
Codon bias shapes bacterial small RNA binding sites within protein-coding sequences
- Department of Microbiology and Molecular Genetics, IMRIC, Faculty of Medicine, The Hebrew University of Jerusalem, Jerusalem 9112102, Israel
- Corresponding author: hanahm{at}ekmd.huji.ac.il
-
Handling editor: Peter Stadler
Abstract
Bacterial small RNAs (sRNAs) regulate gene expression by base-pairing with target mRNAs, affecting their stability and translation. While sRNA binding sites were initially identified in 5' untranslated regions of mRNAs, consistent with their role as translation-initiation regulators, recent large-scale studies have revealed sRNA binding sites within protein-coding sequences, suggesting additional regulatory mechanisms. It is intriguing to explore how the latter sRNA binding sites are adjusted with the reading frame and what selection forces maintain them within the coding sequence through evolution. Using RIL-seq data, we determined prime sRNA binding positions within coding sequences, which are positions within the inferred binding-site motif that show exceptionally high conservation across target sequences (≥95%), indicating their putative importance for sRNA–mRNA base-pairing. We found that these positions are mostly adjusted with the reading frame and correspond to the most frequent codons, high above random expectation. This suggests that frequent codons may facilitate sRNA–mRNA encounters and that codon usage bias influences binding site formation via selective pressures. Conservation analysis across genomes in the Enterobacterales order revealed that prime positions show relatively high conservation of base-pairing interactions. However, in some genomes base-pairing in these positions may be hampered due to the degeneracy of the genetic code. This is often compensated for by other positions that conserve the base-pairing interactions, ensuring the maintenance of a requisite number of base pairs for sustaining the sRNA–target interaction. Our findings highlight the importance of distinct interacting positions as well as an adequate number of base pairs for sustaining sRNA–target interactions.
Keywords
INTRODUCTION
Bacterial small RNAs (sRNAs) play major roles in posttranscriptional regulation of gene expression. These 50–400 nt-long RNA molecules act mainly in trans by base-pairing directly with their target mRNAs. Bacterial sRNAs usually function in association with the protein chaperone Hfq, a ring-shaped hexamer that is abundant in Gram-negative bacteria (De Lay et al. 2013; Updegrove et al. 2016; Santiago-Frangos and Woodson 2018). As more and more sRNAs were identified in a wide range of organisms (Axmann et al. 2005; Vogel 2009; Schluter et al. 2010; Liang et al. 2011; Tesorero et al. 2013), it has become evident that gene expression regulation by these molecules is widespread and plays a role in many cellular mechanisms and especially in response to stress conditions.
sRNAs control protein expression by acting as positive or negative regulators (Waters and Storz 2009; Storz et al. 2011; Wagner and Romby 2015; Hör et al. 2020). They can regulate translation initiation by affecting ribosome binding. As negative regulators, they base pair with the mRNA near the ribosome binding site (RBS), preventing ribosome binding, thus inhibiting translation initiation. As positive regulators, they can bind the mRNA and change its structure, exposing an otherwise occluded RBS, thus enhancing translation initiation. Through the sRNA effect on the ribosome, it also has an indirect effect on the mRNA stability, by influencing the exposure of the mRNA to endoribonucleases. sRNAs can also affect cleavage by endoribonucleases in a translation-independent manner, by base-pairing with the mRNA and blocking or exposing the endoribonuclease binding or cleavage site (e.g., Huntzinger et al. 2005; Frohlich et al. 2013). When repressing translation by inhibiting ribosome binding, often the sRNA binding site on the mRNA was found to be in the vicinity of the RBS (Masse and Gottesman 2002; Sharma et al. 2007). However, to induce a structural change in the mRNA in order to expose an occluded RBS or to affect endoribonuclease binding and/or cleavage, the sRNA binding site may be located in various regions along the target transcript.
Recent advances in elucidation of global sRNA–target maps, such as RIL-seq (Melamed et al. 2016), GRIL-seq (Han et al. 2016), CLASH (Iosub et al. 2020), or iRIL-seq (Liu et al. 2023), and their application to various bacteria, have allowed the determination of several sRNA–target interactomes along with the putative regions of base-pairing involved in each interaction, opening the door to comprehensively study sRNA binding strategies from genomic and evolutionary points of view.
RIL-seq data provide clues to the regions involved in base-pairing in the target transcript and in the sRNA. The determination of these regions defines the sequences in which the putative sRNA–target binding sites may reside and allows more focused analysis toward their identification. Since it is conceivable that most target sequences of a certain sRNA will interact with it through the same binding site, a common practice has been to search for a shared motif among the target sequences and verify that it is complementary to a subsequence of the sRNA. This way, large-scale RIL-seq data have been exploited to determine sRNA binding sites on the targets, many of which were either consistent with previous experimentally based knowledge or further validated experimentally (e.g., Melamed et al. 2016). Since the motif representing the binding site compiles information from multiple targets, it is often presented by a position-specific scoring matrix (PSSM) and is visualized by a Logo, capturing the frequency and/or enrichment of each nucleotide in every position along the motif. Examination of the motifs indicates that there are positions that maintain a specific nucleotide in most target sequences, while other positions are more variable, suggesting that the preserved positions may be more important for the binding and regulation by the sRNAs (Fig. 1). Hereinafter, the highly preserved positions are termed “prime positions.”
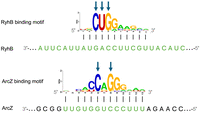
sRNA binding motifs. Common sequence motifs identified in E. coli RIL-seq target sets of RyhB and ArcZ (Melamed et al. 2016), which are complementary to the respective binding sites on the sRNAs (marked in green). The y-axis depicts the information content of a position, in bits. Black vertical lines denote base-pairing interactions. The blue arrows mark highly preserved positions, hereinafter, termed “prime positions.”
From RIL-seq data it is clear that while there are putative binding sites at 5′ UTRs, as expected for regulation of translation initiation, most of the putative binding sites were identified in the coding sequences (CDSs) of the targets. This raises an intriguing question about the locations of prime positions in regard to the reading frames. In case of one prime position per binding motif, it could align with the first, second, or third position of a codon. Nucleotide changes in the first or second position are more detrimental to the identity or nature of the encoded amino acid, respectively, and therefore it is expected that there might be evolutionary pressure to keep them preserved. Such preservation should also maintain the base-pairing between the sRNA and target and keep the posttranscriptional regulation intact. The nucleotide at the third codon position is more flexible due to the degeneracy of the genetic code, and therefore, alignment of a prime position with the third codon position might be less favorable for maintaining the base-pairing and the regulation. In case of two or three primary positions, they could align either adjusted with the reading frame or shifted off the reading frame (Fig. 2). What strategy has actually taken place in nature? The wealth of data on sRNA binding sites allows addressing this question systematically.
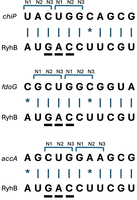
Possible alignments of prime positions with codon positions. Possible adjustments of RyhB binding sites in reference to the reading frames of the targets are demonstrated. For RyhB, three prime positions were identified (Fig. 1) and are underlined. (A) Example of RyhB interaction with a target where the first prime position aligns with position N3 of a codon, and the next two prime positions align with positions N1 and N2 of the next codon. (B) Example of RyhB interaction with a target where the two prime positions align with positions N2 and N3 of a codon, and the last prime position aligns with position N1 of the next codon. (C) Example of RyhB interaction with a target where the three prime positions are adjusted with the reading frame.
In the current study, we investigate binding sites located in CDSs from genomic and evolutionary points of view, paying special attention to the prime positions. Our genomic view is codon-centered, aiming to learn from sRNA binding site data whether there is a favorable arrangement of the prime positions in regard to the reading frame. The evolutionary view takes advantage of the rich bacterial genome data to examine the evolutionary conservation of nucleotides and positional base-pairing in the binding sites in general, and in particular at prime positions.
RESULTS
A reliable data set of sRNA–target interactions
In this study, we focus on sRNA–mRNA interactions for which the binding site on the target was identified in the CDS. We based our study on Escherichia coli RIL-seq sRNA–target interaction data (Melamed et al. 2016) and extracted from it interactions obeying at least one of the following criteria: (1) The interaction was identified in E. coli K-12 as statistically significant in at least three individual libraries for at least one of the three tested conditions or growth phases in the original RIL-seq study (Melamed et al. 2016): exponential phase, stationary phase, or exponential phase under iron limitation. (2) The E. coli interaction was also determined as statistically significant in at least one other of the following organisms to which RIL-seq was applied: enteropathogenic E. coli (EPEC) (Pearl Mizrahi et al. 2021), Salmonella enterica (Matera et al. 2022), or Klebsiella pneumoniae (Goh et al. 2024). We believe that this set of interactions is of high reliability. In total, 554 sRNA–target interactions complied with at least one of the above criteria. Encouragingly, 182 out of the 554 interactions complied with both criteria, further supporting their validity.
Of this set, we included for further study only targets in which the sRNA binding site was located within the coding sequence (CDS), as previously identified (Melamed et al. 2016), resulting in a total of 402 sRNA–target interactions involving 20 different sRNAs (Supplemental Table S1). Among these, 121 interactions met only the first selection criterion, 158 met only the second, and 123 interactions satisfied both criteria.
General characteristics of the binding sites
In the analyses in this section, only sRNAs with at least five targets were included.
Number of fulfilled base pairs between the sRNA and target
For each sRNA, a binding motif was defined by searching for a common motif among the sequences of its targets (Fig. 1; Melamed et al. 2016). These motifs ranged in length between 8 and 15 nt (Fig. 3). For each sRNA, we recorded the putative binding sites in E. coli target sequences (Materials and Methods), and counted in all the targets of each sRNA both the number of putatively fulfilled base pairs and the number of consecutive base pairs involved in the sRNA–target interaction (Fig. 3). Not all positions in the binding site on the sRNA necessarily perform base-pairing with positions within the binding site on the mRNA, as there are positions with mismatches. The term “fulfilled base pairs” refers to the nucleotides within the sRNA and mRNA binding sites that are complementary and can be engaged in base-pairing. As shown, both sRNAs with short binding motifs and sRNAs with long binding motifs showed comparable numbers of putatively fulfilled base pairs (involving 8–10 nt on average across the targets of an sRNA). Likewise, for all sRNAs, the average number of consecutive base pairs is independent of the length of the binding site and involves 4–8 nt (showing some tendency to be located at the 5′ part of the binding site [Fig. 3]). This may suggest that a minimum number of base pairs as well as of consecutive base pairs is necessary for establishing sRNA–target interactions, regardless of the binding site length.
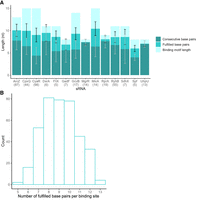
Number of positions fulfilling sRNA–target base-pairing interactions. (A) Bars represent the motif length for each sRNA (light green), the number of fulfilled base pairs (darker green), and the number of consecutive fulfilled base pairs (darkest green). Number in parentheses indicates the number of targets of each sRNA in the data (sRNAs with at least five targets were included in this analysis). Black lines and gray lines indicate the standard deviations of the number of fulfilled base pairs and consecutive base pairs, respectively. (B) Number of fulfilled base-pairing interactions per binding site across all studied sRNAs. The y-axis shows the number of sRNA–target pairs that fulfilled a particular number of base pairs.
Distribution of base pair types in the interaction sites
Nucleotides A and C can each base pair with only one nucleotide, A with U and C with G, while G and U can base pair with two nucleotides, G with C and U and U with A and G. As shown in Supplemental Figure S1, the most frequent base pairs in the interaction sites are satisfied by C and U in the sRNA interacting with G and A, respectively. While U in the sRNA can also base pair with G, evidently in most cases, it is involved in the canonical interaction with A. Likewise, when G in the sRNA is involved in the base-pairing, it most often interacts canonically with C rather than with U. Base-pairing between G and U is the least frequent between the sRNA and the target. Also, there is divergence in prevalent base pair types among different sRNAs, with U-A (U in the sRNA and A in the target) exhibiting prominence in the interactions of six distinct sRNAs.
Location of the binding sites
For each sRNA, we analyzed the location of the binding sites within the target CDS and found that in 50% of the targets, the binding site is located in the first third of the CDS (Fig. 4A). In contrast, when examining the position of the binding sites along the sRNA molecules themselves, we did not observe a consistent positional preference (Fig. 4B). This result is in agreement with previous reports. While some studies have reported that binding sites reside close to the 5′ end of sRNAs (e.g., Papenfort et al. 2010), such positional bias was demonstrated only for a few sRNAs. In a previous systematic analysis of 14 major E. coli sRNAs, binding sites were found at various positions along the sRNAs (Peer and Margalit 2011). Supporting this broader distribution of binding site locations, Gorski et al. (2017) highlighted the heterogeneity in both sRNA sizes and the positioning of their seed sequences, in contrast with the more defined organization observed in miRNAs and crRNAs (Gorski et al. 2017). Thus, the observed variability in binding site positions along sRNAs likely reflects genuine biological diversity, and highlights the different structural and functional constraints of bacterial sRNAs compared to miRNAs.

Distribution of binding site locations on targets and sRNAs. (A) For each sRNA, the relative positions of its binding sites along target mRNAs are shown. Target coding sequences were divided into 10 equal-length bins, and the fraction of binding sites per bin is shown. (B) Binding site locations along the sRNAs themselves are similarly binned into 10 segments. Only sRNAs with at least five targets are shown.
Prime positions
As described above, the binding motif of each sRNA is described by a PSSM, representing the positional frequency (or preference relative to background frequency) of each of the 4 nt along the motif positions. High frequency at a position indicates that a certain nucleotide at that position was present in many of the target sequences, hinting at its possible importance for sRNA–target interaction and for regulation by the sRNA. As noted above, we term these positions “prime positions.” We arbitrarily and strictly defined a prime position as a position in which one of the nucleotides has a frequency of at least 0.95. For instance, among RIL-seq targets of RyhB in E. coli K-12, positions 3, 4, and 5 within the RyhB binding motif exhibit a frequency ≥0.95 for C, U, and G, respectively, determining them as prime positions of RyhB binding site (Fig. 1). While we refer to the prime positions by their location in the binding site on the target mRNA, we correspondingly define the prime positions on the sRNA.
Six sRNAs in our data had less than five targets and one sRNA (GcvB) did not have any position with a nucleotide frequency ≥0.95 in the PSSMs representing its R1 binding site (Sharma et al. 2011), and therefore these seven sRNAs were not included in this analysis. For the remaining 13 sRNAs, we identified a total of 26 prime positions in the PSSMs (Table 1). For most sRNAs, the prime positions are preferentially located at the center of the binding site (Supplemental Fig. S2). There is overrepresentation of C and underrepresentation of U in the prime positions (10 and 3, respectively). These Us in the sRNAs always base pair canonically with A in the targets. Likewise, when G is present at a prime position in the sRNA, it almost exclusively base pairs with C in the target sequence, while in other positions along the binding sites, base-pairing of G:U is also observed, although to a small extent. Apparently, 98% of the interactions involving the prime positions present canonical base-pairing in all targets.
Prime positions in the binding sites of sRNAs
Location of the prime positions in reference to the reading frame
To learn about the location of the prime positions in regard to the reading frame, we turned back to the individual targets of an sRNA and for each sequence recorded the alignment of a prime position with a reading frame position, whether N1 (first nucleotide in codon), N2 (second nucleotide in codon), or N3 (third nucleotide in codon) (Fig. 2). We first regard sRNAs for which we identified only one prime position in the binding site (CyaR, DsrA, FliX, MicA, and UphU) and sRNAs where the prime positions are at a distance of at least 3 nt and hence cannot align to the same codon (RprA and Spf position 7) (Table 1). In quite a few targets of these sRNAs, we find the prime positions aligned with N1, which is crucial for the identity of the amino acid and therefore is highly conserved. Only in a few targets, the prime positions align with N2, the preservation of which preserves the characteristics of the amino acid. Exceptions are RprA and Spf (prime position 7), where in 50% of the targets, the prime positions align with the second position of a codon. Interestingly, for a substantial number of targets of these sRNAs, the single prime positions align with N3, high above random expectation (P ≤ 10−4 by χ2 test), often when the specific nucleotide involved in the sRNA binding defines the most frequent codon of the encoded amino acid (Table 1). The frequent codon was defined as the most commonly used codon per amino acid, or the sole codon in cases where only one exists. Conceivably, there is selective pressure to keep the nucleotide of the frequent codon intact, and this also preserves the binding capability of the sRNA in these positions.
Next, we turn to sRNAs with two prime positions (ArcZ, CpxQ, GadF, and SdhX) (Table 1). For three of these sRNAs, the prime positions are consecutive (CpxQ, GadF, and SdhX). The prime position of most targets of CpxQ and SdhX aligns with the second and third positions of a codon, while in most targets of GadF, they span two codons (the third position of a codon and the first position of the next codon). As to ArcZ, its prime positions are 1 nt apart, and in most targets, they align with the first and third positions of a codon. The number of targets in which the two prime positions are adjusted with a codon is statistically significantly above random expectation (P ≤ 10−11 by χ2 test). Furthermore, in almost all of these targets, the nucleotide aligned to the third codon position is the one defining the most frequent codon.
RyhB has three consecutive prime positions, positions 3, 4, and 5 of the binding site on the target. Out of 53 targets, in 40 targets, these positions are aligned respectively with the CUG codon, the most frequent codon of leucine (Fig. 5). MgrR has five prime positions, three of them are consecutive, and in 50% of the targets, they align to the first, second, and third codon positions, where the nucleotide in the third position defines the most frequent codon of the encoded amino acid.
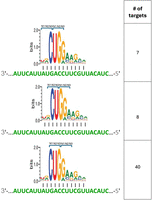
Alignment of RyhB prime positions with the reading frame. As shown in Figure 2, RyhB has three consecutive prime positions, which imply its possible positioning in three locations in regard to the reading frame (N3, N1, N2, upper lane; N2, N3, N1, middle lane; N1, N2, N3, bottom lane). On the right side, the number of targets with the positioning shown schematically on the left is indicated.
All in all, for nine out of the 13 sRNAs discussed above, the prime positions are adjusted with the reading frame. Compiling the data for all prime positions across all the targets of all the sRNAs in Table 1 (714 in total), we find that 296 of the prime positions coincide with the third codon position, above random expectation (P ≤ 10−5 by χ2 test). In 228 of the 296 incidences (77%), the nucleotide at the prime position is that of the frequent codon. Furthermore, in our target data, one-third of the codons where a prime position overlapped with N3 are of the codon CUG, encoding leucine, the most frequent of the 61 codons (Supplemental Table S2). To assess whether our results randomly reflect the codon usage bias in E. coli, we compiled all prime positions aligning with N3, extracted the codons they are part of, and organized the data per encoded amino acid (Supplemental Table S2). We then turned to compare the distribution of codons per amino acid in prime positions and in the E. coli genome by a χ2 test. For some amino acids, the numbers of prime positions associated with them were too small to allow a statistical comparison. However, some codons are associated with a large number of prime positions of targets interacting with several sRNAs, including CUG, encoding leucine, CAG, encoding glutamine, and ACC, encoding threonine, allowing the statistical test. For each of these amino acids, we compared the distribution of codons among the prime positions to its codon distribution in the genome (Supplemental Table S2) and found that the counts of prime positions associated with the frequent codons are high above random expectation (P ≤ 0.0001 for CUG and ACC and P ≤ 10−6 for CAG by χ2 test). Our results may suggest an evolutionary scenario by which initially the high prevalence of these frequent codons in the binding sites on the target mRNAs led to their frequent encounters with sRNAs with complementary sequences and to their interaction. Since there are selection forces acting to maintain the frequent codons, they indirectly guaranteed also the maintenance of sRNA–target base-pairing at the prime positions, awarding preference for prime positions in these locations through evolution (reflected in the a high above random coincidence of prime position with frequent codons). This revelation underscores the dominance of the codon frequency in dictating the alignment of the prime positions within the reading frame.
To study if the codon adjustment is associated with a functional output, we examined whether targets that were affected by the sRNA overexpression and showed a change in their expression levels (Faigenbaum-Romm et al. 2020) differ from unaffected targets in terms of reading frame adjustment of the prime positions. This analysis was conducted for the targets of ArcZ, CyaR, and RyhB for which such expression data were available (Faigenbaum-Romm et al. 2020). No statistically significant difference was observed in regard to reading frame adjustment of prime positions between the affected and unaffected targets of ArcZ and RyhB (P > 0.4 by χ2 test, Materials and Methods; Supplemental Tables S1, S3). For ArcZ with two prime positions and RyhB with three prime positions, in both affected and unaffected targets, most prime positions were adjusted with the reading frame, and the nucleotide aligned with N3 coincided with that of the frequent codon. For CyaR, with one prime position, in the majority of targets it aligned with N3 and coincided with the nucleotide of the most frequent codon, even more so in the unaffected targets compared to affected targets (P ≤ 0.05 by χ2 test). These results indicate that alignment of prime positions with the reading frame and frequent codon preference are prevalent in sRNA–target interactions, independent of the regulatory outcome.
Conservation of sRNA–target base-pairing
sRNA and target orthology data in the Enterobacterales order
The wealth of whole genome sequence data of ample bacterial species enables studying the conservation of the base-pairing between the putative binding sites of the sRNAs and their targets. Here we analyzed a total of 2027 genomes included in the eight families of the Enterobacterales order. For each of the 20 E. coli sRNAs included in our study, we identified orthologs. For seven sRNAs, ArcZ, CyaR, DsrA, MicA, RprA, Spf, and RyhB, we identified orthologs by relying on homologs previously detected with Infernal and deposited in Rfam (Nawrocki et al. 2009; Nawrocki and Eddy 2013) (currently integrated into RNAcentral). For sRNAs not represented in Rfam along with their homologs, we searched for homologs in our genome database using BLAST. Importantly, we also applied BLAST to the former six sRNAs and obtained results highly similar to those provided by Infernal, adding further support for the orthologous relationships relying on BLAST.
Genomes encoding an sRNA were further analyzed by BLAST to identify the homologs of the 402 reliable E. coli targets with binding site within the CDS (Materials and Methods; Supplemental Table S1). At the end of this process, for each sRNA and target in our E. coli data set, we generated a list of genomes in which the pair is conserved. The sequences of sRNAs and targets in those genomes form the basis for our analysis of the base-pairing conservation along the binding sites and, in particular, at the primary positions.
The conservation of each sRNA is presented in Figure 6, showing the number of genomes it is conserved in, for each family in the Enterobacterales order. Most sRNAs are highly conserved in the Enterobacteriaceae family. Certain sRNAs, like RyhB, exhibit substantial conservation across all families within the Enterobacterales order. Other sRNAs, such as ArcZ and ChiX, are abundant in some families but are missing from others. For example, ChiX is abundant in the Erwiniaceae family but is almost absent from the Pectobacteriaceae. Some sRNAs, such as DsrA, MgrR, GadF, FliX, and SdhX, are found only in the Enterobacteriaceae. In general, the newly discovered sRNAs, which are often encoded in the 3′UTR of protein-coding genes (e.g., GadF, UphU), are missing from most of the families, following the absence of their hosting genes from these genomes (Supplemental Table S4). Exceptions are CpxQ and AceK-int, which are preserved in most bacterial families, probably due to the high conservation of their parent gene cpxP and aceK, respectively, from which they are derived. Yet, there are hosting genes that are highly conserved, but the sRNAs derived from them are only poorly conserved, such as sucD and the sRNA derived from it, SdhX.
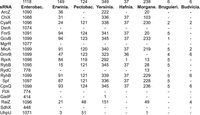
Conservation of sRNAs. For each sRNA, the number of organisms in which it is conserved in each family of the Enterobacterales order is noted. The top numbers indicate the number of organisms in each family. Names of families: Enterobac.- Enterobacteriaceae; Erwinia.- Erwiniaceae; Pectobac.- Pectobacteriaceae; Yersinia.- Yersiniaceae; Hafnia.- Hafniaceae; Morgane.- Morganellaceae; Bruguieri.- Bruguierivoracaceae; Budvicia.- Budviciaceae. First presented are canonical sRNAs (in alphabetical order) and next are 3′UTR-derived sRNAs (in alphabetical order). For the sRNAs ArcZ, CyaR, DsrA, MicA, RprA, Spf, RyhB, the orthologs and binding site region were identified by Infernal and for the other sRNAs by BLAST.
For each sRNA, we studied the conservation of its targets in the genomes where it is encoded. Most targets of all sRNAs are highly conserved in Enterobacteriaceae, but show various conservation levels in the other families (Supplemental Table S1). Figure 7 showcases a heat map delineating the conservation of RyhB targets beyond Enterobacteriaceae. Although some targets are hardly conserved beyond Enterobacteriaceae, there is sufficient body of data to follow the conservation of the base-pairing interactions in the Enterobacterales order. Since the conservation in genomes of the Enterobacteriaceae family is very high, the following analyses were done separately for genomes in the Enterobacterales order beyond Enterobacteriaceae and for genomes belonging to Enterobacteriaceae.
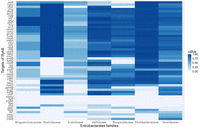
Conservation of targets. As an example, we present a heat map displaying the conservation level of each RyhB target across the different Enterobacterales families. The conservation of the targets of all sRNAs, including RyhB, is detailed in Supplemental Table S1.
Base-pairing conservation in the binding sites
In addition to identifying sRNA or target homologs by Infernal or BLAST, we performed multiple sequence alignments for all target and sRNA homologs in all genomes except for Enterobacteriaceae in reference to the E. coli K-12 gene sequences. This has allowed us to determine the sRNA binding site (sBS) and mRNA binding site (mBS) regions in the homologous sequences in the various genomes. We prioritized minimizing gaps in defining the sBS/mBS for each homolog. In instances where alignment posed challenges, we submitted to BLAST the sBS/mBS sequence of E. coli as query and searched for it in the homologous sequences in the other genomes, or utilized the MAST algorithm (Bailey and Gribskov 1998) that identifies known motifs in the sequences.
For each organism, we meticulously scrutinized base-pairing between the sRNA and the target at each position within the binding site region and gauged the degree of base-pairing conservation within the binding site regions. Theoretically, base-pairing could be preserved by conservation of the nucleotides in both the sRNA and target, or by changing a G:C interaction to G:U, or, by compensatory mutations in the sRNA and target. In practice, in most organisms where the base-pairing defined in E. coli was conserved, it was due to the conservation of the sRNA sequences and of the coding sequences (where the binding sites reside), maintaining exactly the same base-pairing. The fraction of compensatory mutations in our data was negligible (0.2%), found in only 2665 base-pairing interactions out of all the 1,206,952 base-pairing interactions in our data. This is due to the high conservation of the sRNA sequences and of coding sequences in the organisms where they are found to be conserved. We conducted the same analysis on genomes in the Enterobacteriaceae family and obtained an even lower percentage of compensatory mutations (0.05%).
Conservation of base-pairing interactions at prime positions
Recall that the prime positions were determined by compiling information of all targets of an sRNA in E. coli, and were found to be largely associated with the most frequent codons of the respective amino acids. This was evident for binding sites with one, two, or three prime positions. Studying the evolutionary conservation of the nucleotides and base-pairings at the prime positions sheds light on them from a different direction. We first address this question for nine E. coli sRNAs that were conserved beyond Enterobacteriaceae (Fig. 6), had at least five targets, and prime positions could be defined in them (Supplemental Fig. S3). For example, the prime positions in RyhB binding site are 3, 4, 5, overlapping the CUG codon, which is the most frequent E. coli codon for leucine. The fraction of E. coli RyhB targets in which these 3 nt are apparent is close to 1. These 3 nt perfectly fit base-pairing with CAG of the sRNA RyhB (Fig. 1). Since the sequence of RyhB is highly conserved in the other genomes (Supplemental Fig. S4), maintaining the base-pairing with RyhB in the other organisms would require that the corresponding leucine would be encoded by CUG, independent of the codon usage of the organism. In Figure 8A, we examine the positional evolutionary sequence conservation of RyhB target fabZ and of the base-pairing interactions it makes with RyhB. At position 4, all the organisms that have homologs of fabZ exhibit U, enabling base-pairing with A in RyhB. At position 3, there is variation between C and U. Yet, both maintain the base-pairing with G in RyhB, as well as the encoding of leucine (encoded by six codons, including UUG and CUG). At position 5, however, there is variation between G and A, both conserving the encoding of leucine, but only G is capable of base-pairing with C in RyhB. This suggests that in some genomes CGA is the preferred codon of leucine, and while RyhB in all genomes has C in the corresponding position, the selection force to maintain the respective frequent codon in those genomes is stronger than the one that acts to maintain the base-pairing.
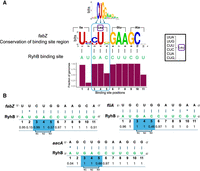
Conservation of base-pairing interactions between an sRNA and its targets. (A) Conservation is exemplified by fabZ–RyhB base-pairing interactions. At the top is the logo of RyhB binding sites based on E. coli targets (Melamed et al. 2016). Conservation of RyhB binding site across the different genomes (except for Enterobacteriaceae) is presented below in bits of information, computed based on equal frequency of the four bases. The logo representation was generated using WebLogo (http://weblogo.berkeley.edu/logo.cgi). Blue arrows mark the prime positions, and the reading frame and encoded amino acids are marked as well. RyhB binding site is shown below in green. The black lines represent base-pairing between the RyhB and fabZ nucleotides. The height of the bars below represents the fraction of genomes in which the positional base-pairing could be fulfilled. On the right, the six codons encoding Leu are shown. (B) Examples of conservation of positional base-pairing in RyhB–target interactions. Shown below each base pair is the fraction of genomes in which this putative base pair interaction could be fulfilled. The reading frame corresponding to the dominant alignment of the prime positions of RyhB binding site is marked (N1, N2, N3).
To examine how codon usage patterns are manifested across species at sRNA binding sites, we performed a focused analysis of four well-conserved sRNAs with extensive target interactions: RyhB, ArcZ, CpxQ, and CyaR. For each sRNA and each organism in which both the sRNA and its target were conserved, we analyzed those targets where a prime position aligns with the third nucleotide of a codon. Across the four analyzed sRNAs, we observed that in most targets, this position follows codon usage bias, even when this results in a mismatch with the sRNA. For instance, in RyhB, 95% of the analyzed sites (687 out of 724) coincide with the most frequently used codon in the corresponding genome, despite 128 of these positions not maintaining base-pairing (Supplemental Table S5). Notably, in most of the remaining cases, where the codon associated with the prime position was not the frequent one, the third nucleotide supported base-pairing with the sRNA. These findings suggest that codon optimization is a dominant constraint at sRNA binding sites, but in some cases, organisms may prioritize the maintenance of base-pairing potential over strict adherence to codon optimality (Supplemental Table S5), suggesting that in some organisms, preserving the potential for effective base-pairing may take precedence over codon optimality.
Interestingly, the analysis of positional evolutionary conservation of the base-pairing interactions suggests that other positions may compensate for the mismatches at prime positions (often due to variation in codon preferences). For example, as shown in Figure 8B for three RyhB targets, including fabZ, although there is decrease in base-pairing interactions at position 5 due to the degeneracy of the genetic code, they seem to be compensated by other base-pairing interactions along the binding site, at positions that were not defined in the analysis of E. coli targets as prime positions. While the degree of conservation may vary between corresponding positions of different targets, the number of positions with high degree of base-pairing conservation is highly similar among various targets and is around 8–9 positions (Fig. 8B). When examining the overall degree of positional base-pairing conservation across all targets of RyhB and all genomes (Fig. 9), it can be seen that the three prime positions along with positions 2 and 6, which were not determined as prime position among E. coli targets, show the highest degrees of base-pairing conservation, further supporting their important roles in establishing the sRNA–target interaction. There are positions that fulfill base-pairing in some targets but not in others, and therefore their overall degrees of base-pairing conservation are medium (Fig. 9).
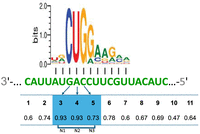
Overall conservation of positional base-pairing across all targets of RyhB. We counted for each position of the binding site the number of putatively fulfilled base-pairing interactions across all RyhB targets and across all genomes in all families (except for Enterobacteriaceae, which were analyzed separately). Presented are the fractions of genomes putatively fulfilling a positional base-pairing interaction out of all genomes in the analysis. Prime positions are shaded in blue. The reading frame corresponding to the dominant alignment of the prime positions of RyhB binding site is marked (N1, N2, N3).
Similar results were obtained for other sRNAs. For quite a few sRNAs in our analysis, even when a prime position coincided with the third position of a codon where degeneracy is anticipated, it showed high conservation of the base-pairing interaction (see, e.g., MicA in Supplemental Fig. S3 and FliX, GadF, MicA, and UphU in Supplemental Fig. S5). At positions where degeneracy took place in a fraction of the genomes, base-pairing conservation was reduced, but for most sRNAs, these positions still were among the positions with the highest degrees of base-pairing conservation. Similar to the results for RyhB, other positions, which were not determined in E. coli as prime positions, were also found to highly conserve their base-pairing interactions through evolution (Supplemental Fig. S3). Compiling the evolutionary data for each sRNA across all its targets, we find that for five out of the nine sRNAs, more than seven base-pairing interactions were conserved on average (Supplemental Table S6). Repeating this analysis for genomes of the Enterobacteriaceae family, we found that for 11 out of 14 sRNAs conserved in Enterobacteriaceae, more than seven base-pairing interactions were conserved on average (Supplemental Table S6). These results support the conjecture that keeping an appropriate number of base pairs plays a role in maintaining the sRNA–target interaction.
DISCUSSION
Bacterial genome sequences encompass various layers of information that coaffect their evolution. The DNA sequence contains information of transcription and translation initiation and termination sites, of the protein coding sequence, as well as of regulatory elements embedded and inherent in the sequences, which may play a role at the DNA, RNA, or protein level. These various sequence signals overlap, and they mutually affect the evolution of the sequence. For example, while the specific usage of codons in a bacterial sequence was shown to affect the expression level of the encoded protein (Lithwick and Margalit 2003; Plotkin and Kudla 2011), at the same time these sequences determine the secondary structure at the translation initiation site, affecting its accessibility to the translation machinery (Kudla et al. 2009; Gu et al. 2010). Conceivably, the selection forces shaping the coding sequence and those shaping the regulatory elements embedded in it act in concert to guarantee the desired gene expression level and its functionality (Weatheritt and Babu 2013). Here, we address this theme, focusing on the adjustment of sRNA binding sites located within protein coding sequences with the reading frame.
Traditionally, bacterial sRNAs have been considered as regulators of translation initiation. This regulation is manifested by sRNAs base-pairing with their target mRNAs in upstream regions to the CDS, preventing or enhancing ribosome binding in the vicinity of translation initiation sites and thus, repressing or accelerating translation initiation (Wagner and Romby 2015). Data accumulated in large-scale studies in search of sRNA targets have demonstrated many putative binding sites of the sRNAs that reside in the mRNA within protein coding sequences (e.g., the RIL-seq study [Melamed et al. 2016]). This may suggest that sRNAs also affect translation elongation by binding within the coding sequence. However, this possibility is questionable because the ribosome is known to function as a helicase (Takyar et al. 2005) and to dissociate the mRNA–sRNA hybrid (Pfeiffer et al. 2009), probably overcoming the possible effects of the sRNA on translation elongation. Yet, it was shown that sRNAs may impact the expression level of their targets while binding inside the coding sequence by affecting their cleavage by RNase E (Pfeiffer et al. 2009; Bandyra et al. 2012), or by locally affecting translation elongation (Thongdee et al. 2025).
In the current study, we aimed to characterize the positional and codon usage principles underlying sRNA binding within coding regions. We first characterized the putatively fulfilled base-pairing interactions in the binding site for each sRNA and found that regardless of the length of the binding site (determined in previous studies), the average number of fulfilled base pairs is 8–10 for most sRNAs (Fig. 3). These base-pairing interactions are not necessarily consecutive. If we regard only consecutive base pair interactions, we find that these account for 4–8 consecutive interactions for the various sRNAs included in our analysis (Fig. 3). Notably, the identified base pairs are mostly canonical base pairs (A-U, U-A, G-C-, C-G), with an underrepresentation of G-U and U-G (Supplemental Fig. S1). To assess the location of the binding site in reference to the reading frame, we focused on “prime positions,” positions that showed high preservation of a certain nucleotide across all E. coli targets of an sRNA, capable of base-pairing with the corresponding sRNA nucleotides (Fig. 1). These positions might be anchors of sRNA–target interactions, which with adequate nucleotide sequences around them establish the interaction between an sRNA and its targets. Considering the potential importance of the specific nucleotides at the prime positions, and given the fact that they are located within reading frames, it is conceivable that analyzing their location in reference to the reading frame in the individual targets of an sRNA would provide clues as to their establishment within the coding sequences (Fig. 2).
Our analysis has led to the finding that for one prime position per binding site, it is aligned in most targets with the third codon nucleotide, and often it is the nucleotide defining the most frequent codon of the amino acid (Table 1). For two prime positions per binding site, the prime positions align preferentially with the reading frame, either at positions 1 and 3 or 2 and 3 of the codon, and they, once again, mostly coincide with the most frequent codon. For three consecutive prime positions, as determined for RyhB, they mostly coincide with the most frequent codon of leucine, CUG (Supplemental Table S2). Notably, high-throughput approaches, such as RIL-seq, may capture many transient interactions or RNA–RNA encounters that are not known to generate regulatory effects. We previously showed that many sRNA–target interactions identified in the RIL-seq experiment do not result in detectable changes in gene expression under various growth conditions (Faigenbaum-Romm et al. 2020). Using these data, we examined whether affected and unaffected targets differ in the adjustment of the prime positions with the reading frame. The results of this analysis indicated that codon adjustment and preference for the most frequent codon were evident among all targets, affected and unaffected (Supplemental Table S3). Thus, many interactions that are not known as regulatory contribute to the detected association between prime positions and frequent codons. Yet, the lack of an effect manifested in an expression change does not necessarily imply biological insignificance. Functional output can be condition-dependent, as sRNA activity is influenced by competition for Hfq binding among multiple transcripts expressed under specific physiological contexts. Furthermore, alluded nonfunctional interactions may serve alternative biological roles, such as buffering or titrating sRNA availability, thereby indirectly modulating the regulation of other targets (Jost et al. 2013).
Independent of the mechanism, it seems that encounter of transcripts with complementary sequences on Hfq often results in an interaction, and since sequences with frequent codons are prevalent and selected for, they are more prone to interact with sRNAs with complementary sequences.
Previous studies already suggested that when complementary sequences meet on Hfq, they often interact (Faigenbaum-Romm et al. 2020), where the interaction was shown to be more substantial as the nucleotides of the two RNAs are more cognate (Roca et al. 2025). The high fractions of prime positions of different sRNAs aligning with the frequent codons may suggest that the relatively high probability of an encounter with nucleotides of a frequent codon plays a role in determination of the interactions between binding sites within the reading frames and the sRNAs. Furthermore, our observation that the coincidence of prime positions with the frequent codons of several amino acids is statistically significantly high above random expectation may suggest that evolutionary pressures have underlined the alignment of prime positions with frequent codons. Finally, prime positions that align with frequent codons are maintained through the selection to maintain the frequent codon. Once an interaction between the sRNA and target transcript is formed and has some function (affecting the target levels or buffering the sRNA molecules), the selection force to maintain this interaction may join the selection force acting on the coding sequence, leading to establishment of this interaction.
Our evolutionary analysis adds insights into the properties of the binding sites. While the prime positions of an sRNA binding site were determined in E. coli targets by their almost complete nucleotide conservation, in the evolutionary analysis across genomes in families of the Enterobacterales order, some of these positions showed only partial nucleotide conservation, despite high conservation of the sRNA sequences (Supplemental Fig. S3), and consistent with a previous study (Richter and Backofen 2012). We observed the lack of conservation especially at positions coinciding with the third position of the codon, due to the degeneracy of the genetic code and due to possible constraints in the other genomes to maintain another nucleotide in the codon. Thus, while the overall base-pairing at the prime positions is highly conserved across targets and genome families (Fig. 9; Supplemental Fig. S3), there are genomes where base-pairing interactions at these positions are not fulfilled. Yet, we find that positions that have not been defined as prime positions in E. coli may exhibit increased evolutionary conservation of base-pairing interactions (Fig. 9; Supplemental Fig. S3), in order to compensate for the loss of base-pairing at the prime positions. This is consistent with the results of the initial analysis of E. coli binding sites (Fig. 3), showing that there is a characteristic number of base-pairing interactions per the binding sites of the various sRNAs. This strategy ensures the maintenance of a requisite number of base-pairing interactions essential for sustaining the sRNA–target interaction, which compensates for a lost interaction at a primary position, while keeping a codon that might be crucial for the translated protein. In general, it seems that the selection forces acting on the reading frame are stronger than the selection forces to maintain the sRNA binding site, and the sRNAs adjust their binding accordingly.
MATERIALS AND METHODS
Organization of sRNA–target interaction data
The technology of RIL-seq involves immunoprecipitation of Hfq and bound RNAs, ligation of the RNAs, and sequencing of the RNA fragments (Melamed et al. 2016). RNA fragments identified as involving two different RNAs were considered by RIL-seq as chimeric fragments. These chimeric fragments were determined as representing putative interacting RNAs if their counts were statistically significantly above random expectation. The statistical analysis was applied to the unified libraries per condition, as well as to the individual libraries. In this study, we included sRNA–target pairs that followed one of two criteria: (1) The chimeric fragments representing the pair passed the statistical threshold in E. coli analysis both for a unified library of at least one condition or growth phase and for at least three individual libraries of at least one condition or growth phase; (2) the sRNA–target pair passed the statistical threshold in an E. coli RIL-seq unified library and in a RIL-seq unified library of another bacterium (enteropathogenic E. coli [EPEC] [Pearl Mizrahi et al. 2021], Salmonella enterica [Matera et al. 2022], or Klebsiella pneumoniae [Goh et al. 2024]).
Defining prime positions
To identify prime positions within sRNA binding motifs, we analyzed their position-specific scoring matrices (PSSMs), which represent the positional frequency of each nucleotide across motif positions. A prime position was stringently defined as a position where one nucleotide exhibited a frequency ≥0.95.
Testing the adjustment of prime positions in regard to the reading frame
In each target we determined the binding site based on Melamed et al. (2016), and labeled the prime positions based on their alignment to the reading frame as N1 (first nucleotide in codon), N2 (second nucleotide in codon), and N3 (third nucleotide in codon). We tested whether there is a bias toward a specific arrangement in regard to the reading frame for cases of one prime position, two prime positions that are at most at a distance of 1 nt separating them, and three consecutive prime positions. There are three possible alignments of the prime positions in regard to the reading frame, and if it were random, we would expect the prime positions to distribute randomly between the possible arrangements (1/3, 1/3, 1/3). We applied a χ2 test to test the null hypothesis that the distribution is random.
Comparing the adjustment of prime positions with the reading frame between affected and unaffected targets
Based on the data of Faigenbaum-Romm et al. (2020), for each of the sRNAs ArcZ, CyaR, and RyhB, we determined two sets of targets: targets that showed a change in expression level under overexpression of the sRNA and targets that did not show such a change. For each set we counted the number of targets with prime positions adjusted with the reading frame. For ArcZ and RyhB, we then compared the distributions of targets with adjusted prime positions and targets with nonadjusted prime positions between these two target sets by a χ2 test. For CyaR we compared the numbers of prime positions aligned with N3 that coincide with the nucleotide of the frequent codon between the two sets by a χ2 test.
Evolution of RNAs and binding sites
Searching for sRNA and target orthologs
To study the conservation of the binding sites and base-pair interactions, we first determined a set of orthologs of the sRNAs and their target genes across genomes belonging to families within the Enterobacterales order (genomes were obtained from NCBI's Assembly database). For seven sRNAs, ArcZ, CyaR, DsrA, MicA, RprA, Spf, and RyhB, orthologs were identified based on homologs previously detected using Infernal and deposited in Rfam (Nawrocki et al. 2009; Nawrocki and Eddy 2013) (currently integrated into RNAcentral). These homologs were detected in the Enterobacterales genomes using the Infernal cmscan program (with the ‐‐rfam and ‐‐cut_ga options) against the Rfam covariance model database (Rfam.cm), and hits were filtered to retain high-confidence homologs.
For sRNAs that were not represented in Rfam, or whose homologs were not detected by Infernal, we used our in-house ConservationProfiler tool (https://github.com/YairGatt/ConservationProfiler), which relies on BLAST with several filtering thresholds (identity ≥59%, E-value ≤0.05, and query coverage ≥0.5) to identify orthologs relative to E. coli K-12 substr. MG1655.
Recognizing binding sites
The binding sites in E. coli were recognized previously by applying MEME (https://meme-suite.org/meme/) (Bailey et al. 2015) to the E. coli binding regions identified by RIL-seq and verifying the base-pairing potential with the sRNA (Melamed et al. 2016). To identify for each ortholog (sRNA or target) the region corresponding to the E. coli binding site, we performed multiple sequence alignments (Edgar 2004) for all sRNA or target orthologs. Analyzing the multiple sequence alignment in regard to the binding site in E. coli has allowed us to determine the binding site regions on the sRNA (sBS) and the binding site on the mRNA (mBS) in the various genomes. We prioritized minimizing gaps in defining the sBS/mBS for each ortholog. In instances where alignment posed challenges, we performed BLAST using the binding site sequence of E. coli as a query and the homologous sequences in the other organisms as the data set. For this we used standalone BLASTn+ version 2.8.1 (Camacho et al. 2009). A binding site was declared as present in the homolog if there was a hit with an E-value ≤10 and an identity of at least 80% between the query and the sequence identified by BLAST. In cases where the mBS was not detected by BLAST, we used the MAST algorithm that identifies input motifs in the sequences (Bailey and Gribskov 1998). Genomes where no match was found using BLAST or MAST, and the mBS region identified through multiple sequence alignment contained more than 30% gaps, were excluded from the analysis for that specific interaction.
Base-pair conservation analysis
Conservation of base-pairing per target
To assess the conservation of base-pairing within sRNA–mRNA interactions, we analyzed each sRNA–target interaction across all genomes in the data set. For each genome and for each position, we determined whether the base pair was feasible. The number of genomes with a possible base pair at a given position was then divided by the total number of genomes included in the analysis (i.e., genomes where both the sRNA and the target gene were conserved, and the binding site region was identified). This calculation yielded the fraction of organisms in which a specific position within the binding region exhibited a possible base-pairing interaction, considered as conservation degree.
Conservation of base-pairing of an sRNA across all its targets
To evaluate the conservation of positional base-pairing across all targets of a specific sRNA, we performed a similar analysis as above, this time computing the fraction of genomes fulfilling a positional base-pairing interaction, when all targets are considered. For each position within the binding sites, we counted the total number of genomes exhibiting a possible base-pairing interaction across all targets of the sRNA, and divided it by the total number of genomes included in the analysis across all these targets. This calculation yielded the fraction of organisms, across all targets of the given sRNA, in which a specific position exhibited possible base-pairing—representing the overall conservation degree of that position across the sRNA's entire target set.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Meshi Barsheshet and Yair E. Gatt for technical assistance, Yael Altuvia for helpful comments, and all our laboratory members for useful discussions. This study was supported by an Advanced Grant of the European Research Council (833598 granted to H.M.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080839.125.
-
Freely available online through the RNA Open Access option.
- Received October 29, 2025.
- Accepted January 1, 2026.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








