
Novel trinucleotide mRNA capping reagents: improved synthetic route and efficient cotranscriptional incorporation in mRNA
- Chunping Xu,
- Russell Cousins,
- Ilya Ilichev,
- Jesus Ceja,
- Paul Ludford,
- Vagarshak Begoyan,
- Marc Turner,
- Maria Santos,
- Coleen Vo,
- Farinaz Rezvani,
- Andrew Ujita,
- Jordana Henderson,
- Michael Houston,
- Chanfeng Zhao and
- Alexandre V. Lebedev
- Corresponding author: cxu{at}trilinkbiotech.com
-
Handling editor: Adrian Ferre-D'Amare
Abstract
The 5′-N7-methylated guanosine triphosphate cap structure plays a critical role in mRNA translation and mRNA stability. The recent invention of cotranscriptional capping of mRNAs using trinucleotide capped primers (TCPs) allows for development of large-scale in vitro transcription (IVT) synthesis of mRNA carrying a eukaryotic Cap 1 structure (TCP-mRNA). Here we present a novel “one-pot-two-step” methodology for the synthesis of TCPs that improves the yield and simplifies the isolation and purification of the TCPs. Over 70 different modified TCPs, the analogs of a 7mGpppAmpG trimer, were synthesized, characterized, and tested for their ability to initiate IVT reaction. The results demonstrate that full complementarity of TCP to a template strand of dsDNA template at transcription initiation (start) site, at positions +1 and +2, is required and sufficient to obtain capped TCP-mRNA with high capping efficiency (>98%) and high yield (>5 mg/mL). This approach can be applied from small- to large-scale mRNA synthesis carrying various 5′-cap structures.
Keywords
INTRODUCTION
Remarkable success in development and applications of messenger RNA (mRNA) has revolutionized the field of molecular medicine (Chavda et al. 2022; Li et al. 2023; Liu et al. 2023; https://www.nobelprize.org/prizes/medicine/2023/press-release/). In vitro transcription (IVT) is a key enzymatic step employed in the manufacturing of therapeutic mRNAs for biomedical applications. Incorporation of a Cap structure (7mG5′ppp) on the 5′-end of an mRNA molecule (Fig. 1) is a critical IVT step defining the success of a manufacturing process. The presence of the 7mG5′ppp is essential since it protects the mRNAs from degradation by exonucleases and plays a key role in assembly of the translation initiation complex in eukaryotic cells (Shatkin 1976, 1985; Furuichi et al. 1977; Filipowicz 1978; Sonenberg 1988; Gingras et al. 1999; Rhoads 1999). Only those mRNAs that carry the cap structure are active in cap-dependent translation. “Decapitation” of mRNA results in an almost complete loss of their template activity for protein synthesis (Both et al. 1975; Muthukrishnan et al. 1975; Chu et al. 1978).
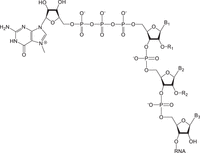
Natural cap structures on 5′-end of mRNA. Cap 0: R1 and R2 = H; Cap 1: R1 = methyl; R2 = H; Cap 2: R1 = methyl; R2 = methyl; B1, B2, and B3—nucleoside bases.
Another critical structural element, that is characteristic of eukaryotic mRNA, is the 2′-O-methyl group on the nucleoside residue at position 1 (Cap 1; Fig. 1, R1 is methyl; R2 is hydroxyl) and, in some cases, at positions 1 and 2 (Cap 2; Fig. 1, both R1 and R2 are methyl groups). The 2′-O-methylation of mRNA is required for higher efficacy of mRNA translation in vivo (Bouloy et al. 1980; Hyde and Diamond 2015), and it further improves nuclease stability of the 5′-end of capped mRNA. Also, the presence of Cap 1 (and Cap 2) is a distinctive mark that allows cells to recognize the bona fide mRNA 5′ end and, in some instances, to discriminate against transcripts originating from infectious agents (Kulasegaran-Shylini et al. 2009; Daffis et al. 2010; Züst et al. 2011; Lundstrom 2018). The mRNAs lacking a Cap 1 or Cap 2 structure are recognized as foreign and activate innate immune responses that shut off translation in vivo (Hyde and Diamond 2015).
Until recently, the conventional way to synthesize capped mRNA in vitro was to use a synthetic dinucleotide 7mGmpppG as an initiator of transcription (cotranscriptional capping). In 7mGmpppG, which is also known as antireverse cap analog (ARCA), the 3′-OH group of the 7mG residue is replaced with the 3′-O-methyl group (7mGm). Several other variants of ARCA analogs are known (Stepinski et al. 2001; Peng et al. 2002; Jemielity et al. 2003; Kore et al. 2007, 2009, 2015; Kowalska et al. 2008; Kore and Shanmugasundaram 2014; Shanmugasundaram et al. 2021; Cesaro et al. 2023). ARCA initiates mRNA synthesis only in the correct “forward” orientation (Stepinski et al. 2001; Peng et al. 2002) in contrast to an original in vitro transcription method, with 7mGpppG dinucleotide, where a bidirectional initiation of mRNA synthesis is a major problem (Fig. 2, compare entries 2, 3, and 4; Pasquinelli et al. 1995; Stepinski et al. 2001; Peng et al. 2002). Specifically, when 7mGpppG is used for in vitro transcription, there is a tendency of the 3′-hydroxyl group of either the G or 7mG moiety of 7mGpppG molecule to serve as an initiation point for transcriptional elongation with nearly equal probability. This leads to synthesis of two isomeric forms of mRNAs: 7mGpppG-mRNA (natural, forward orientation) and Gppp7mG-mRNA (unnatural, reversed orientation) in approximately equal amounts (Fig. 2; entries 2 and 3). While 7mGpppG-mRNA is active in translation, there is a consensus that Gppp7mG-mRNA isomer is not (Pasquinelli et al. 1995; Jemielity et al. 2003; Grudzien-Nogalska et al. 2007).

Structure of “d(CCCT)” DNA template, T7 promoter, and alternative forms of mRNA that could potentially form on this template with or without TCPs or ARCA. Entries 7 and 9 show the 7mGpppGmpG and 7mGpppAmpG structures in reverse orientation; however, formation of respective reverse TCP-mRNAs was not detected (see below). Positions of transcription start site and DNAzyme cleavage site are shown with arrows.
RNA polymerase from bacteriophage T7 is a commonly used enzyme for IVT production of mRNAs. Most promoters of T7 phage direct the initiation of RNA synthesis with guanosine at start position +1 (common T7 promoter sequence shown in Fig. 2). There is one significant technical obstacle when mRNA is synthesized cotranscriptionally using ARCA (7mGmpppG)—a strong competition between ARCA and GTP for initiation of mRNA synthesis (Stepinski et al. 2001) (note: GTP is an essential part of any transcription mixture). The competition leads to the formation of the mixture of two types of mRNA molecules. Thus, when mRNA synthesis is initiated with ARCA (7mGmpppG), the resultant RNA product is 7mGmppp-mRNA (Fig. 2, entry 4), which is active in translation. However, when mRNA synthesis is initiated with GTP, the resultant product is 5′-triphosphorylated uncapped mRNA (ppp-mRNA; Fig. 2, entry 1), which is not only inactive in translation but, in addition, may be immunogenic due to the presence of the 5′-triphosphate group (Hornung et al. 2006; Abbas et al. 2013). The IFN-induced protein with tetratricopeptide repeats-1 (IFIT-1) recognizes and binds to triphosphate RNAs and sequesters them from the translational machinery (Abbas et al. 2017). To reduce the immunogenicity of mRNA synthesized with ARCA, a treatment with Antarctic phosphatase is commonly used to remove 5′-triphosphate fragments from the immunogenic ppp-mRNA molecules. Consequently, uncapped (5′-dephosphorylated) 5′-OH-mRNA molecules, obtained after Antarctic phosphatase treatment, are not immunogenic but inactive and do not participate in the translation process.
To improve the ratio of capped 7mGmppp-mRNA molecules to uncapped 5′-ppp-mRNA molecules, while performing IVT, a large molar excess of 7mGmpppG (ARCA) over GTP (from 4:1 to 10:1) must be used to favor production of capped mRNA transcripts (Stepinski et al. 2001). At optimal conditions, up to 70%–80% of mRNA molecules can be capped. Consequently, since the typical initial concentration of ARCA in the mixture must be high (up to 6–8 mM), whereas the initial concentration of GTP must be low (1.5 mM or less), the final yield of capped mRNA is inevitably low (1.5 mg/mL or less) due to a rapid depletion of GTP during the IVT reaction. The fast depletion of GTP may also lead to a large fraction of shorter inactive mRNA molecules with aborted (incomplete) sequences.
Since mRNA synthesized with ARCA has Cap 0 structure, an additional step is needed to convert Cap 0 mRNA to Cap 1 mRNA by treatment with nucleoside-2′-O-methyltransferase (Myette and Niles 1996; Egloff et al. 2002). Typically, after appropriate optimization of the 2′-O-methylation protocol, mRNA with a high percentage of molecules carrying Cap 1 structure can be prepared (Beverly et al. 2016; Fuchs et al. 2016; Vlatkovic et al. 2022). Since the enzymes for converting Cap 0 to Cap 1 are commercially available, enzymatic 2′-O-methylation can be conveniently used for preparation of milligram quantities of Cap 1 mRNA. However, gram-kilogram quantities of Cap 1 mRNAs are needed for therapeutic applications and/or for worldwide vaccination, such as SARS-CoV-2 mRNA vaccine. At a large scale, the enzymatic 2′-O-methylation becomes laborious, expensive, and it may be quite difficult to fully control. Moreover, for large-scale manufacturing, to the best of our knowledge, no preparative methods have been developed for separation of Cap 1 mRNAs from residual Cap 0 mRNA retained in the mixture due to incomplete enzymatic 2′-O-methylation. Therefore, a substantial fraction of unmethylated Cap 0 mRNA could be present (and expected) in preparations of Cap 1 mRNA made by enzymatic 2′-O-methylation.
Ishikawa and coworkers described a direct synthesis of Cap 1 mRNA by IVT, without a need for enzymatic 2′-O-methylation (Ishikawa et al. 2009). They showed that trinucleotide capped primers (TCPs) of the structure 7mGpppAmpG and 7mGppp6mAmpG could initiate RNA synthesis on “d(CCCT)” DNA template strand containing T7 promoter that starts with “dC” nucleotide at +1 template position of transcription start site (TSS). They suggested that transcription, in this example, begins at −1 template position because of the base-pairing between the adenosine and deoxythymidine in T7 promoter at −1 position. Consequently, the capped RNA transcript (TCP-mRNA) contained one untemplated nucleotide (Am or 6mAm, respectively) at the 5′-end of the RNA (Fig. 2, entries 8 and 10). As in cotranscriptional capping with ARCA, a large molar excess (6.67-fold) of 7mGpppAmpG or 7mGppp6mAmpG (at 6 mM) over GTP (at 0.9 mM) was used to improve capping efficiency. However, the actual level of mRNA capping was not quantitatively determined in that publication. While this approach eliminated the need for enzymatic 2′-O-methylation step of mRNA, it still suffered from a limited yield of TCP-mRNA (as in the case with ARCA, see above) due to limited concentration of GTP used in transcription reaction (the actual yields were not reported).
The critical improvement that transformed TCP-mRNA synthesis from a research scope to a large-scale manufacturing process was an invention introduced by TriLink Biotechnologies in 2016 (Hogrefe et al. 2016). A full-scale production of Cap 1 TCP-mRNAs became attainable both with “high capping efficiency” and “high yield.” The key element of the invention is a requirement that TCP must contain an “initiation dinucleotide” fragment that is complementary to the DNA template strand at TSS positions +1 and +2. Thus, a cotranscriptional mRNA capping with 7mGpppAmpG (Am and G are +1 and +2 transcribed nucleotides, respectively) requires the use of complementary DNA template strand with “d(TC)” at TSS (Fig. 3). Full complementarity between TCP and DNA template at TSS allows TCP to outcompete the NTP substrate(s) for binding to DNA template strand and initiate efficient synthesis of Cap 1 mRNA even when concentrations of TCP and any NTP are equal (no limited NTP). As a result, Cap 1 mRNA transcript (TCP-mRNA) is synthesized with high yield, high level of capping (low ppp-mRNA by-product), and it contains a correct 5′-end mRNA sequence without incorporation of nontemplated nucleotides (compare Fig. 2, entries 8 and 10, with Fig. 3, entries 2 and 5).

Structure of “d(TCCT)” DNA template, T7 promoter, and alternative forms of mRNA that could potentially form on this template with or without TCP (7mGpppAmpG or 7mGppp6mAmpG). Entries 4 and 6 show the 7mGpppAmpG and 7mGppp6mAmpG structures in reverse orientation; however, formation of respective reverse TCP-mRNAs was not detected (see below). Positions of transcription start site and DNAzyme cleavage site are shown with arrows.
Despite the obvious advantage of in vitro synthesis of mRNA using cotranscriptional TCP capping method, a major problem of this approach remains: It is limited availability of high-quality TCP derivatives and a lack of TCP quantities necessary for preparation of TCP-mRNAs at scales required for biomedical applications. The common “classical” approaches to the synthesis of TCP (and their variants) are laborious, expensive, often inefficient, and regularly lead to a mediocre yield and low quality of the TCP. Moreover, the modified versions of TCP are more difficult to prepare and isolate, even in small quantities. Therefore, one of the main goals of the study was to develop convenient and reliable routes to the synthesis of common and modified TCPs containing various substitutions in 7mG unit, triphosphate bridge, and/or in a dinucleotide fragment of the TCP.
Here we present a novel methodology for the synthesis of TCP that allowed us to improve the yield and simplify the isolation and purification of TCPs. Over 70 different TCP derivatives were synthesized, characterized, and tested for their ability to initiate IVT reaction. A full complementarity of TCP to a template strand of dsDNA template at transcription initiation site at positions +1 and +2 is required and sufficient to obtain capped mRNA with high capping efficiency (>98%) and good yield (3–10 mg/mL). The developed approach can be applied for small- and large-scale mRNA synthesis.
RESULTS
Synthesis of trinucleotide capped primers
To improve the entire process of preparation of TCPs, we developed a convenient “one-pot-two-step” approach for the synthesis of TCPs (Fig. 4). We found that activation of 7mGDP derivatives and conversion to the imidazolide intermediate can be performed with 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) in a water/DMSO mixture, and the EDC can fully substitute for triphenylphosphine/2,2-dipyridyl-disulfide pair commonly used in “classical” route (Jemielity et al. 2003) shown in Supplemental Figure S1. The activation reaction of 7mGmDP proceeded smoothly with 1.6 equiv. of EDC and 3.0 equiv. of imidazole at room temperature. Anion exchange HPLC analyses (Fig. 5A) and 31P NMR spectra (Fig. 6A) of the reaction mixture showed that the imidazolide intermediate formed within several hours with a high yield (typically, at >90%) and, surprisingly, it was stable in the reaction mixture for at least 24 h without noticeable degradation. Next, without isolating the imidazolide intermediate, dinucleotide 5′-monophosphate and MgCl2 solution were added to the reaction mixture. After overnight reaction at room temperature, a nearly quantitative conversion of the dinucleotide to respective TCP was achieved in most cases as evident from HPLC analyses (Fig. 5B) and 31P NMR spectra (Fig. 6B). The gradual changes in 31P signals shape (and noticeable line broadening) are typically observed for reversible interaction (exchange) of Mg2+ ions with the phosphate residues in the nucleotide intermediates and the final TCP product (Vasavada et al. 1984; Glonek 1992).
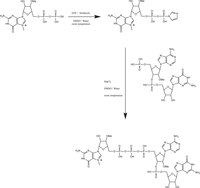
Scheme for the synthesis of TCP using standard “one-pot-two-step” approach. Synthesis of 7mGmpppAmpG is shown as an example.

AX-HPLC analysis of (A) a conversion of 7mGmDP into imidazolide intermediate using EDC activation (from top to bottom: starting 7mGmDP, and formation of 7mGmDP-imidazolide after 1, 2, 4, 8, and 24 h at room temperature); (B) a synthesis of 7mGmpppAmpG (compound 1-2A) by coupling 7mGmDP-imidazolide and pAmpG (from top to bottom: starting mixture of 7mGmDP-imidazolide and pAmpG, and formation of 7mGmpppAmpG after 1, 2, 4, 8, and 24 h at room temperature).
31P NMR spectra of (A) a conversion of 7mGmDP into imidazolide intermediate using EDC activation (from top to bottom: starting 7mGmDP, and formation of 7mGmDP-imidazolide after 1, 2, 4, 8, and 24 h, at room temperature); (B) the synthesis of 7mGmpppAmpG (compound 1-2A) by coupling 7mGmDP-imidazolide and pAmpG (from top to bottom: starting mixture of 7mGmDP-imidazolide and pAmpG before MgCl2 addition, and formation of 7mGmpppAmpG after 1, 2, 4, 8, and 24 h after MgCl2 addition; all at room temperature).
Using the described “one-pot-two-step” approach, we synthesized 29 TCPs, analogs of 7mGpppAmpG and 7mGppp6mAmpG containing various modifications of sugar 7mG-residue (Table 1; see also Supplemental Tables S1, S2). All TCP products were isolated and purified (up to 98%–99+% purity) by anion exchange QFF chromatography and then converted to a sodium salt form. During this purification/processing protocol, all TCP derivatives were quite stable, and no noticeable decomposition of TCP molecules was observed. The typical TCP isolated yields ranged from 40% to 70+%, depending on the chromatographic resolution of all components of the mixture (see Supplemental Section S1; Supplemental Table S1). In few cases, the isolated yields were intentionally sacrificed (even below 30%; compounds 1-8A, 1-11B) in favor of a desired purity of TCP (98+%) by discarding most of the chromatography fractions contaminated with unreacted materials and some side products. Although we did not attempt to recover TCP from contaminated fractions by an additional chromatographic AX or RP purification, it would be a reasonable approach to improve the isolated yields.
Synthesized TCPs with 7mG substituted at 2′- and/or 3′-ribose positiona
The next series of synthesized TCP derivatives contain modifications in triphosphate bridge (Table 2). In most cases, a standard synthetic route (Fig. 4) worked well; however, in other cases, a development of alternative routes was required. Thus, an alternative “one-pot-two-step” route 1 (Supplemental Fig. S2) was designed for the synthesis of TCP derivatives containing a bridging αβ-NH or αβ-CH2 substitutions in triphosphate fragment (2-8A and 2-9A, respectively). This route requires a coupling of 7mGMP-imidazolide to a respective modified 5′-dinucleotide derivative (such as pNHpAmpG or pCH2pAmpG; see Supplemental Section S1). Generally, the conversion of 7mGMP derivative into activated imidazolide with EDC/imidazole mixture was not as efficient (∼70%) as a conversion of 7mGDP into respective imidazolide (>80%) used in our standard route. Moreover, the activated 7mGMP-imidazolide was not stable if left in aqueous reaction mixture for more than 10–15 h, and it hydrolyzed slowly back to 7mGMP derivative when a supply of EDC in the reaction mixture was fully consumed. However, by using an appropriate excess of active imidazolide intermediate over dinucleotide derivative, it was possible to obtain the 2-8A and 2-9A TCPs although in a moderate yield (44% and 16%, respectively; Supplemental Table S1).
Synthesized TCPs with triphosphate bridge modifications
For the synthesis of γ-thio-TCP derivative 2-4B12, we used alternative “one-pot-two-step” route 2 where an efficient conversion of dinucleotide 5′-diphosphate into a respective imidazolide intermediate (Step 1) was achieved using EDC activation in the presence of imidazole, similarly to a conversion of 7mGDP into its imidazolide (compare Supplemental Fig. S3 to Fig. 4). Then, the imidazolide of dinucleotide 5′-diphosphate was reacted with 7mGmMP-thio derivative (Step 2) to give a desired γ-thio-TCP derivative in a good yield (62%, Supplemental Table S1). The same alternative route 2 was used for preparation of compounds 2-2A and 2-2B containing tetraphosphate bridge (7mGmppppAmpG and 7mGmpppp6mAmpG, respectively).
Finally, for the synthesis of β-thio-TCP derivatives (2-5A12 and 2-5B12), we used alternative “one-pot-two-step” route 3 where a conversion of dinucleotide 5′-phosphate into respective imidazolide intermediate (Step 1) was achieved using EDC activation in the presence of imidazole, similarly to a conversion of 7mGMP into its imidazolide (compare schemes in Supplemental Figs. S2, S4). At Step 2, the imidazolide of dinucleotide 5′-phosphate was reacted with excess of 7-methyl-3′-O-methylguanosine 5′-O-(2-thiodiphosphate). The respective β-thio-TCP derivatives, 2-5A12 and 2-5B12, were obtained only in moderate yield (37% and 22%, respectively; Supplemental Table S1) due to the difficulties in isolation and separation of the TCP derivatives from the unreacted 7-methyl-3′-O-methylguanosine 5′-O-(2-thiophosphate) and some other by-products.
A group of TCP derivatives that contain other modifications/substitutions in the dinucleotide fragment (Table 3) was prepared using standard “one-pot-two-step” route and purification protocol as described above for 7mGmpppAmpG and 7mGmppp6mAmpG without further changes or optimization. Significant difficulties had arisen only during a purification of 7mGpppGmpG and 7mGmpppGmpG. These TCP molecules showed a strong tendency to aggregate and form a mixture of monomeric and oligomeric forms, and this problem was not fully resolved in this study. As a result, a significant loss of the yield of 7mGpppGmpG and 7mGmpppGmpG occurred during the purification/isolation process.
Synthesized TCPs with nucleotide and/or internucleotide linkage (p) modifications
A separate but important issue for several TCPs was the formation of two diastereomeric forms due to the presence of chiral phosphorus atom. Specifically, these TCPs contained phosphorothioate fragments either in triphosphate bridge or in internucleotide linkage. In some cases (2-5A12, 2-5B12, 3-4A12, 3-8A12, and 3-6B12), we were able to separate diastereomers and obtain individual forms (D1 and D2, as a first and a second eluted compound). The separation was performed by reverse phase chromatography as described in the Materials and Methods section and in Supplemental Section S1 and Supplemental Figure S5. In other cases (2-6A12, 2-4B12, and 3-2B12), the separation of diastereomers was not performed, and a mixture of the diastereomers was used for further mRNA synthesis. Individual diastereomeric TCPs, 3-7B1 and 3-7B2, were synthesized by standard route (Fig. 4) using the individual diastereomers of pAmp(S)G dinucleotide separated by reverse phase chromatography.
Using the main standard “one-pot-two-step” approach and three alternative routes, we synthesized, isolated, and characterized over 70 TCP analogs containing different modifications in the nucleotide units and/or in triphosphate bridge (Tables 1–3; full structures and analytical data for TCPs are summarized in Supplemental Section S1 and Supplemental Tables S1, S2). All TCP compounds were purified and isolated as sodium salt and contained <0.1% of a residual (unreacted) dinucleotide component.
IVT synthesis, purification, and analysis of TCP-mRNAs carrying modified caps
Next, using T7 RNA polymerase and cotranscriptional IVT capping, we synthesized, purified, and characterized over 50 FLuc mRNAs containing various TCPs on the 5′-end of mRNA (TCP-mRNA) according to the described protocol (Mandell et al. 2025; see also Henderson et al. 2021). After purification, all TCP-mRNAs went through the analytical test panel including TCP-mRNA integrity, capping efficiency, dsRNA content, residual DNA template, and residual proteins (Supplemental Section S2; Supplemental Tables S3, S4). According to our tests, most TCP-mRNAs contained only small residual amounts of DNA template (<2 ng/μg of purified mRNA) and proteins (<0.2 ng/μg of purified mRNA). The integrity of TCP-mRNAs was between 71% and 97% of full-length product (Supplemental Section S2; Supplemental Tables S3, S4; Supplemental Fig. S6). The TCP-mRNA integrity was slightly better for TCP-RNAs isolated by oligo(dT) affinity chromatography compared to RNeasy column purification (87.5% average with σ = 6.3% and 79.1% average with σ = 3.0%, respectively). Detection of dsRNA in TCP-mRNA showed the dsRNA level of <5 ng/μg of TCP-mRNA in most cases (full experimental dsRNA data are reported in Supplemental Tables S3, S4; Supplemental Figs. S7, S8).
Efficiency of capping is a critical parameter that characterizes the potency of IVT-synthesized TCP-mRNA. To determine capping efficiency, we used a DNAzyme-mediated cleavage of 5′-oligonucleotide fragment of TCP-mRNA following with analytical HPLC-MS for mass identification and quantitative analysis of the cleavage products. We have found that DNAzyme was compatible and worked equally well with TCP-mRNAs starting with AG, AA, GG, or AU at positions +1 and +2 of TSS. The results showed that a high level of TCP-mRNA capping was achieved in every IVT reaction reaching >95% capping in most cases (Supplemental Table S3). An example of capping assay analysis is shown in Supplemental Figure S9.
All IVT reactions were performed using a DNA template containing the T7 promoter sequence where the first transcribed nucleotide (+1) was coded for adenosine (or guanosine) and the second one (+2) for guanosine (or adenosine, or uridine, depending on the TCP nucleotide sequence). For non-6mAm TCPs, a full complementarity of TCP and DNA template at TSS positions +1 and +2 allowed for a synthesis of TCP-mRNA with high yield and high capping efficiency (Supplemental Section S2; Supplemental Tables S3, S4), and there was no need to use a large excess of the capping reagent (TCP) as typically used for cotranscriptional capping with ARCA (“sixfold over concentration of competing GTP” [Stepinski et al. 2001]). Quite the opposite, while in our experiments the initial concentration of competing ATP and all other NTPs in IVT reaction was 5 mM, the concentration of TCP we used was 4.0 mM (i.e., 1.25-fold lower than the concentration of GTP, ATP, CTP, or N1MeΨTP). Under these conditions, the capping efficiencies for the large majority of obtained TCP-RNAs ranged from 95% to 100%. Interestingly, a slightly lower capping efficiency of TCP-mRNA (down to 91%–94%) was typically observed for some diastereomeric TCPs that contained either the phosphorothioate internucleotide linkage in the dinucleotide fragment or the phosphorothioate linkage in the 5′-5′-triphosphate bridge (Supplemental Section S2; Supplemental Tables S3, S4).
The role of N6-methyladenosine (6mA) nucleoside in the regulation of mRNA translation was discussed in several publications (Akichika et al. 2019; Liu et al. 2021; Benak et al. 2023; Boulias and Greer 2023; Cesaro et al. 2023; Höfler and Duss 2023). Since many TCPs prepared in this report contained an adenosine nucleoside as the first transcribed nucleotide (+1) in TCP-mRNA, it was important to evaluate the effect of 6mA for A substitution on the efficiency of capping of the synthesized TCP-mRNAs. Initially, using standard IVT conditions, we observed relatively low capping efficiency (∼65%) for TCPs carrying 6mA for A substitution at the +1 position (e.g., for 7mGmppp6mAmpG). Therefore, the IVT reaction for TCP derivatives containing the 6mA base did require an additional search for optimal conditions. It was found that the increase of TCP concentration up to 10 mM while maintaining a concentration of all NTPs (including ATP) at 5 mM and optimization of reaction buffer allowed for the synthesis of 6mA-containing TCP-mRNAs with high capping efficiency over 95% (Mandell et al. 2025). The optimized IVT conditions (see Materials and Methods) were used in this study for a synthesis of all TCP-mRNAs containing 6mA residue at +1 position.
Next, the probability of TCP initiation of RNA synthesis at −1/+1 position of DNA template, proposed by Ishikawa et al. (2009), was evaluated in comparison with +1/+2 position concept (Hogrefe et al. 2016). We performed a detailed analysis of the 5′-end products obtained after DNAzyme treatment of three TCP-mRNAs containing 3-12A, 3-12B, and 3-14B cap structures. These TCP-RNAs were prepared using 7mGppp6mAmpA, 7mGmppp6mAmpA, or 7mGmpppAmpA, respectively, in conjunction with DNA template containing the T7 promoter sequence where the first transcribed nucleotide (+1) was coded for adenosine (or 6mA) and the second one (+2) also for adenosine (Fig. 7). Note that the T7 promoter has thymidine at −1 position of DNA coding strand. Theoretically, in this case, there were two possibilities for 7mGmppp6mAmpA (or 7mGppp6mAmpA or 7mGmpppAmpA) to bind at TSS and initiate mRNA synthesis either at positions −1/+1 or at positions +1/+2. Consequently, after DNAzyme treatment of TCP-mRNA, a formation of 15-mer cleavage product (Cap + 14-mer; Fig. 7, entry 3) would indicate the initiation of the mRNA synthesis at −1/+1 position, while the initiation at +1/+2 position should result in a formation of 14-mer (Cap + 13-mer; Fig. 7, entry 2). The HPLC-MS assay data for 7mGmppp6mAmpA capping clearly showed the presence of two main capped products: 14-mer and 15-mer and some minor related products. A calculated molar ratio of all (+1/+2 start):(−1/+1 start) capped products was 12.1:1 (Supplemental Fig. S9A). It means that >92% of the initiation of mRNA synthesis occurred at +1/+2 position of TSS in agreement with the proposition of +1/+2 concept. Similarly, results showing a stronger preference for IVT initiation at +1/+2 position were obtained in the case of TCP-mRNAs prepared with 7mGppp6mAmpA (85% starts at +1/+2; Supplemental Fig. S9B) or 7mGmpppAmpA (81% starts at +1/+2; Supplemental Fig. S9C). In other words, the efficient IVT initiation of TCP-mRNA synthesis requires a full complementary of TCP and DNA template strand, and it starts at positions +1/+2.

Structure of “d(TTCT)” DNA template, T7 promoter, and alternative forms of mRNA that are being synthesized on this template with or without TCP (where xAm is either 6mAm or Am). Positions of transcription start site and DNAzyme cleavage site are shown with arrows and indicated for each product.
Another important question was whether the TCP-initiated mRNA synthesis proceeded only in correct “forward” orientation. Obviously, TCPs that contain sugar-modified 7mG fragment (like in ARCA or most of TCPs) cannot initiate transcription reaction in “reverse” orientation. However, this argument cannot be applied to TCPs that contain 7mG with unmodified ribose. Thus, the TCPs such as 7mGpppAmpG (1-1A) or 7mGppp6mAmpG (1-1B), in theory, could initiate IVT synthesis either in “forward” or “reverse” directions, and the resulting RNA molecules would have two alternative cap orientations (Fig. 3, entries 2, 4, 5, and 6). Consequently, in this case, the capping assay should reveal the presence of two isomeric forms of cleaved 5′-fragment of mRNA (13-mer + 7mG cap) with the same mass in HPLC-MS analysis. However, in all cases studied (1-1A, 1-1B, and Series 3-A compounds), the HPLC-MS data showed the presence of only one major product with correct predicted mass (Supplemental Section S2; Supplemental Fig. S10 as an example). Therefore, we concluded that all TCPs initiate IVT reaction only in natural “forward” orientation in agreement with published results for similar compounds (Ishikawa et al. 2009; Sikorski et al. 2020).
DISCUSSION
Several TCP variants of parent 7mGpppAmpG were recently synthesized by us and others (Ishikawa et al. 2009; Hogrefe et al. 2016; Vaidyanathan et al. 2018; Sikorski et al. 2020; Henderson et al. 2021; Senthilvelan et al. 2021, 2023; Warminski et al. 2021, 2023; Shanmugasundaram et al. 2022; Inagaki et al. 2023). These TCPs contained different nucleotide substitutions and/or modifications of triphosphate bridge, sugars, or bases of the nucleotide units and were synthesized using “classical” route (Supplemental Fig. S1). This route requires preparation of two key intermediate components: 7mGmDP imidazolide derivative (or analog) and modified (or nonmodified) 5′-phosphorylated dinucleotides. These two key intermediates are prepared through multistep chemical processes where nearly each reaction step requires isolation, purification, and characterization of the products. The final step of the TCP syntheses is accomplished using a common approach, where the activated 7mGmDP-imidazolide derivative is coupled to a 5′-phosphorylated dinucleotide (Hogrefe et al. 2016; Sikorski et al. 2020). This approach works relatively well for a small-scale synthesis (up to a few grams of TCP). However, for large-scale (up to 100 g or more) synthesis of TCPs, which is required for vaccine production (e.g., Covid-19), the process faces several principal challenges. (a) Using a large excess of activating reagents is not economical. (b) The precipitation and purification of imidazolide intermediate with multiple acetone washes are increasingly laborious, and it is difficult to obtain the 7mGmDP-imidazolide intermediate in high purity. (c) Using large volumes of flammable acetone is not desirable for large-scale manufacturing. (d) The solubility of 7mGmDP imidazolide as a sodium salt in DMSO and/or DMF is quite poor, and the reaction needs a continuous powerful mechanical mixing/stirring and, sometimes, a long sonication to completely dissolve the sodium salt of activated 7mGmDP imidazolide. (e) In our experience, the reactive behavior of various batches of 7mGmDP-imidazolide was unpredictable, and each production reaction set required a continuous real-time full control and “run-on” adjustment of process conditions to allow the reaction to proceed at a reasonable rate. (f) The overall yields of the TCPs are typically moderate at best, and the final TCP product needed a double (sometimes a triple) round purification process to bring the final TCP to an acceptable quality. (g) The whole process produces a large amount of dangerous waste. (h) Finally, the whole process is long, laborious, and expensive.
By designing and implementing a novel “one-pot-two-step” approach, we successfully synthesized over 70 variants of TCPs containing different modifications in nucleoside units and/or in triphosphate bridge (Tables 1–3). All TCP compounds were prepared using either a standard route (Fig. 4) or alternative routes (Supplemental Figs. S2–S4). The key elements of the novel approach are as follows: (a) First, a reactive imidazolide intermediate of 7mGDP (or 7mGMP, or respective dinucleotide) is prepared in aqueous/organic media (Step 1) and (b) next, without isolation/purification, the imidazolide intermediate reacts with 5′-phosphate (or thiophosphate) monoester group of the respective mono- or dinucleotide counterpart in the presence of magnesium chloride catalyst (Step 2; Fig. 4; Supplemental Figs. S2–S4). Since both reaction steps are very efficient and proceed with high yield, there is no need for a large excess of reactants and activating reagent (EDC; ∼1.6 equivalent), thus making isolation and purification of the final TCP product easier. All side- and by-products can be removed by anion exchange chromatography and, if necessary, by additional reverse phase chromatography. The approach reported here shortens the overall chemical process, eliminates several intermediate synthetic and purification steps, and opens the opportunity for economical preparation of high-quality TCPs at a wide range of scales (from a few milligrams to 100 g).
During our work, special attention was paid to the quality (purity) of synthesized TCPs. Typically, TCPs were isolated and purified by anion exchange chromatography resulting in most cases up to 98%–99+% nucleotide purity as determined by 1H and 31P NMR, AX, and RP HPLC analysis (see Supplemental Section S1). In some cases, where a full separation of the TCP peak from side- and/or by-products was not achieved, the “hard” cutoff of the contaminated fractions had to be taken; thus, the isolated yields were sacrificed in favor of purity of TCP. Especially, all necessary precautions were made to purify TCPs from remaining unreacted dinucleotide (e.g., pAmpG, ppAmpG, or other modified versions of the dinucleotide) to the level of 0.1% or less. Our separate experiments showed that uncapped dinucleotide is a strong competitor for the initiation of IVT reaction. For example, ppAmpG is more than ninefold more efficient in initiating IVT reaction compared to 7mGpppAmpG (see Supplemental Section S2). Disregarding a possible contamination of TCP with the dinucleotide (or other by-products containing dinucleotide fragment) may significantly affect the outcome of the IVT reaction and could result in substantial contamination of TCP-mRNA with inactive “uncapped” mRNA molecules that will have a dinucleotide fragment on the 5′-end, for example, ppAmpG (Fig. 3, entry 7).
While this work was in progress, several studies on the application of capped oligonucleotide trimers (TCPs) for the synthesis of mRNAs by in vitro transcription were published (Vaidyanathan et al. 2018; Sikorski et al. 2020; Henderson et al. 2021; Senthilvelan et al. 2021, 2023; van Dülmen et al. 2021; Warminski et al. 2021, 2024a,b; Inagaki et al. 2023; Mandell et al. 2025). Overall, the use of cotranscriptional TCP capping technology enabled researchers to prepare TCP-mRNAs with proposed cap structure and with good quality that was sufficient for most biological and biomedical applications (e.g., for preparation of the Covid-19 vaccine, the primary sequence was published [Gau et al. 2023]). In several reports, however, when TCPs (or their analogs) were used for cotranscriptional capping, a high level of TCP-mRNA capping was not achieved (Sikorski et al 2020; Depaix et al. 2022; Kozarski et al. 2023; Warminski et al. 2024a,b). Specifically, for the 55-mer-long dsDNA template model system, based on ϕ6.5 promoter, authors reported a moderate level of capping efficiency (54%–90%) despite sixfold higher concentration of each tested TCP over a competing GTP (Sikorski et al. 2020). This result is not surprising, since the 55-mer dsDNA template construct selected for the study had “d(CCCT)” sequence at TSS (Fig. 2) and was used for all TCPs independently of their nucleotide sequence. Consequently, for eight out of 10 TCPs tested in that study, the DNA template strand was not fully complementary to TCP at critical +1 and +2 template positions of the TSS. If fully complementary DNA templates and TCPs had been used, the capping efficiency of the mRNAs, the mRNA yield (and likely their biological efficiency), could be as high as demonstrated in this and in other publications (Henderson et al. 2021; Strezsak et al. 2022; Vlatkovic et al. 2022; Inagaki et al. 2023; Mandell et al. 2025). Moreover, the use of proposed “complementary DNA template—TCP” format can significantly reduce or suppress a commonly observed heterogeneity of 5′-RNA ends caused by incorporation of untemplated nucleotides and/or deletion of templated nucleotides (Axelrod and Kramer 1985; Milligan and Uhlenbeck 1989; Pleiss et al. 1998; Huang 2003; Kuzmine et al. 2003; Coleman et al. 2004; Sikorski et al. 2020; Inagaki et al. 2023; Warminski et al 2024b).
Alternative base-pairing between 7mGpppAmpG and “d(CCCT)” DNA template strand (at −1 and +1 positions; −1/+1 concept) was proposed by Ishikawa et al. (2009) and later argued for by another group (Sikorski et al. 2020; Depaix et al. 2022; Kozarski et al. 2023; Warminski et al. 2024a). The proposal postulated the existence of base-pairing not only between 2′-deoxycytidine of DNA template strand at position +1 and guanosine residue of 7mGpppAmpG but also a base-pairing between thymidine of DNA template strand (dT at position −1) and 2′-O-methyladenosine nucleoside unit of the TCP (Fig. 2, entries 8 and 10). The −1/+1 concept was brought to explain a better capping efficiency observed for RNAs prepared on “d(CCCT)” DNA template using adenosine containing TCPs, such as 7mGpppAmpG and 7mGppp6mAmpG, compared to cytidine and uridine containing TCPs, such as 7mGpppCmpG and 7mGpppUmpG (Sikorski et al. 2020). Our data (Supplemental Fig. S9) clearly show that initiation of RNA synthesis at positions −1/+1 is highly inefficient compared to initiation at +1/+2 positions. It may indicate that the probability of classical Watson–Crick hybridization of 5′-adenosine residue of TCP and thymidine residue of the DNA template strand at −1 position is low, if it happens at all.
We suggest an alternative explanation for the substantial differences in capping efficiency for TCP-mRNA synthesized on “d(CCCT)” DNA template using 7mGpppAmpG and 7mGppp6mAmpG vs 7mGpppCmpG and 7mGpppUmpG (observed by Sikorski et al. 2020). We believe that it is a consequence of the differences in stability of TCP ternary IVT complexes with “d(CCCT)” DNA template strand and T7 RNA polymerase. Apparently, the purine-only TCPs, such as 7mGpppAmpG and 7mGppp6mAmpG, can form more stable complex with DNA template strand at TSS thus allowing for a more efficient competition of these TCPs with GTP for transcription initiation. The origin of complex stabilization lies in a favorable intramolecular base stacking of Am nucleoside of the 7mGpppAmpG with a neighboring 3′-G residue, which is involved in base-pairing with dC residue of at +1 position of DNA template strand. It is known that a single unpaired nucleotide at the end of the double-stranded oligonucleotide duplex, termed a dangling end, contributes significantly to duplex stability (Petersheim and Turner 1983; Sugimoto et al. 1987). The most effective stabilization of the oligonucleotide duplex occurs when the dangling end is a purine nucleoside (e.g., adenosine); the pyrimidine nucleosides (U or C) stabilize duplex significantly less efficiently. Based on this consideration, 7mGppp6mAmpG is expected to form a more stable ternary IVT complex and, consequently, can lead to a higher TCP-mRNA capping efficiency compared to 7mGpppUmpG or 7mGpppCmpG. Similarly, N6-methyladenosine base, as a purine dangling end, also stabilizes the oligonucleotide duplexes, even more efficiently than an adenine base (Roost et al. 2015). Therefore, the 7mGppp6mAmpG is expected to provide a higher TCP-mRNA capping efficiency compared to 7mGpppUmpG and 7mGpppCmpG, as observed in cited work (Sikorski et al. 2020).
While most TCPs, studied in this work, showed a robust IVT initiation and high mRNA capping efficiency, the TCPs composed of guanosine-only nucleotides, such as 7mGpppGmpG and 7mGmpppGmpG, demonstrated an abnormal IVT behavior. According to previous reports (Hogrefe et al. 2016; Sikorski et al. 2020), a lower level of TCP-mRNA capping (∼90%) was detected despite the fact that “several-fold higher concentration” of the TCP (7mGpppGmpG and 7mGmpppGmpG) over pppG was used. Such reduced level of TCP-mRNA capping was unexpected since there was a full “G-C” complementarity between 7mGmpppGmpG (or 7mGpppGmpG) and “d(CCCT)” DNA template strand at +1 and +2 positions.
We believe that abnormal IVT behavior of 7mGmpppGmpG and 7mGpppGmpG is a result of unique physical properties that are characteristic of the TCPs composed of guanosine-only nucleotides. For example, in some conditions, even guanosine 5′-monophosphate can form stable oligomeric self-structures (Gellert et al. 1962). Indeed, we observed a formation of stable self-structures (aggregates) of 7mGpppGmpG (or 7mGmpppGmpG) especially when a concentrated aqueous solution was stored for prolonged time at or below room temperature. The aggregates formed from guanosine-only composed TCPs were detected and identified by AX-HPLC and/or RP-HPLC-MS analysis as dimeric, trimeric, and even tetrameric structures (see Supplemental Table S2, compounds 3-15A and 3-15B). It appears that these TCP-aggregates, when they form, not only decrease an effective concentration of active monomeric form of TCP, but they themselves are inefficient in initiation of the IVT RNA synthesis thus making pppG more competitive. An additional “preheating” step to mitigate a 7mGmpppGmpG and 7mGpppGmpG aggregation issue was later added to the IVT protocol allowing for TCP-RNA synthesis with improved capping efficiency (will be published separately).
Important quality characteristic of IVT-synthesized TCP-mRNA is a level of so-called “double-stranded RNA” (dsRNA). The presence of dsRNA contaminants in TCP-mRNA may trigger cellular innate immune response and consequently suppress protein expression up to 1000-fold (compare HPLC purified vs. nonpurified mRNA in Karikó et al. 2011). Fortunately, the negative effect of dsRNA can be suppressed almost entirely by using N1-methylpseudouridine for uridine substitution in synthesized mRNA (Andries et al. 2015; Svitkin et al. 2017; Baiersdörfer et al. 2019; Kim et al. 2022). Therefore, there was no need to apply HPLC, a gold standard in purification of mRNAs from dsRNA contaminants (Karikó et al. 2011; Sikorski et al. 2020). The use of affinity oligo(dT) chromatography or RNeasy column purification was sufficient to obtain N1-methylpseudouridine–modified TCP-RNAs with good quality (study on translation of TCP-mRNAs in cell culture will be published separately).
Since we used the affinity oligo(dT) or RNeasy column chromatography and did not use HPLC for TCP-mRNA purification from dsRNA contaminants, the observed levels of dsRNA contamination may reflect a true impact of TCPs on the formation of dsRNA contaminants during IVT reaction. Interestingly, a higher level of dsRNA contaminants (up to 8 ng/μg) was generally detected in TCP-mRNAs prepared with TCPs containing either LNA-adenosine (3-3A, 3-5A, 3-5B) or 2,6-diaminopurine riboside (3-1A, 3-1B) substitution for adenosine residue at position +1 of the TCP (Supplemental Section S2; Supplemental Fig. S8). While these TCPs are expected to have a higher affinity to the DNA template strand at TSS and, consequently, higher IVT initiation power (compared to the other TCP derivatives containing unmodified 2′-O-methyladenosine), they also have a higher potential to bind any complementary 5′-CU-3′ sequence(s) on synthesized TCP-mRNA strand, and they could initiate synthesis of dsRNA by-products. Earlier, it was shown that T7 RNA polymerase can initiate second strand RNA synthesis on ssRNA template (Cazenave and Uhlenbeck 1994; Arnaud-Barbe et al. 1998). Note: It was reported that some dsRNA molecules may contain TCP caps (Sikorski et al. 2020); this agrees with our unpublished observations.
Since the capping efficiency for all TCP-mRNAs studied was typically high (>95%), the influence of uncapped ppp-mRNA contaminants (at <5% level) on the protein synthesis should not be a determining factor. However, TCP-mRNA translation efficiency may depend substantially on the integrity of TCP-mRNA. There were noticeable variations in the TCP-mRNAs integrity, particularly the percentage of the full-length product (FLP) (Supplemental Section S2; Supplemental Tables S3, S4). Two different analytical instruments (methods) were used to assess the RNA FLP quality: fragment analyzer (FA) and reverse phase HPLC. The observed percent of FLP was generally high but not identical for prepared TCP-mRNAs (from 71% to 97%). The reason for such variation is not clear, and it may reflect that IVT reaction and TCP-mRNA isolation/purification protocols still need further improvement. Thus, we noticed that oligo(dT) affinity chromatography may have an advantage over the RNeasy column for TCP-RNAs purification since the average percent of FLP isolated by oligo(dT) affinity chromatography was modestly higher (by ∼8%).
It is worthwhile to mention that inside the cell, the TCP-mRNA will be a substrate for cytoplasmic decapping enzymes, such as Dcp2 (Grudzien-Nogalska and Kiledjian 2017). The TCP-mRNA can (and will) lose TCP-cap resulting in an accumulation of inactive decapped 5′-p-mRNA. Consequently, a level of susceptibility of each TCP-mRNA to decapping enzymes in the cell culture and in vivo may (and should) affect the translation efficiency of the TCP-mRNA (Mandell et al. 2025). We speculate that not only the chemical nature of TCP modification but also a conformational status of 7mGppp fragment of TCP-mRNA may play a significant role in efficiency of TCP-mRNA decapping and consequently may affect its translation efficiency.
At this time, without further detailed structural studies (NMR, crystallography, and molecular modeling simulation) and biological testing, it is not possible to make solid predictions and/or recommendations regarding a specific nature of modifications in TCP-fragment (as a part of TCP-mRNA) that would lead to a higher eIF4E affinity, higher decapping resistance, lower dsRNA contaminants formation and, consequently, to a higher translation efficiency of TCP-mRNA. Also, a closer look may be required to assess the influence of the 5′ untranslated region (UTR) neighboring nucleotide sequence, next to TCP fragment, on the translation properties of the TCP-mRNA.
Conclusion
We have designed and prepared over 70 modified TCPs, derivatives of the parent 7mGpppAmpG molecule, using a novel “one-pot-two-step” synthetic approach. At optimized conditions, the use of TCP derivatives and complementary DNA templates allowed for a robust IVT synthesis of many TCP-mRNAs with a high yield and high capping efficiency. Using appropriate purification/isolation methods, we prepared high-quality TCP-mRNAs necessary for a future cellular experiment. Most importantly, the development of this technology allowed for the large-scale (multi-Kg level) production of the TCP compound 1-2A that was used in the preparation of the SARS-CoV-2 mRNA vaccine by Pfizer–BioNTech during the Covid-19 pandemic.
MATERIALS AND METHODS
Analytical
Mass spectra were recorded with LTQ Orbitrap LX or Q Exactive Orbitrap Mass Spectrometers (both Thermo Fisher Scientific) using negative mode detection and ESI mode of ionization. Proton and phosphorus NMR spectra of TCPs were recorded at 25°C with Bruker Avance II 500 MHz spectrometer; some phosphorus NMR spectra were recorded with Magritek Spinsolve 80 Benchtop NMR spectrometer operating at 32.4 MHz (phosphorus).
Analytical anion exchange HPLC of TCP derivatives and analogs was performed on Vanquish Horizon Flex UHPLC system (Thermo Scientific) using a DNAPac 200 RS column (4.6 × 50 mm; Thermo Scientific DX082508). Analytical reverse phase HPLC and HPLC-MS TCP derivatives and analogs were performed on Vanquish Horizon Flex UHPLC system (Thermo Scientific) coupled to Orbitrap Exploris 120 mass spectrometer (when required) using ACQUITY UPLC BEH C18 column (2.1 × 100 mm, 130 Å, 1.7 µm, Waters part ID:186002352). For further details, see Supplemental Section S1, Supplemental Table S2, Supplemental Figures S11 and S12.
Reagents
All common solvents, chemical reagents, supplies, and starting materials, including nucleosides, nucleotides, and their phosphoroamidites, were purchased from commercial sources. The dinucleotides (pAmpG, pAmpGm, pAfpG, pAfpU, pAmpA, pAmpU,p6mAmpU, p6mAmpA, pGmpG, pALNApG, and pDmpG) were either synthesized on solid support using commercially available phosphoramidites and purified by TriLink Biotechnologies oligonucleotide manufacturing or purchased from commercial sources.
General approach to the synthesis and purification of trinucleotide capped primers (TCP)
General information on the TCP structures and analytical data is presented in Tables 1–3 and in Supplemental Section S1 and Supplemental Tables S1 and S2.
Standard “one-pot-two-step” route to TCP
Step 1: Synthesis of imidazolide of 7mGDP derivative
To a stirred solution of 7mGDP derivative (0.2 M; triethylammonium salt form, 1 mol equiv.) in water:DMSO = 1:9 (v/v) at room temperature was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.6 mol equiv.) followed by imidazole (3 mol equiv.). The resulting mixture was allowed to stir at room temperature. The progress of formation of 7mGDP-imidazolide was monitored using AX-HPLC and/or 31P NMR spectroscopy. The reaction can take up to 24 h to complete.
Step 2: Synthesis of TCP derivative
After Step 1 reaction was completed, the magnesium chloride (3.15 M in water, 1.3 mol equiv.) was added to the stirred solution, followed by 5′-phosphorylated dinucleotide derivative (triethylammonium salt form, 0.75 mol equiv.). The resulting solution was allowed to stir at room temperature, and the progress of formation of 7mGppp-dinucleotide was monitored with AX-HPLC. Typically, the reaction was completed within 10–24 h, depending on the type of 7mGDP-imidazolide derivative and dinucleotide derivative used.
Alternative route 1
Step 1: Synthesis of imidazolide of 7mGMP derivative
To a stirred solution of 7mGMP derivative (0.2 M; triethylammonium salt form, 1 mol equiv.) in water:DMSO = 1:9 (v/v) at room temperature was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.6 mol equiv.) followed by imidazole (3 mol equiv.). The resulting mixture was allowed to stir at room temperature. The progress of formation of 7mGMP-imidazolide was monitored using AX-HPLC and/or 31P NMR spectroscopy (up to 24 h).
Step 2: Synthesis of TCP derivatives containing bridging αβ-NH or αβ-CH2 substitutions in triphosphate fragment
After Step 1 reaction was completed, the magnesium chloride (3.15 m in water, 1.3 mol equiv.) was added to the stirred solution, followed by 5′-pNHp- or pCH2p-modified dinucleotide derivative (triethylammonium salt form, 0.6 mol equiv.). The resulting solution was allowed to stir at room temperature, and the progress of formation of 7mGppNHp- or 7mGppCH2p-dinucleotide product was monitored with AX-HPLC. Typically, the reaction was completed within 10–48 h, depending on the type of dinucleotide derivative used.
Alternative route 2
Step 1: Synthesis of imidazolide of 5′-diphosphorylated dinucleotide derivatives
To a stirred solution of 5′-diphosphorylated dinucleotide derivative (0.2 M; triethylammonium salt form, 1 mol equiv.) in water:DMSO = 1:9 (v/v) at room temperature was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.6 mol equiv.) followed by imidazole (3 mol equiv.). The resulting mixture was allowed to stir at room temperature. The progress of formation of a respective imidazolide derivative of dinucleotide was monitored using AX-HPLC and/or 31P NMR spectroscopy (up to 24 h).
Step 2: Synthesis of γ-thio TCP derivatives
After Step 1 reaction was completed, the magnesium chloride (3.15 M in water, 1.3 mol equiv.) was added to the stirred solution, followed by phosphorothioate 7mGMP derivative (triethylammonium salt form, 0.5 mol equiv.). The resulting solution was allowed to stir at room temperature, and the progress of formation of 7mGp(s)pp-dinucleotide was monitored with AX-HPLC. Typically, the reaction was completed within 10–48 h, depending on the phosphorothioate 7mGMP derivative and/or the dinucleotide imidazolide derivative used in the coupling step.
Alternative route 3
Step 1: Synthesis of imidazolide of 5′-phosphorylated dinucleotide derivatives
To a stirred solution of 5′-phosphorylated dinucleotide derivative (0.2 M; triethylammonium salt form, 1 mol equiv.) in water:DMSO = 1:9 (v/v) at room temperature was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.6 mol equiv.) followed by imidazole (3 mol equiv.). The resulting mixture was allowed to stir at room temperature. The progress of formation of a respective dinucleotide imidazolide derivative was monitored using AX-HPLC and/or 31P NMR spectroscopy (up to 24 h).
Step 2: Synthesis of β-thio TCP derivative
After Step 1 reaction was completed, the magnesium chloride (3.15 M in water, 1.3 mol equiv.) was added to the stirred solution, followed by N7-methyl-3′-O-methylguanosine 5′-O-(2-thiodiphosphate) derivative (triethylammonium salt form, 2.0 mol equivalent). The resulting solution was allowed to stir at room temperature, and the progress of formation of 7mGpp(s)p-dinucleotide derivative was monitored with AX-HPLC. Typically, the reaction was completed within 15–48 h, depending on the dinucleotide imidazolide derivative used in the coupling step.
Purification of TCPs by anion exchange chromatography and preparation of 100 mM solution
The crude reaction mixture was then diluted with 10× volume of water and loaded on an anion exchange column equilibrated in buffer A (Q Sepharose Fast Flow Resin, QFF; 1 mmol of crude product per 100 mL of QFF). Elution was performed using a linear gradient from 25% to 45% buffer B for 4 column volumes and holding at 45% for an additional 1.5 column volumes. Buffer A: 20% acetonitrile in water; buffer B: 1.5 m triethylammonium acetate in water, pH 6.8. The fractions containing the desired product were pooled and concentrated under vacuum, and the final product was precipitated as a sodium salt with 95% ethanol containing 0.3 m sodium acetate. The precipitate was washed with 95% ethanol and redissolved in water, and the final product was concentrated on rotavapor at 25°C and reduced pressure to a dried solid. The solid TCP product was placed under a high vacuum at room temperature until a constant weight was reached. The 100 mM solution of TCP in water (at pH 6.8) was prepared from the dried solid based on the molecular weight of TCP (as disodium salt) and accounting for residual triethylammonium and/or acetate ions contamination when they were detected in the 1H NMR spectrum (the frozen solution was stored at −20°C for several months). A typical nucleotide purity for TCP was 98%–99+%; the isolated yields after AX-purification were reported in Supplemental Section S1 and Supplemental Table S1.
When it was necessary, a repurification of TCP was performed by anion exchange chromatography using Q Sepharose High Performance Resin (QHP) using the same buffer system and chromatography gradient profile but at fivefold reduced flow rate. After QHP purification, the product was converted into sodium salt form as described above.
Purification of TCPs and/or separation of diastereomers of TCPs by reverse phase chromatography
In a few cases, a reverse phase chromatography using AKTA Avant 25 chromatography system on Nova-Pak HR C18 column (e.g., 19 × 300 mm, 6 µm, 60 Å; Waters WAT025822 or similar column of appropriate size) was necessary to further repurify the TCP sample. Chromatography was performed using buffer A of 100 mM triethylammonium acetate in water, pH 6.8 and buffer B of acetonitrile; linear gradient: from 0% to 20% of buffer B over 10 column volumes at 25 mL/min flow rate.
Separation of diastereomers of phosphorothioate TCP derivatives was performed using the above-mentioned Nova-Pak HR C18 column. As an example, the mixture of 2-5A1 and 2-5A2 TCP diastereomers (200 mg in 200 mL of buffer A) was loaded on the column, washed with 200 mL of buffer A, and eluted with linear gradient of buffer B (from 0% to 5% over 800 mL). Buffer A: 50 mM ammonium acetate, pH 6.0; buffer B: acetonitrile; flow rate 20 mL/min. Pure TCP diastereomers of 2-5A were isolated: Diastereomer 1 (80 mg) and Diastereomer 2 (95 mg); for further details and HPLC analyses, see Supplemental Section S1 and Supplemental Figure S5. Using the same approach, the mixtures of other diastereomers (2-5B1/2-5B2, 2-6B1/2-6B2, 3-4A1/3-4A2, 3-8A1/3-8A2, 3-6B1/3-6B2, and 3-7B1/3-7B2) were separated.
Synthesis of pp6mAmpG dinucleotide
To a stirred solution of p6mAmpG dinucleotide (2× TEAH+ salt; 5.0 g, 5.42 mmol) in 20 mL of water:DMSO = 1:9 (v/v) at room temperature was added 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride EDC (2.59 g, 16.66 mmol) followed by imidazole (2.20 g, 32.27 mmol). The resulting mixture was allowed to stir at room temperature, and the progress of reaction was monitored using AX-HPLC and/or 31P NMR spectroscopy. When formation of active imidazolide derivative was completed (∼24 h), the 1 M tributylammonium phosphate in DMF (85 mL, 85 mmol) was added. The resulting solution was allowed to stir at room temperature for 16 h. The crude reaction mixture was then diluted with 10× water and purified by QFF anion exchange chromatography, as described above for TCP, followed by reverse phase chromatography in 100 mM triethylammonium bicarbonate (pH 8.0) using a linear gradient of acetonitrile. The fractions containing pure (98%–99%, by AX-HPLC at 260 nm) pp6mAmpG were pooled, evaporated, and coevaporated three times with methanol. The isolated yield of 2× triethylammonium salt of pp6mAmpG was 3.46 g (3.45 mmol; 63%). 1H NMR (500 MHz, D2O): δ: 8.49 (s, 1H), 8.20 (s, 1H), 7.93 (s, 1H), 6.12 (d, 1H, J = 6.2 Hz), 5.83 (d, 1H, J = 6.2 Hz), 4.97 (m, 1H), 4.82 (t, 1H, J = 5.6 Hz), 4.57 (t, 1H, J = 5.2 Hz), 4.54 (t, 1H, J = 2.6 Hz), 4.49 (m, 1H), 4.35(t, 1H, J = 2.3 Hz), 4.19 (m, 4H), 3.42 (s, 3H). 31P NMR (200 MHz, D2O): δ: −0.34 (1P), −5.75 (1P), −10.55 (1P). MS: m/z = 799 [M-H].
Synthesis of other modified dinucleotide derivatives
The synthesis, isolation, purification, and characterization of other modified dinucleotide derivatives are described in Supplemental Section S1.
Synthesis of N7-methyl-3′-O-methylguanosine 5′-phosphorothiolate
3′-O-methylguanosine (1.00 g, 3.36 mmol) was dissolved in anhydrous N-methyl-2-pyrrolidone (10 mL), followed by the addition of imidazole (1.38 g, 20.2 mmol), triphenylphosphine (2.65 g, 10.1 mmol), and iodine (2.55 g, 10.1 mmol). The reaction was stirred at 25°C for 3 h and then transferred to a 120 mL dichloromethane/water mix (3:1). The heterogenous mixture was kept at 4°C overnight, at which point a white precipitate formed. The precipitate was filtered off under reduced pressure and dried overnight in a vacuum desiccator to yield 5′-iodo-3′-O-methylguanosine. The material was used without further purification.
5′-Iodo-3′-O-methylguanosine (1.00 g, 2.45 mmol) was dissolved in 15 mL of 100 mM NaOH, followed by the addition of sodium thiophosphate (2.21 g, 12.30 mmol). The reaction mixture was heated at 50°C for 3 h. The reaction progress was followed by reverse-phase HPLC in 100 mM TEAA-acetonitrile gradient system. The isolation and purification of 3′-O-methylguanosine 5′-phosphorothiolate was performed by preparative reversed phase chromatography in 100 mM triethylammonium bicarbonate using a linear gradient of acetonitrile.
3′-O-methylguanosine 5′-phosphorothiolate (1.00 g, 2.54 mmol) was dissolved in 10 mL of 0.5 M sodium acetate buffer (pH 4.0), followed by the portion-wise addition of dimethyl sulfate (0.64 g, 5.09 mmol) over an hour. The pH of the solution was maintained at 4.0 using the addition of 1 M NaOH as needed. The reaction mixture was diluted 10× using deionized water and then extracted twice with 100 mL of ethyl acetate. The 7-methyl-3′-O-methylguanosine 5′-phosphorothiolate was purified by anion exchange chromatography using QFF resin and linear gradient of triethylammonium bicarbonate (0%–30%; pH 8.0) over 3 column volumes. The fractions containing pure product (98+%) were pooled, evaporated, and coevaporated three times with methanol. The isolated yield of triethylammonium salt of 7-methyl-3′-O-methylguanosine 5′-phosphorotholate was 0.86 g (2.05 mmol; 64%).
Synthesis of N7-methyl-3′-O-methylguanosine 5′-O-(2-thiodiphosphate)
Synthesis of triethylammonium salt of N7-methyl-3′-O-methylguanosine 5′-O-(2-thiodiphosphate) was performed similarly as described for N7-methyl-2′-O-methylguanosine 5′-O-(2-thiodiphosphate) (Kowalska et al. 2008).
IVT synthesis, purification, and analysis of TCP-mRNAs
Synthesis and analysis of TCP-mRNAs were performed using previously described protocols (or similar) using 1-methyl-pseudo-UTP substitution for UTP (Mandell et al. 2025). For TCP derivatives containing N6-methyl-2′-O-methyladenosine in the dinucleotide fragment, modified IVT conditions were used (Mandell et al. 2025; see also https://www.trilinkbiotech.com/cleancap-reagent-m6.html). Structures of synthesized TCP-mRNAs and full nucleotide sequences of FLuc-mRNA and eGFP-mRNA are shown in Supplemental Section S2. The TCP-mRNAs were treated with DNase I and then isolated and purified according to the manufacturer suggested protocols either using oligo(dT) affinity column (https://www.biaseparations.com/en/library/technical-notes/1084/purification-of-mrna-with-cimmultustm-oligo-dt); see also Mandell et al. (2025) or RNeasy Midi/RNeasy Maxi mRNA Purification Kit (https://www.qiagen.com/de/resources/download.aspx?id=a58c7b03-a25f-4ba7-9825-8fa473ba784f&lang=en). After purification, all TCP-mRNAs went through the analytical test panel including TCP-mRNAs integrity, capping efficiency, dsRNA content, residual DNA template, and residual proteins (details are presented in Supplemental Section S2).
Evaluation of TCP-mRNA capping efficiency
The analysis of capping efficiency for IVT-synthesized TCP-mRNAs was performed using a protocol adapted from Vlatkovic et al. (2022). The main modification to the protocol was a replacement of RNAzyme with 8–17 DNAzyme (Santoro and Joyce 1997). A short oligonucleotide sequence(s) was cleaved from the 5′-end of TCP-mRNA (see Figs. 2, 3, 7) using treatment with DNAzyme: 5′-d(TTACTCTTCTTTTTCCGAGCCGGACGACTCTTATTTCCC). The 5′-triphosphates and 2′,3′-cyclic phosphates were removed with CIP (New England Biolabs M0525) and T4 PNK (New England Biolabs M0201) treatment combining the manufacturer's procedure and published protocol (Schürer et al. 2002), respectively. The cleaved TCP-capped and/or uncapped 5′-OH oligonucleotide fragments, the DNAzyme, and remaining large fragment of mRNA molecule were separated, identified, and analyzed by HPLC-MS method using Vanquish UHPLC that was connected to the Orbitrap Exploris 120 or LTQ Orbitrap LX. The deconvoluted MS signal intensities (calculated by ProMass software for each individual species) corresponding to uncapped and TCP-capped fragments, with correct observed masses (based on calculated predictions), were used in the calculation of TCP-mRNA capping efficiency using the following formula: capping efficiency, % = [(Σ mass intensities of identified TCP-capped species)/(Σ mass intensities of identified 5′ TCP-capped + identified 5′ uncapped species)] × 100%. The capping efficiency data are presented in Supplemental Section S2 and Supplemental Tables S3 and S4; examples of capping assay results are shown in Supplemental Figures S9 and S10.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
All authors are current or former employees of TriLink Biotechnologies, and they own or have owned stock in Maravai Life Sciences.
ACKNOWLEDGMENTS
We thank all our current and former colleagues at TriLink Biotechnologies and Maravai Life Sciences (San Diego, CA) who supported us during this study. We thank Inna Koukhareva for help and advice on analytical assays for TCP-mRNA capping efficiency and Evan Myers for adapting the oligo(dT) mRNA-purification protocol. We also thank Kate Broderick, Stephanie Ramos, Ben Hudson, Gali Steinberg-Tatman, and Inna Koukhareva for helpful discussions and advice on the manuscript. We appreciate Anton McCaffrey, Krist Azizian, Dongwon Shin, and Richard Hogrefe for their role in creating the TCP-mRNA technology.
Author contributions: C.X., C.Z., M.H., and A.V.L. designed the study; R.C., C.X., and A.V.L. designed and developed the main and alternative transient activation methods for the synthesis of TCPs; R.C., I.I., M.T., C.X., J.C., M.S., V.B., and P.L. synthesized, purified, and analyzed TCPs and intermediate compounds; M.S., C.V., F.R., A.U., and J.H. performed IVT TCP-mRNA synthesis, analysis, and qualification of mRNAs; A.V.L. wrote the first and revised drafts of the manuscript. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080634.125.
-
Freely available online through the RNA Open Access option.
- Received June 10, 2025.
- Accepted November 6, 2025.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Chunping Xu is the first author of this paper, “Novel trinucleotide mRNA capping reagents: improved synthetic route and efficient cotranscriptional incorporation in mRNA.” Chunping is the senior director of research and development at TriLink Biotechnologies, part of Maravai Biosciences, where she has been working for close to 9 years, with a main focus on research on mRNA capping technology development. Over the years, they have developed three generations of cotranscriptional mRNA capping reagents that have been used broadly in the mRNA field. One of the mRNA capping reagents (CleanCap AG 3′OMe) has also been used in the Pfizer/BioNTech Covid vaccine.
What are the major results described in your paper, and how do they impact this branch of the field?
This paper mainly describes our unique and highly efficient synthesis of trinucleotide mRNA cap analogs. This is the first time that the one-pot-two-step synthesis using EDC/imidazole as an activation reagent was published. This in general will greatly improve the synthetic process for mRNA cap analogs and will streamline any large-scale synthesis of mRNA capping reagents. Also, it is a very versatile synthesis: We used it to introduce various modifications at N7-G, triphosphate linkage, and AG dimer to mRNA cap analogs.
What led you to study RNA or this aspect of RNA science?
When I first joined TriLink Biotechnologies back in 2018, developing and optimizing the mRNA capping reagent was one of the key projects for our team. I was fascinated by the high efficiency of IVT initiation from our first-generation mRNA capping reagent, CleanCap AG, but the chemical synthetic route for CleanCap AG was not scalable to make it a commercial product for wide application in mRNA for clinical study at that time, which led us to optimize the chemistry.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
At the beginning of the mRNA capping innovation, we were working on optimizing the process for CleanCap. We developed a very efficient and scalable synthetic process for CleanCap. During the pandemic, when our process was used in making hundreds of kilograms of cap for the Pfizer/BioNTech Covid mRNA vaccine, that was the moment I felt very rewarded as a scientist to see our science turned into real-life applications.
If you were able to give one piece of advice to your younger self, what would that be?
As scientists, we do our best to make science ready for applications. When the right time comes, it will find its application.








