regimes: kinetic and thermodynamic. Transcriptional riboswitches typically function kinetically since the window for ligand binding is necessarily short (Wickiser et al. 2005; Quarta et al. 2012; Steinert et al. 2017; Scull et al. 2021). The aptamer must sense the appropriate ligand before the polymerase has transcribed past the terminator. Translational riboswitches, on the other hand, can operate thermodynamically to control gene expression over the lifetime of the transcript (Lemay et al. 2011). The conformational dynamics of the 5′ UTR populate binding-competent states that can respond to ligand and alter the rate of ribosome association post-transcriptionally. Given the equilibrium nature of the assay, we cannot observe the kinetic ramifications of these mutations, only the thermodynamic effects. As seen with a Type 1 glycine singlet (Torgerson et al. 2018), we have identified a single GU-to-GC transition in the P. aeruginosa gdm-II riboswitch that constitutes the difference between a broken and a functional switch. While a translational guanidine riboswitch and a transcriptional glycine riboswitch are obviously dramatically different elements in sequence, mechanism, and kinetics, the present data indicate that, regardless of regime, the thermodynamics of riboswitch expression platforms are precisely tuned—a single hydrogen bond can make or break them.
These data also indicate that direct pairing of the SD sequence is not strictly necessary for controlling gene regulation in this translational riboswitch system. Interestingly, both single and double mutations to the SD sequence have very little effect on translation initiation efficiency in this in vitro assay, indicating a significant tolerance for deviation from canonical 16S rRNA pairing. We see no evidence of covariation within P0a, which comprises the SD and aSD. The U36C mutant breaks the riboswitch OFF by enhancing the stability of the P0a helix, so P0a can still play a functional role. However, the only variants capable of breaking the riboswitch ON reside in P0b. Pairing in this region exhibits covariation, and structure in between the SD and the start codon is sufficient for effecting ligand-dependent gene regulation. Formation of the P0b helix in the OFF state abrogates ribosome binding and translation initiation. Rather than utilizing the formation of an alternate helix in the ON state, ligand-induced dimerization of the P1 and P2 helices physically separates the two strands of the P0b helix in space to allow for loading of the ribosome on the SD. Thus, the true expression platform, instead, resides in the P0b helix. This finding may assist in the categorization of additional translational riboswitches that do not include obvious anti-SD sequences.
Mutations to the flexible linker region of the P. aeruginosa gdm-II riboswitch highlight variants that are not functionally tolerated in this fixed sequence background. Bioinformatic consensus diagrams illustrate the conservation of key nucleotides and positions in sequences derived from diverse organisms. These sequences are the product of extensive evolutionary pressure, so conflicting mutations, especially those close in sequence space, would be rapidly eliminated. This comprehensive mutational analysis has experienced no functional selective pressure and instead offers an unbiased view of mutational interactions in this fixed background: Both positive and negative interactions are seen within this data set. The biochemical consensus, therefore, reveals the rugged functional landscape within two mutations of this defined sequence. Mutations to the highly bioinformatically conserved nucleotides are predictably detrimental. The repressive conformational rearrangements we hypothesize are induced by the U33G and U36C mutations indicate that single point mutants outside of the aptamer and expression platform can severely inhibit riboswitch function. Of the possible single mutations to the Pae gdm-II riboswitch, ∼50% retain function. Previous work has shown that ∼30% of random single amino acid mutations in a given protein were inactivating substitutions (Guo et al. 2004). This comparison suggests that within a fixed sequence background, single nucleotide substitutions to a functional RNA may be more detrimental than single amino acid substitutions to a functional protein.
These data highlight the important functional role of the GU wobble base pairs in the P1 and P2 helices of this riboswitch. Single mutations restoring canonical Watson–Crick pairings reveal that these positions fine-tune the sensitivity and apparent cooperativity of the riboswitch. Explaining the loss of apparent cooperativity with mutations at these positions is more elusive. Cooperativity has been observed in multiple classes of riboswitches including glycine (Kwon and Strobel 2008), THF (Trausch et al. 2011), cyclic-di-AMP (Gao and Serganov 2014), and, more recently, PreQ1 (Schroeder et al. 2022). Dual ligand binding may provide the riboswitch with a more sensitive dial, allowing a more digital response to its ligand. Binding two ligands, especially for small metabolites like glycine and guanidine, may provide more energy for conformational switching as well. The loss of apparent cooperativity with a single mutation is therefore an intriguing dimension to the Pae gdm-II functional landscape. It could simply reflect a divergence from the energetic assumptions of the Hill equation, including that each binding site has a similar affinity (Stefan and Le Novère 2013). Cooperative responses are observed when binding to the first site improves the affinity of the second site. This results in a steeper response, since binding a second equivalent is more favorable after the first binding event. However, these mutations to the GU wobble positions may stabilize one binding site to such an extent that the second binding event is no longer significantly more favorable than the first thereby reducing the Hill coefficient. In a tandem glycine system, remediation of a GU wobble to a GC pair stabilized the weaker aptamer enough to restore cooperativity as evidenced by a Hill coefficient of 1.4 (Torgerson et al. 2020). We may be observing the inverse in this guanidine system, however, where stabilization of one binding site over the other with a GU to GC transition destroys apparent cooperativity by increasing the discrepancy between binding site affinities. Alternatively, these mutations may also indicate a disruption of the communication network between the two binding pockets. Further study is needed to make the distinction.
The use of RelE in high-throughput mutational analysis has allowed us to generate a vast amount of functional data about a translational riboswitch, but this method can be readily applied to other translational regulators. We have previously shown the ability of RelE to report specificity changes in variant riboswitches and quantitative translation initiation differences in yeast 5′ UTR isoforms. Application of this method to RNA thermometers (Kortmann and Narberhaus 2012), viral IRESes (Jaafar and Kieft 2019), variant riboswitches (Weinberg et al. 2017), and T-box RNAs among others will increase our understanding of the biology, evolution, and therapeutic potential of these elements.
MATERIALS AND METHODS
Design of riboswitch constructs
The wild-type sequence for the P. aeruginosa gdm-II riboswitch has been reported previously (Reiss and Strobel 2017). For sequencing purposes, a 22-nt handle was added to the 5′ end of the riboswitch, and ∼20 nt past the endogenous start codon were taken from the P. aeruginosa PAO1 genome. The second codon was mutated to a TAG stop codon for efficient RelE cleavage, and GTA was added to the 3′ terminus such that the cleaved and full-length products contained the same 3′ triplet to eliminate potential ligation bias. The mutant library was created via doped oligos, where each mutated position was mutated at the frequency of 4.2% (1.4% for each substituting nucleotide).
RNA preparation and labeling
RNA was transcribed directly from oligonucleotides ordered from Keck Oligo Synthesis Resource at Yale University. Briefly, the in vitro transcription reaction contained 80 mM HEPES NaOH (pH 7.5); 1 mM spermidine; 120 µg/mL BSA; 5 mM DTT; 33 mM MgCl2; 4 mM each f ATP, CTP, GTP, and UTP; 20 ng/µL template oligo; 10 ng/µL T7 duplex oligo; 1 U/mL iPPase; and in-house prepared T7 RNA polymerase. After incubating at 37°C, the RNA was purified via denaturing PAGE. For 5′ radiolabeling, RNA was dephosphorylated with Antarctic Phosphatase (NEB) and labeled with 32P-γ-ATP (PerkinElmer) via T4 PNK (NEB). Radiolabeled RNA was similarly purified via denaturing PAGE.
Translation initiation and RelE cleavage
RNA refolding solutions contained the following: 100 nM RNA, 1× 219H Buffer (50 mM HEPES-KOH at pH 7.5, 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2) and 0 µM, 125 µM, 500 µM, 1.25 mM, 5 mM, 12.5 mM, 25 mM, 50 mM, 125 mM, or 250 mM guanidine hydrochloride. For gel-based assays, refolding solutions also contained 5′-radiolabeled RNA in trace. RNA was refolded in a thermocycler by heating for 2 min to 90°C and slow cooling in a thermocycler with a cooling rate of 0.1 C/sec. Translation initiation solutions contained the following: 1× 219H Buffer, 100 nM IF1, 100 nM IF2, 100 nM IF3, 1 mM GTP, 100 nM fMet-tRNAfMet, 100 nM 70S E. coli ribosomes, 10 nM RNA, and 0 µM, 12.5 µM, 50 µM, 125 µM, 500 µM, 1.25 mM, 2.5 mM, 5 mM, 12.5 mM, or 25 mM guanidine hydrochloride. IF1, IF2, and IF3 were purified from E. coli as previously described (Brunelle et al. 2006). fMet-tRNAfMet was charged with S-100 lysate as previously described (Walker and Fredrick 2008). Translation initiation solutions were incubated for 15 min at 37°C before incubating with 1 µM RelE. Cleavage reactions were quenched with the addition of an equal volume of either FLB (25 mM EDTA, <0.1% Xylene Cyanol, <0.1% Bromophenol Blue, ∼93% formamide) for gel-based assays or SPRI Binding Solution (2.5 M NaCl, 20% PEG) for sequencing-based assays.
Preparation of RNA for high-throughput sequencing
An equal volume of SPRI beads (Bulldog Bio) was added to each quenched RelE reaction tube and mixed thoroughly. Bead purification of the RNA was performed by incubating for 15 min, washing the pelleted beads twice with 70% ethanol, and eluting into water. The 2′,3′ cyclic phosphate left by RelE cleavage was healed with T4 PNK (NEB) to restore the 3′-OH. The T4 PNK master mix was added to each RNA sample, and the reaction was performed with-bead at 37°C for 1 h (Fisher et al. 2011). An equal volume of SPRI Binding Solution (2.5 M NaCl, 50% PEG) was added to each sample, and bead purification was performed as previously described.
A preadenylated DNA adaptor (/5rApp/NNNNNCTGTAGGCACCATCAAT/3ddC/; ordered from IDT) was ligated onto the 3′ end of the eluted RNA via T4 RNA Ligase II KQ (NEB). The ligation mixture contained 1× T4 Ligase Buffer, 1 µM DNA adaptor, 25% PEG8000, 10 U/µL T4 RNA Ligase II KQ and was incubated at room temperature overnight. An equal volume of SPRI binding solution was added to each sample, and bead purification was performed as previously described.
Priming off the ligated adaptor, reverse transcription (RT) was used to convert the RNA into cDNA. The RT primer was annealed to the RNA during a 5-min incubation at 65°C in the presence of dNTPs before cooling for 2 min to 4°C. The remaining reagents were then added such that the final RT reaction contained 1× SSIV buffer, 500 µM dNTPs, 25 nM primer, 0.2 U/µL RNaseOUT (Invitrogen), 10 U/µL SuperScript IV (Thermo Fisher), and RNA. The reaction was incubated for 45 min at 55°C and then heated for 5 min to 85°C to heat-inactivate the SSIV enzyme. RNaseA (0.5 µg/µL; Thermo Fisher) and RNaseH (0.1 U/µL; Invitrogen) were added, and the mixture was incubated for 1 h at 37°C to degrade the RNA. The crude RT mixture was then used as the template for 12 rounds of PCR, during which the barcoded Illumina adaptors were added to the 5′ and 3′ termini. SPRI beads (1.2×) were added to the PCR reactions, and the DNA was bead-purified as previously described, except the DNA was eluted into 10 mM Tris-HCl (pH 7). The DNA concentrations were measured via Qubit, and samples were combined at approximately equal ratios for submission to the Yale Center of Genome Analysis (YCGA) for sequencing.
High-throughput sequencing
Samples from two complete replicates were sequenced at the YCGA on an Illumina HiSeq 4000 (2 × 150 bp). For each replicate, samples were pooled and sequenced using 30% of a lane for (∼3%–4% of a lane per sample). Demultiplexing was done by the YCGA.
Analysis of sequencing results
The 5′ constant region and the 3′ adaptor were removed from the sequencing reads using CutAdapt (Martin 2011). The remaining sequences were aligned to a full-length WT sequence using Bowtie2 (Langmead and Salzberg 2012). Discordant alignments were not permitted. The first replicate generated 50,906,513 aligned reads, and the second replicate
generated 105,873,887 aligned reads. Custom Python scripts previously reported (Torgerson et al. 2018) were used to determine the fraction of cleaved RNA for all variants with zero to two mutations at each ligand concentration
tested. Reads were classified according to the individual nucleotide mutations they contained between position 1 and position
72 (the last nucleotide before the start codon). Reads were labeled as cleaved if the last nucleotide of the sequence was
between positions 75 through 77, consistent with RelE cleavage of the transcript at the second codon. Reads were labeled as
full-length if the last nucleotide fell after position 85, indicating that the RelE had not cut the RNA and the message remained
full-length. The number of full-length and cleaved reads were then counted for each variant to calculate the fraction of cleaved
RNA. Fraction cleaved was calculated for each mutant at each ligand concentration. The data were fit with the Hill Equation
in R,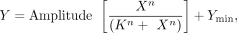 (1)
where X is the concentration of ligand; Y is the percent cleaved; K is the K1/2; Ymin is the minimum percent cleaved; amplitude is the difference between Ymax and Ymin; n is the Hill coefficient. Standard deviations of all fit values were calculated as previously described in Torgerson et al. (2018). Curves were plotted and visualized in Prism 9.
(1)
where X is the concentration of ligand; Y is the percent cleaved; K is the K1/2; Ymin is the minimum percent cleaved; amplitude is the difference between Ymax and Ymin; n is the Hill coefficient. Standard deviations of all fit values were calculated as previously described in Torgerson et al. (2018). Curves were plotted and visualized in Prism 9.
Values were mapped onto secondary structures using custom Python scripts. Double mutant heat maps were generated in Prism 9.
Calculation of a function parameter
Fits were excluded if the standard deviation was greater than the value for the K1/2 and amplitude. The apparent K1/2 value was converted into a free energy according to the following equation: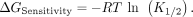 (2)
(2)
The ΔΔGSensitivity was calculated by subtracting the WT value from the variant value. The amplitude was transformed into a pseudoenergy according
to the following equation: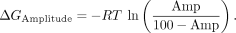 (3)
(3)
The ΔΔGAmplitude was similarly calculated by subtracting the WT value from the variant value. The function parameter F was calculated from the sum of the two energies as follows: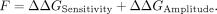 (4)
(4)
To calculate epistasis, the average function parameter for each individual mutation was calculated (Fi). The sum of the individual mutations was then subtracted from the double mutation function: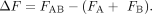 (5)
Propagated error was calculated according to the appropriate transformations, and functional parameter values were excluded
if the standard deviation was greater than the parameter value.
(5)
Propagated error was calculated according to the appropriate transformations, and functional parameter values were excluded
if the standard deviation was greater than the parameter value.
Generating a biochemical consensus
The frequency distribution of function parameters was determined in Prism 9. Sequences were removed as nonfunctional if the function parameter was one standard deviation above the mean (Supplemental Fig. 6). This generated a pool of functional single mutants and double mutants. The number of times a single mutation (e.g., A21G) appeared in the pool of functional single and double mutants (Ni,j) was then determined, where i indicates the position (e.g., A21) and j indicates the mutation (e.g., G) such that i,j identifies the specific variant (e.g., A21G).
For the WT nucleotide at each position, the number of functional single mutants at that position was subtracted from the total
number of functional singles: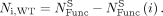 (6)
(6)
The proportion of each nucleotide at each position was calculated by dividing its number of functional variants by the sum
of the functional number for all mutations and WT at that position.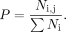 (7)
(7)
Cutoffs for nucleotide identity and purine/pyrimidine distinctions were made by plotting the frequency distribution of nucleotide proportions in Prism and visually assigning cutoffs (Supplemental Fig. 8).
Modeling minimum free energy conformations
The MFE and structure for WT and select mutant constructs were determined by RNAfold (Gruber et al. 2008) using the following command: RNAfold -p -d2 –noLP.
DNA oligonucleotides and chemicals
All DNA oligos were synthesized by the W.M. Keck Oligonucleotide Synthesis Facility at Yale University unless noted otherwise. Guanidine hydrochloride was obtained from Sigma.
DATA DEPOSITION
Raw sequencing data analyzed in this manuscript were deposited in the Sequencing Read Archive (BioProject ID PRJNA912765). An Excel file containing all the fit parameters for the single and double mutants from both replicates is included in the Supplemental. An Excel file containing the biochemical consensus calculations is also included in the Supplemental. Any additional data pertinent to this work are available from the author upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Kathryn Barth, Chad Torgerson, and Matthew Simon and members of his laboratory for insights during data analysis. We also thank the Yale Center for Genome Analysis, particularly Chris Cristaldi, for their assistance with sequencing, and the Yale Center for Research Computing for their infrastructure. C.M.F. was supported by the National Institutes of Health Cellular and Molecular Biology Training Grant (T32GM7223-44) and the National Institutes of Health Molecular Medicine Training Grant (T32GM100884-07). S.G.G. was supported by the National Institutes of Health Chemical Biology Training Grant (5T32GM067543-19). This work was supported by the National Institutes of Health grant GM136969 to S.A.S.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079560.122.
-
Freely available online through the RNA Open Access option.
- Received December 18, 2022.
- Accepted April 5, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Caroline Focht is the first author of this paper, “Translation regulation by a guanidine-II riboswitch is highly tunable in sensitivity, dynamic range, and apparent cooperativity.” Caroline did this work as a graduate student in Molecular Biophysics and Biochemistry at Yale University performing research in Scott Strobel's laboratory. Her research focused primarily on studying translation regulation by elements of the 5′ UTR.
What are the major results described in your paper and how do they impact this branch of the field?
We extended our previous gel-based translation initiation assay to a next-generation sequencing output, which allowed us to simultaneously analyze over 23,000 mutations to the Pseudomonas aeruginosa guanidine-II riboswitch and how these variants affected the riboswitch's function. These data revealed how the riboswitch can access amplitudes from 0% to 100% and tighten its sensitivity 100-fold within two mutations. This work dramatically expands the toolbox for comprehensive examination of translational riboswitches and translational regulators in general.
What led you to study RNA or this aspect of RNA science?
I actually have a distinct memory of doodling “I love protein synthesis” at the top of a worksheet in high school biology, but when I began pursuing my PhD at Yale—one of the premier institutions for RNA biochemistry—I really became intrigued by the chemical versatility of this macromolecule. Scott's laboratory provided the perfect environment for examining the intersection of structured RNAs and translation machinery.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
Ironically, I think the moment that impacted my development as a scientist the most actually came during office hours for a history course I took in undergrad. I had asked my professor something about this population of ancient Greeks I couldn't find much information on, and he responded, “Those are the best types of questions: Noticing what sources aren't saying is as important as what they are. The negative space is the most exciting.” Honestly, that really helped me think comprehensively and pay attention to what I didn't know as much as what I did know.
If you were able to give one piece of advice to your younger self, what would that be?
The people you are surrounded by are just as impactful as the work that you're doing. It's extremely hard to perform good science in a bad environment. Everyone will celebrate when the experiment works, but you want to be in a laboratory that helps you move forward when it doesn't. I had fantastic labmates during my PhD, and they made a world of difference in how much I enjoyed grad school.








