
Constructing an active chimeric pRNA ring with a stoichiometry of six and identifying 12 domains of the pRNA ring binding to the 12-subunit channel of phi29 DNA-packaging motor
- 1College of Pharmacy, Division of Pharmaceutics and Pharmacology, Center for RNA Biology, The Ohio State University, Columbus, Ohio 43210, USA
- 2Center for RNA Nanobiotechnology and Nanomedicine, James Comprehensive Cancer Center, The Ohio State University, Columbus, Ohio 43210, USA
- 3Department of Biochemistry and Molecular Biology, Indiana University School of Medicine, Indianapolis, Indiana 46202, USA
- Corresponding authors: guo.1091{at}osu.edu; wz15{at}iu.edu
-
Handling editor: Eric Westhof
Abstract
During the last stage of replication of double-stranded RNA or DNA viruses, their genome is packaged into a preassembled protein capsid. The bacterial virus phi29 dsDNA-packaging motor uses a noncoding packaging RNA (pRNA) molecule to gear its genomic DNA translocation. In this study, we constructed chimeric pRNAs by fusing the pRNA of bacterial virus M2 and that of phi29. The chimeric pRNAs can form dimers or trimers. The dimeric or trimeric pRNAs were active in the packaging of the phi29 dsDNA genome into the purified procapsid, which was subsequently converted into the infectious viruses, as proven by counting plaque-forming units (PFUs). These data show that the stoichiometry of the chimeric pRNAs on the motor is six subunits, a multiple of 2 and 3. Furthermore, AFM studies on pRNA fused to an RNA-triangle revealed hexamer formation. But how do the six identical RNAs anchor on the 12-subunit connector with the double stoichiometry? Structural analysis in combination with enzymatic and chemical probing data revealed that each native pRNA contributes two domains to bind to the 12-subunit DNA-packaging channel at three positively charged residues RKR, proving the formation of the hexameric ring. Resolving the hexamer versus pentamer debate clarifies the mechanism of dsDNA translocation in living organisms.
Keywords
- revolving motor
- hexameric ATPase
- asymmetrical biomotor
- DNA-packaging motor
- DNA translocation
- noncoding RNA
INTRODUCTION
One of the basic features of life is motion, which is realized by the ubiquitous motors that belong to the ATPase family, including the DNA translocating enzymes. All living organisms contain genomic double-stranded DNA (dsDNA) that is transported to subsequent progeny cells. DsDNA translocation is ubiquitous in living systems, such as cell mitosis, bacterial binary fission, DNA replication, genome transport, DNA repair, homologous recombination, viral dsDNA packaging, and intracellular trafficking. A fundamental question is how living systems translocate their lengthy helical, dsDNA genomes without coiling and tangling. If translocation of a dsDNA genome uses a rotational mechanism, the resulting lengthy dsDNA genome will be supercoiled or intertwined. Thus, the mechanism of dsDNA translocation has been of interest for many years. A sequential revolving mechanism (Fig. 1) without rotation using asymmetric ATPase motors has been recently reported in phages, bacteria, Streptomyces, and human viruses, thereby overcoming the coiling and tangling of long dsDNA genome during translocation.
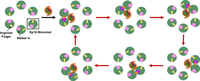
Model of the asymmetrical hexameric ATP-driving revolving motion. Sequential motion is driven by a series of conformational changes regulated by ATP binding and hydrolysis. (Green) Gp16 subunit, (pink) Walker A domain, (yellow) arginine finger.
The DNA-packaging motor of phi29 is a prototypical model of dsDNA translocation. It uses an RNA ring to regulate the motion of the dsDNA-packaging motor. During bacteriophage replication, phages use DNA-packaging motors to translocate DNA genomes into procapsids. The simplicity of bacteriophage DNA-packaging motors has provided intriguing models for the study of dsDNA translocation. These DNA-packaging motors can generate over 50 pN of force, making them among the strongest molecular machines in nature (Smith 2011). The components of the biomotor can be synthesized artificially and can be studied in vitro with great efficacy (Lee et al. 2009). The phi29 biomotor contains a funnel-shaped channel that connects the viral capsid to the tail. This channel is composed of a dodecameric ring of gp10 protein (Simpson et al. 2000; Guasch et al. 2002), the packaging RNA (pRNA) (Guo et al. 1987b; Shu et al. 2007; Hill et al. 2016), and the ATPase gp16 (Guo et al. 1987a; Cai et al. 2019).
The debate over whether the RNA-driven DNA-packaging motor is a rotating pentamer or a revolving hexamer has persisted for more than two decades (Bohmer et al. 2023). The rotary mechanism is analogous to the Earth spinning on its own axis (Thomsen and Berger 2009). In contrast, the revolving mechanism is analogous to the Earth revolving around the sun. Initially, it was popularly believed that DNA-translocation motors moved DNA through a rotating “nut and bolt” mechanism, wherein the portal operated as a nut and the DNA operated as the bolt that was propelled into the procapsid via rotational force (Hendrix 1978; Simpson et al. 2000). This rotary mechanism was later contested by the fact that the T4 and phi29 DNA-packaging motors could pack immobilized dsDNA without rotating (Baumann et al. 2006; Chang et al. 2008). Fluorescence polarization studies found no rotational movement of the phi29 biomotor during DNA transport (Hugel et al. 2007). However, extensive investigation demonstrated that rather than a rotary mechanism, dsDNA-packaging motors and many long-dsDNA translocation motors use a revolving mechanism with a DNA strand revolving around the inner wall of the channel. Additionally, the diameter of the narrowest pore of the phi29 and T4 biomotors are ∼3.6–3.8 nm, which is much larger than the dsDNA (∼2 nm) and therefore unlikely to employ a rotating nut and bolt mechanism, which would require that both strands make contact with the channel wall at the same time (Guo et al. 2014a).
Biomotors confirmed to have a rotation mechanism have proven to have right-handed channels to gear the right-handed dsDNA, matching the bolt-and-nut screwing mechanism since one of the strands of the dsDNA move outside the channel. Contrary to the right-handed chirality of dsDNA, the lengthy-dsDNA translocation motor channels using revolution mechanisms all have left-handed chirality. The left-handed chirality channel includes phi29 (Xu et al. 2019), SPP1 (Lebedev et al. 2007), P22 (Olia et al. 2011), T4 (Sun et al. 2015), T7 (Cuervo et al. 2013), herpesvirus (Liu et al. 2019), FtsK (Massey et al. 2006), and TraB (Amado et al. 2019). Rotating biomotors have pore diameters similar in size to the diameter of DNA, ensuring a tight fit, but the revolving motors listed above all have the channel size from 2.8 to 3.5 nm larger than the dsDNA (Bohmer et al. 2023). Through X-ray crystallography studies, it was discovered that the subunits of the connector wall were tilted 30° with respect to the central axle (Schwartz et al. 2013; Guo et al. 2014b; Bhullar et al. 2022). Cryo-electron microscopy (Cryo-EM) studies have uncovered that the dsDNA of the T7 phage moved along the wall of the connector instead of going through the center (Agirrezabala et al. 2005; Guo et al. 2013).
Examples of rotary motors include the bacterial translocase TrwB (Gomis-Ruth et al. 2001), DNA helicase channel of RepA (Niedenzu et al. 2001), the bacterial transcription factor Rho (Thomsen and Berger 2009), papillomavirus helicase E1 (Enemark and Joshua-Tor 2006), Vps4 ATPase (Caillat et al. 2015), human 26S proteasome (Wehmer et al. 2017), Cdc48/p97 ATPase (Banerjee et al. 2016), and katanin hexamer (Zehr et al. 2020).
The revolving motors reported include phi29 (Schwartz et al. 2013; Zhao et al. 2013, 2016). T7 DNA-ejection motor (Cuervo et al. 2019), conjugative plasmid transfer enzyme TraB (Amado et al. 2019), Gram-negative bacterial translocase FtsK (Jean et al. 2020), Gram-positive bacterial translocase SpoIIIE (Liu et al. 2015), and herpesvirus DNA-packaging motor (Cuervo et al. 2019; Yang et al. 2020).
The debate of pentameric rotation and hexameric revolving leads to one difficult question: How do six copies of pRNA form a stable structure with its anchor base containing 12 subunits? Thus, it has been proposed that pentameric RNA binds to the pentameric capsid vertex to turn the motor core.
In this study, we constructed double-sized chimeric RNAs to investigate the stoichiometry of active pRNA on the motor. We used atomic force microscopy (AFM) images to confirm the hexamer structure. We also use structural details, various probing techniques, and AlphaFold reconstruction to assess the pRNA structure and stoichiometry. Our new structural analysis shows that the stoichiometry of phi29 pRNA ring is six subunits, common multiples of 2 and 3. Phi29 pRNA hexamer contains 12 negatively charged protruding regions, with each pRNA contributing two protruding regions to interact with the three positively charged amino acids Arg–Lys–Arg extending from the N-terminal of each motor subunit. Resolving the hexamer versus pentamer debate will clarify the mechanism of dsDNA translocation in living organisms.
RESULTS
Formation of pRNA dimers and trimers
The formation of the dimer and the trimer as the assembly intermediate of the RNA hexameric ring between the upper and lower loops of different pRNA molecules (visualized in Fig. 2) was confirmed by the shift of pRNA migration rate in native PAGE gels, where pRNA Ab′M2 was mixed with Ba′M2 (Fig. 3). The gel and running buffer contained magnesium (1× TBM), which is indispensable for proper pRNA folding (Chen and Guo 1997; Zhang et al. 2013). A shift to a slower-migrating band was seen when both pRNAs were run together, indicating dimerization. Similarly, the formation of trimers was assessed with Ab′M2, Be′M2, and Ea′M2. When all three pRNAs were combined, a trimer band was observed in the native gel (Fig. 4).
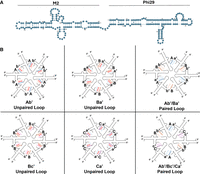
pRNA structure and ring formation. (A) Secondary structure of phi29-M2 pRNA. (B) Complementary right- and left-hand loops interact to form hexameric rings that were proved to be active (paired) and inactive (unpaired, with a red X).
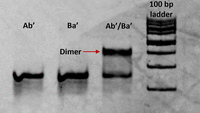
Assay for dimerization by hand-in-hand interaction of the left-hand Ab′ and right-hand Ba′ designed via complementary modifications (8% native PAGE in 1× TBM).
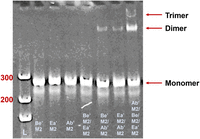
Assay for trimerization by hand-in-hand interaction of Ab′M2/Be′M2/Ea′M2 pRNA designed via complementary modifications (8% native PAGE in 1× TBM; [L] DNA ladder).
The number of pRNA molecules is a multiple of 2
Two hybrid chimera pRNA systems were constructed, with both of them approximately doubling the size of the pRNA. The first one is the fusion of the phi29 RNA with the M2 phage pRNA, conjoined by a 5 nt linker made up of 5 As (Bailey et al. 1990; Chen et al. 1999; Hao and Kieft 2016). Phi29 pRNA was at the 5′ end and the M2 pRNA was at the 3′ end. The right- and left-hand regions of the M2 pRNA were mutated to be noncomplementary to prevent dimerization, ensuring that any observed multimerization is due to the phi29 sequence and not the M2 sequence. Two pRNA variants were produced by intruding complementary mutations to the right- and left-hand loops. The subsets are denoted as Ab′M2 and Ba′M2. The capital letter refers to the right-hand loop and the lowercase letter refers to the left-hand loop. For example, an A right-hand loop can bind to an a′ left-hand loop, but not to a b′ left-hand loop. These pRNAs were tested for their ability to facilitate DNA packaging (Guo et al. 1986). Briefly, all the components required for DNA packaging (procapsid, DNA-gp3, gp16, pRNA, and ATP) were combined in vitro. Any unpackaged DNA was digested, followed by digestion of the procapsid. The remaining DNA, the DNA that had been packaged into the procapsid, was visualized on an agarose gel.
The motor pRNA ring is assembled via intermolecular instead of intramolecular interactions. The two loops interact during DNA packaging and only four bases are required in a loop for the interaction. Ab′M2 and Ba′M2 pRNAs, with unpaired right- and left-hand loops, were inactive in the packaging of phi29 genomic DNA when used alone. However, when the two inactive pRNAs, with trans-complementary upper and lower loops, were mixed in a 1:1 molar ratio, DNA packaging (Fig. 5A,B) and virion assembly (Fig. 5C) were observed. Since a closed ring composed of an even number of pRNAs can be produced from two mutant pRNAs with interlocking upper and lower loop sequences, it suggests that Ab′ and Ba′ interact to form a complex by the interaction of the right- and left-hand loops. The observed increase in DNA-packaging activity of a mixture of two inactive mutants indicates that the number of pRNAs in the DNA-packaging complex is a multiple of two, that is, an even number.

Evaluation of the biological function of the dimeric pRNA. (A) Assay for the DNA-packaging activity of the elongated Ab′M2/Ba′M2 pRNA. Positive control (+c) is with Aa′ pRNA. Negative control is Aa′ pRNA but without ATP. 1% agarose gel in 1× TAE. (B) Quantification of the gel, normalized to the positive control. (C) Plaque-forming unit assay of the elongated Ab′M2/Ba′M2 pRNA.
The number of pRNA molecules is a multiple of 3
Two other sets of pRNAs comprised of the three mutant pRNAs: Ab′M2, Bc′M2, and Ca′M2 (Set 1, Figs. 2, 6) and Ab′M2, Be′M2, and Ea′M2 (Set 2, Fig. 4) were designed and tested. When each of the pRNAs in this set was tested alone, each individual pRNA produced little to no activity in DNA packaging (Fig. 6). However, when all three pRNAs from these two sets were mixed in a 1:1:1 ratio, DNA-packaging activity was restored (Fig. 6). This indicates that the pRNAs interact to form a functional complex only when present in the correct stoichiometry, supporting the hypothesis that the active pRNA ring consists of a multiple of 3 pRNAs. The lack of activity of the pRNA mutants alone and the restored activity of mixtures of the three mutants was expected since the mutations in each RNA were engineered such that only the presence of all three RNAs would produce a closed ring. In considering the previous section suggesting that the number of pRNA in the ring was a multiple of two, an even number, a conclusion was reached that the pRNA is hexameric, excluding the possibility of a pentamer; if a pentamer is required for DNA packaging, then the dimeric or trimeric pRNAs by themselves would not result in DNA packaging since a pentamer cannot be made from dimers nor trimers. Thus, the number of pRNAs required for DNA packaging is a common multiple of 2 and 3, which is 6. These results strongly suggest that pRNAs interact intermolecularly through base-pairing via hand-in-hand interactions of the upper right-hand loop and the lower left-hand loops for DNA translocation.

DNA-packaging assay of trimeric pRNA. Assay for the DNA-packaging activity of the elongated Ab′M2/Bc′M2/Ca′M2 pRNA. (A) Gel result. (Positive control) Aa′ pRNA. (Negative control) Same as positive control but with no ATP. 1% agarose gel in 1× TAE. (B) Quantification of the gel, normalized to the positive control.
Confirmation of pRNA hexamer via AFM
We also sought to observe the pRNA hexamer via AFM imaging. This involved adding an RNA-triangle onto the 3′ end of each pRNA (Fig. 7A) to make the pRNA easier to visualize (Khisamutdinov et al. 2014; Bohmer et al. 2023). However, due to the complementary regions containing only 4 nt, the pRNA cannot form a ring without the presence of the connector. Therefore, to enable higher-order multimerization of the pRNA, the triangle pRNAs were extended from four to seven complementary nucleotides in each loop, resulting in a set of triangle-RNA via interacting loops of Ab′Δ, Bc′Δ, Cd′Δ, De′Δ, Ef′Δ, and Fa′Δ (Shu et al. 2013). Their combination produced a pRNA hexamer as revealed by AFM imaging (Fig. 7). All these data support that pRNA is a hexamer produced by the interaction of the right- and left-hand loops (Guo et al. 1998) and the procapsid-binding domain falls between nt 23 and 97 (Garver and Guo 1997).
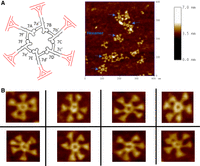
AFM images of the pRNA hexamer showing the structurally flexible arms to meet the sequential action requirement of each hexameric subunit to execute the revolving motion. (A, left) A model of the pRNA-triangle hexamer used for AFM imaging. (Right) An example of the full-sized AFM images. (B) Zoomed-in AFM images of the pRNA hexamer showing the flexible arms for cooperative inter-subunit shifting and revolving.
The structure of the phi29 connector–pRNA complex constructed from the published crystal structure
The first structure of the pRNA ring was computationally derived (Hoeprich and Guo 2002). Later, two crystal structures of the pRNA monomer were reported (Ding et al. 2011; Zhang et al. 2013). The structure of the pRNA hexameric ring was updated and published (Fig. 8A; Zhang et al. 2013) to include the crystal structure of the pRNA three-way junction (3WJ) (Zhang et al. 2013) and the crystal structure of the pRNA right- and left-hand loop (Ding et al. 2011). This published pRNA hexameric ring was used to build the connector–pRNA complex (Fig. 8; Supplemental File S1).

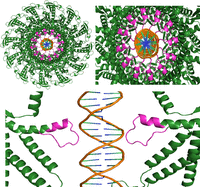
The structure of the Phi29 RNA–connector complex. (A) The pRNA hexamer based on the published crystal structure. Each monomer is a different color. (B) A top-down view of the computationally derived gp10 structure, which is made of 12 subunits. (C) Side and bottom views of the connector–pRNA complex. (Green) Connector; (yellow/red) pRNA. (D) The complex showing the relative position of the N-terminal Arg–Lys–Arg (RKR) motif (blue) and the two gp10-binding regions (red and yellow) of pRNA for interaction with the RKR motif.
On the other hand, the crystal structure of the phi29 connector (the channel of the DNA-packaging motor) was also published (Simpson et al. 2000, 2001; Guasch et al. 2002; Xu et al. 2019). The published crystal structures of the connector are missing three domains: the 14 N-terminal amino acids, the internal loops, and the C-terminal residues (Simpson et al. 2000; Guasch et al. 2002). These three domains were not deflected due to the flexibility resulting from the motion-property. Here, a full-length structure of the connector was computationally constructed via AlphaFold to include these three missing domains of the connector.
The phi29 connector displays a truncated cone shape containing a wide end and a narrow end (Fig. 8). The wide end is embedded within the procapsid, while the narrow end features three essential positively charged amino acids at residues 3–5, lysine–arginine–lysine (RKR), which protrude from the procapsid and are positioned to interact with RNA (Fig. 8D). It has been reported by two labs that the protruding RKR is indispensable for pRNA binding (Atz et al. 2007; Cai et al. 2008). When any one of the three RKR residues was mutated, the pRNA failed to bind the mutant procapsid. Our connector/RNA docking model, based on these experimental data, is used to demonstrate how the pRNA ring attaches to the protruding RKR motif (Fig. 8D, shown in blue).
Previous X-ray crystallography data showed that the connector contains three regions. The diameter of the wider C-terminal region was 13.8 nm, not including the missed 12-flexible loops; a central region was 9.4 nm; and a narrower N-terminal region was 6.6 nm, not including the missed 14 amino acids at each subunit (Simpson et al. 2000; Guasch et al. 2002). The pRNA ring contains a central channel with a diameter of 7.6 nm, which sheaths onto the N-terminus of the connector, interacts with the RKR of each subunit, and is anchored via the physical aid of the connector's central 9.4 nm region. As shown in Figure 8D, the positively charged RKR residues protrude from the connector's narrow end, allowing direct interactions with the negatively charged areas of the RNA ring to facilitate the proper alignment necessary for DNA-translocation activity. Additionally, the stem regions of the pRNA extend outward in six directions, creating a space that accommodates the binding and function of the ATPase gp16. This conformation illustrates the stable interface between pRNA and gp10. If the pRNA ring is a pentamer, the diameter of its internal ring would be 6.3 nm, as calculated even without the consideration of the missed protruding 14 terminal domain. Thus, the pentamer ring would be too small to sheath and anchor to the connector and is too tight for flexibility in gearing the motor.
Each pRNA of the hexamer binds to two positively charged gp10 N-termini
Our structural analysis, in combination with chemical and biological probing, revealed that each pRNA has two regions that bind the connector, generating a stable interaction as the 6-pRNA-ring binds to the 12-subunit connector. Thus, altogether, a pRNA hexamer will contain 12 regions where each one binds one subunit of the 12-subunit connector. One of the two protruding regions of each pRNA provides a better fit as a major binding affinity for the connector, and the other one protruding region only serves as an accessory force to bind the adjacent connector to stabilize the pRNA–connector interaction. This finding favors the conclusion that the pRNA ring is a hexamer and not a pentamer. Our successful modeling of the pRNA/connector complex structure in a 1:2 ratio (6-subunit pRNA hexamer binds to 12-subunit connector) also supports the potential binding motifs between pRNA and the connector and further validates our hypothesis of the pRNA hexameric ring.
Local structural analysis revealed that two adjacent connector subunits bind to a pRNA monomer at two distinct regions of the pRNA (Fig. 9A). The overall conformations of the two connector protein subunits are nearly identical, with slight variations observed in the N-terminus, which was expected since these residues are part of an unstructured flexible region. In our model, the first five amino acid residues of each connector subunit align closely with the grooves of the pRNA (Fig. 9B), suggesting potential hydrogen bonding between the polar residues of the connector and the RNA's negative-phosphate backbone.
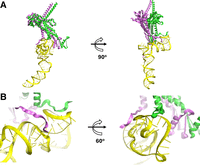
How one pRNA monomer interacts with two gp10 connector subunits. (A) One pRNA monomer with two gp10 subunits. (Yellow) pRNA; (green/magenta) two connector subunit gp10. (B) Zoomed-in view of the pRNA–protein interaction.
In addition to hydrogen bonds, electrostatic interactions occur between the positively charged residues, such as lysine and arginine, within the N-terminus of the connector and the negatively charged phosphates of the RNA. Hydrophobic interactions may also contribute to the stabilization of the complex with less polar regions of the pRNA interacting with hydrophobic patches on the connector. As depicted in Figure 10, the close proximity of these residues and their alignment with the RNA grooves support the hypothesis that multiple types of interactions—hydrogen bonds, electrostatic forces, and hydrophobic contacts—work synergistically to secure the pRNA to the connector.
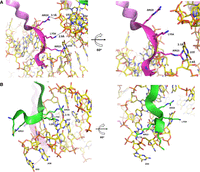
Zoomed-in view showing the location and distance of the interaction regions of pRNA and connector with the N-terminal Arg–Lys–Arg motif. (Ribbons) Connector fragment (three-letter); (wireframe) pRNA (one-letter). Interactions between the RKR of gp10 and (A) U35, U53, and U54 and (B) G55, A56, and G57.
The first 14 residues of gp10 are critical for pRNA binding, as removal of these residues eliminated binding in previous studies (Xiao et al. 2005). Specifically, RNA binds to a positively charged motif, RKR, at residues 3–5 (Donate et al. 1992; Atz et al. 2007). The detailed structural analysis reveals that the first of the gp10 subunits (represented by the pink ribbon in Fig. 10A) is positioned near the U35 residue of pRNA, forming several key hydrogen bonds. Arg3 establishes two hydrogen bonds with the exocyclic oxygens of U35, with distances of 3.1 and 2.8 Å. Additionally, Lys4 and Arg5 form hydrogen bonds with the nonbridging oxygen atoms of the phosphodiester linkages at U53 and U54, measuring 3.1 and 2.9 Å, respectively (Fig. 10A).
The second subunit of the gp10 (shown by the green ribbon in Fig. 10B) primarily interacts with pRNA residues G55, A56, and G57. In this case, Arg3 forms a hydrogen bond with the nonbridging oxygen of the phosphodiester linkage between G55 and A56 at 2.7 Å. Furthermore, both Lys4 and Arg5 engage in hydrogen bonding with the nucleobase of G57, with Lys4 forming a bond with N7 at a distance of 3.2 Å, and Arg5 bonding with O6 at 2.7 Å. These interactions suggest that the second subunit also contributes to the overall stability of the pRNA–connector complex through multiple noncovalent contacts.
Notably, based on our modeled structure, the subunit binding to U35 exhibits a greater quantity of hydrogen bonds between the pRNA and gp10 subunit than the other subunit (4 and 3, respectively). However, this model represents just one possible binding motif for the pRNA/connector complex, as these amino acid residues are dynamic and will consequently adopt different orientations, leading to various noncovalent interactions between the connector and pRNA.
Alignment of structural information with the existing literature on enzymatic probing, chemical probing, and truncation of phi29 pRNA
Reid et al. (1994a) probed pRNA with RNase V1, a ribonuclease that cleaves dsRNA. These footprints found that residues 22–69 were protected, which encompasses both connector binding regions. Residues 37–40 experienced heightened RNase V1 cleavage upon pRNA binding to the prohead, indicating that this region plays a role in gp10 binding. A later study by Garver and Guo (1997) used truncated pRNA mutants and reported that residues 37–91 are required for prohead binding, although at reduced efficiency. Our structure reported here agrees with these findings.
Another study by Zhang et al. (2001) conducted chemical probing (DMS, CMCT, and kethoxal) of phi29 bound and unbound to the procapsid. The entire region-2 residues (53–57) were strongly or weakly modified when unbound to the procapsid but were almost entirely unmodified when procapsid-bound. This indicates that region-2 is protected by the procapsid and therefore is involved in procapsid binding. These data align with our structure model reported here. Since each pRNA has two procapsid-binding regions and gp10 is a dodecamer, each pRNA in the hexameric ring should interact with two gp10 subunits. Since only region-2 showed a difference in chemical modification pattern, it is possible that region-2 is the primary driver of procapsid binding and region-1 is an accessory in binding.
It was previously reported that the pRNA binds to the fivefold procapsid vertex (Simpson et al. 2000). Therefore, it was deduced that the pRNA must be pentameric to bind with the pentameric capsid. However, it was later discovered that pRNA was cross-linked to the connector protein but not to the capsid protein (Garver and Guo 1997; Xiao et al. 2005; Sun et al. 2006). The finding that each pRNA binds to the connector at two regions, generating a stable platform for the 6-pRNA ring to bind to the 12-subunit connector agree with the finding by Garver and Guo (1997), and confirms that the pRNA ring is a hexamer to bind the dodecameric connector but not a pentamer to bind the pentameric vertices.
Assessment of the pRNA–connector complex structure from probing approaches
The computationally derived pRNA–connector complex was validated with a large spectrum of biochemical data. These data include cross-linking with multiple reagents, including phenphi (Mohammad et al. 1999), psoralen (Chen and Guo 1997), and azidophenacyl (Garver and Guo 2000), as well as complementary modification (Reid et al. 1994b; Zhang et al. 1995, 1997; Hoeprich and Guo 2002). As shown in Figures 11⇓–13, the short distances between the highlighted residues align with the probing data and the biochemical data. Therefore, the computational model closely matches and agrees with the empirical data. Furthermore, the AlphaFold model of the gp10 connector included the internal loops, which are absent in the crystal structures (Fig. 14). These loops are required for the one-way traffic of dsDNA during packaging (Zhao et al. 2013). Altogether, the results support the current structure of the pRNA hexamer/connector complex.
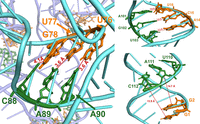
Evaluation of the distance and location of the final structure reveals that the structure agrees with the data from complementary modification showing that C88-A90 (green) pair with U76-G78 (orange), A101-U103 (green) pair with A14-U16 (orange), and U110-C112 (green) pair with G1-G2 (orange).
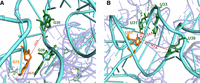
Evaluation of the distance and location of the final structure with experimental probing data. (A) Comparing the structure with the results of psoralen cross-linking revealing an agreement that G75 (orange) cross-linked to G28 and G30 (green). (B) Comparing the structure with the results of phenphi cross-linking revealed the agreement with the data that U69 (orange) cross-linked to U31, U33, and U36 (green).
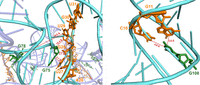
Evaluation of the distance and location of the final structure with the experimental probing data from azido phenacyl cross-linking, revealing an agreement that G75 and G78 (green) cross-linked to A26-U31 (orange). G108 (green) cross-linked to G10 and G11 (orange).
Elucidation of the interaction of the gp10 internal loops with dsDNA. (Gp10) Green; (internal loops) magenta; (dsDNA backbone) orange.
DISCUSSION
Here we successfully constructed pRNA chimeras and confirmed their ability to dimerize, trimerize, and participate in DNA packaging. This indicates that the pRNA forms a hexamer, not a pentamer since six is a multiple of two and three. This hexamer was visualized with AFM imaging. Furthermore, we used a combination of experimental and computational evidence to create a model of the phi29 connector–pRNA complex. This model corroborates the previous evidence that the RKR motif on the N-terminus of gp10 is vital for binding to pRNA, primarily via hydrogen bonding interactions (Atz et al. 2007; Cai et al. 2008). Taken together, these data demonstrate the importance of the hexameric pRNA ring in binding to and gearing the phi29 DNA-packaging motor. The finding that each pRNA binds to the connector at two regions, generating a stable platform for the 6-pRNA ring to bind to the 12-subunit connector, confirms that the pRNA ring is a hexamer and not a pentamer.
The process of molecular motion has been the subject of intense scrutiny. Recent studies on dsDNA or dsRNA translocation motors have shed new light on the migration mechanisms involving nucleic acids. The gate for DNA entry into phage procapsids is the connector. It appears that several similarities exist among the structures and functions of dsDNA-translocating ATPases as reported for dsDNA genome packaging of herpesvirus (Yang et al. 2020); bacterial dsDNA genome translocase FtsK (Crozat and Grainge 2010); the Streptomyces dsDNA plasmid conjugation enzyme TraB (Amado et al. 2019); and the Holliday junction dsDNA recombinase (Rish et al. 2023). The sequential action of the homogenous hexameric subunits via the revolving mechanism ensures translocation of the lengthy genome without coiling and tangling. This elegant revolving mechanism without rotation eliminates the need for a helicase or topoisomerase, which would consume a considerable amount of ATP energy and slow translocation, solving the coiling consequence. The finding that the pRNA is a hexamer, deduced from the common multiple of 2 and 3, and the discovery that the 6-pRNA ring contributes 12 domains to bind the 12-subunit connector, further clarifies the 27-year fervent debate about whether the pRNA is a hexamer with a revolving mechanism or a pentamer with the rotation mechanism. This conclusion is also supported by several key structural and functional features: (1) The motor protein only contacts one strand of the dsDNA (De-Donatis et al. 2014). (2) The motion is regulated via the interaction of the arginine finger with Walker A motif to promote the sequential action regulated by ATPase binding and hydrolysis (Zhao et al. 2016). (3) Crystal structure reveals that the channel wall has a 30° left-handed chirality to match 12 steps for one 360° revolution (Guasch et al. 2002). (4) Cryo-EM reveals an asymmetrical hexamer structure due to the step-size sequential action at any movement during revolving (Bohmer et al. 2023). (5) Channel size is larger than 2.8 nm, failing to contact the 2 nm dsDNA that travels through the center of the channel (De-Donatis et al. 2014). (6) The presence of four positively charged lysine layers to prevent the reverse sliding out during revolving (Fang et al. 2012). (7) The three steps of channel gating (Jing et al. 2010). Confirming the hexamer revolving mechanism will clarify this confusion and facilitate future structural and functional studies on the crucial ATP motors that translocate and traffic the dsDNA genome, a critical life process of humans and other living systems.
MATERIALS AND METHODS
Synthesis of pRNA
The chimeric pRNAs were synthesized by starting with a regular length pRNA DNA template and adding on the additional via a series of PCRs with 3′ templates containing overhangs that contain the 3′ add-on sequence. The T7 promoter sequence was also added to the 5′ end of the DNA template with a 5′ PCR primer containing the corresponding overhang. PCR products were ethanol precipitated then transcribed into RNA using T7 polymerase. The resulting RNA products were purified on an 8% 8M urea-PAGE gel via the crush-and-soak method. The short RNA strands required for the pRNA-triangle were synthesized via phosphoramidite chemistry.
Assay of pRNA activity
The in vitro system for assessing the ability of pRNA to participate in phi29 DNA packaging has previously been published (Guo et al. 1986). Briefly, the following components of the phi29 DNA-packaging motor were combined: prohead (containing connector), gp16, gp3-DNA, pRNA, and ATP-containing buffer. Following a 1 h incubation at room temperature, DNase I was added to remove unpackaged DNA. EDTA and proteinase K were subsequently added to deactivate the DNase I and break down the procapsid, releasing the packaged DNA. The resulting product was run on a 2% Syner gel in 1× TAE and the DNA was visualized with ethidium bromide staining. The bands were quantified with ImageJ (Ouyang et al. 2019). Graphs were made with the ggplot2 R package.
In vitro assembly of phi29 virion
Functional phages were assembled by combining prohead (containing connector), gp3-DNA, gp16, ATP-containing buffer, gp9, and gp11-14. The in vitro formed phi29 phages were added to Bacillus subtilis Su+44 culture and incubated at 37°C overnight. The number of plaques was counted and used to calculate the PFU (Guo et al. 1986).
Atomic force microscopy
The following loop-extended pRNA samples were transcribed (right-hand sequence/left-hand sequence, written 5′ to 3′): Ab′ (AGUGGAC/UGCCUGU), Bc′ (ACAGGCA/AGAACGC), Cd′ (GCGUUCU/CUAGCCU), De′ (AGGCUAG/UGGUGCU), Ef′ (AGCACCA/CACGUCU), and Fa′ (AGACGUG/GUCCACU). To assemble the pRNA-triangle hexamer, equimolar amounts of each component were combined in 1× TMS at 37°C for 1 h. The hexamers were purified on a 4% native PAGE gel in 1× TBM. AFM imaging was done with an APS mica surface with a MultiMode AFM NanoScope IV system (Veeco) in tapping mode.
Construction of connector–pRNA model
The dodecameric model of gp10 was created using AlphaFold Multimer (Evans et al. 2021; Jumper et al. 2021) on the COSMIC2 server (Cianfrocco et al. 2017). The phi29 RNA–connector complex model was built to estimate the potential structure, combining computational predictions with manual modeling to represent the interaction between the pRNA hexamer and the gp10 connector. The RNA component was based on the previously reported high-resolution crystal structure of the pRNA 3WJ (Zhang et al. 2013), which served as a template for modeling the RNA's conformation. The RNA conformation remained fixed throughout the modeling process, with no alterations to the crystal structure.
Traditional docking was not feasible for the entire complex due to its size. Instead, manual alignment in Coot (Emsley et al. 2010) positioned the pRNA to interact with the connector's narrow end. The adjustments aimed to establish a plausible interface based on known structural and biological data, while ensuring that the RNA's conformation remained entirely consistent with the original crystal structure.
Docking was performed with AutoDock (Morris et al. 1998; Huey et al. 2007) to explore possible conformations of the RKR residues as ligand to interact with the fixed RNA structure. The docking grid was set to cover the region around the RNA's known interaction sites and extended to accommodate the flexibility of the RKR residues. Lamarckian Genetic Algorithm (LGA) (Morris et al. 1998) was employed for the docking runs, with the population size set to 150 and the maximum number of energy evaluations to 2,500,000. Docking focused on identifying poses where the RKR residues could form plausible interactions with the RNA, such as hydrogen bonds and electrostatic contacts.
After docking, manual refinement was performed in Coot (Emsley et al. 2010) to adjust the RKR residues’ positions to make them reasonably connected to the rest of the gp10 residues, ensuring reasonable alignment with the RNA. The distances and orientations were fine-tuned based on known structural data and experimental observations.
DATA DEPOSITION
The new pRNA–connector complex can be found as a supplementary file (Supplemental File S1). Other data are available from the corresponding author(s) upon reasonable request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
P.G. is a consultant, grantee, and licenser of Oxford Nanopore Technologies, and is a cofounder of ExonanoRNA.
ACKNOWLEDGMENTS
The work was mainly supported by National Institutes of Health (NIH) grants R01GM141394 and R01EY031452. The content is solely the responsibility of the authors and does not necessarily represent the official views of NIH. Peixuan Guo's Sylvan G. Frank Endowed Chair position in Pharmaceutics and Drug Delivery is funded by the CM Chen Foundation. M.B. is supported by the OSU Center for RNA Biology Fellowship. The authors thank Yi Shu for working on the AFM data and Yudhistira Tesla for helping to prepare Figure 2. The authors also thank Drs. A. Lushnikov and A. Krasnoslobodtsev, as well as Lyudmila S. Shlyakhtenko and Yuri L. Lyubchenko at the University of Nebraska for assistance with AFM imaging.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080383.125.
- Received January 7, 2025.
- Accepted March 18, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Margaret Bohmer is the first author of this paper, “Constructing an active chimeric pRNA ring with a stoichiometry of six and identifying 12 domains of the pRNA ring binding to the 12-subunit channel of phi29 DNA-packaging motor.” Margaret is a PhD student in Peixuan Guo's laboratory at Ohio State University, College of Pharmacy, and Center for RNA Biology. The focus of her research is on the functional properties of noncoding RNA.
What are the major results described in your paper and how do they impact this branch of the field?
We established that the packaging RNA (pRNA) of the phi29 virus DNA-packaging motor exists as a hexamer. This knowledge deepens our understanding of how revolving biomotors translocate dsDNA.
What led you to study RNA or this aspect of RNA science?
I find it fascinating how RNA can serve not only as a carrier of genetic information, but also as a structural component in a biomotor. I believe that RNA is the most versatile material in nature, and there is much left for us to uncover.
If you were able to give one piece of advice to your younger self, what would that be?
While I love the research I do, it is very different from what I thought I wanted to do when I was younger. Therefore, my advice to my younger self would be to keep a more open mind about the different branches of research out there.
What are your subsequent near- or long-term career plans?
My first priority is to graduate with my PhD (projected graduation in spring 2027). After that, I want to embark on a career in academic research.








