
Modulation of diverse biological processes by CPSF, the master regulator of mRNA 3′ ends
- Corresponding author: jlm2{at}columbia.edu
Abstract
The cleavage and polyadenylation specificity factor (CPSF) complex plays a central role in the formation of mRNA 3′ ends, being responsible for the recognition of the poly(A) signal sequence, the endonucleolytic cleavage step, and recruitment of poly(A) polymerase. CPSF has been extensively studied for over three decades, and its functions and those of its individual subunits are becoming increasingly well-defined, with much current research focusing on the impact of these proteins on the normal functioning or disease/stress states of cells. In this review, we provide an overview of the general functions of CPSF and its subunits, followed by a discussion of how they exert their functions in a surprisingly diverse variety of biological processes and cellular conditions. These include transcription termination, small RNA processing, and R-loop prevention/resolution, as well as more generally cancer, differentiation/development, and infection/immunity.
Keywords
- cleavage and polyadenylation
- 3′ end processing
- cleavage and polyadenylation specificity factor
- transcription
- small RNA
- R-loop
- genome stability
- cancer
- differentiation
- development
- infection
- immunity
INTRODUCTION
RNA polymerase II (Pol II) is responsible for the transcription of not only all cellular mRNAs but a variety of noncoding RNAs as well. In all cases, the 3′ ends of these transcripts are not created by transcription termination but instead by an RNA processing reaction dubbed 3′ end formation. This reaction nearly always entails two steps, endonucleolytic cleavage and release of the nascent RNA transcript from Pol II, followed by de novo synthesis of a poly(A) tail using the upstream cleavage product as a substrate. These two reactions are coupled in vivo and are known as cleavage and polyadenylation (CPA).
The existence of a non-DNA-templated poly(A) tail at the 3′ ends of polysome-associated mRNAs was discovered over 50 years ago (Darnell et al. 1971; Edmonds et al. 1971; Lee et al. 1971). Subsequently, Proudfoot and Brownlee identified the conserved AAUAAA motif, known as the polyadenylation signal (PAS), 20–30 nt upstream of the poly(A) tail (Proudfoot 1976; Proudfoot and Brownlee 1976). This motif was found to be required for CPA when Fitzgerald and Shenk (1981) demonstrated that PAS deletion from a model RNA substrate, the simian virus 40 (SV40) late mRNA, prevented poly(A) tail addition. The physiological importance of the PAS was highlighted by the discoveries of two thalassemia-associated PAS point mutations, AATAAG and AACAAA, in the α 2-globin and the β-globin genes, respectively, that cause transcription readthrough and abnormal globin expression (Higgs et al. 1983; Orkin et al. 1985). With the importance of the PAS established, the core mammalian CPA machinery, composed of four multisubunit complexes, now known as cleavage and polyadenylation specificity factor (CPSF), cleavage stimulation factor (CstF), cleavage factors I and II (CFI and CFII) (Fig. 1), began to be identified and characterized, mainly by the Keller and the Manley labs in the late 1980s (Christofori and Keller 1988; Takagaki et al. 1988; Gilmartin and Nevins 1989; Takagaki et al. 1989). Among these protein complexes, CPSF was found to be the PAS-interacting factor that confers the sequence specificity of CPA (Bardwell et al. 1991; Keller et al. 1991; Murthy and Manley 1992), and also contains the essential endonuclease responsible for cleavage (Mandel et al. 2006). A single subunit poly(A) polymerase (PAP), as well as several ancillary protein factors, such as Symplekin and RBBP6, are also required for CPA, and Pol II itself, via the C-terminal domain (CTD) of its largest subunit, plays an important role (Shi et al. 2009). Numerous recent excellent reviews have covered the composition and function of the CPA machinery, which are well conserved throughout eukaryotes (Kumar et al. 2019; Sun et al. 2020; Boreikaitė and Passmore 2023; Rodríguez-Molina and Turtola 2023), but here we will focus on CPSF, highlighting its central and diverse roles in CPA and beyond.
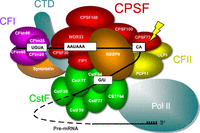
The mammalian CPA machinery consists of four multisubunit complexes and other auxiliary factors. Schematic showing the mammalian CPA machinery. It consists of CPSF, CstF, CFI and CFII. There are six CPSF subunits, CPSP160, CPSF100, CPSF73, CPSF30, FIP1, and WDR33. CPSF160, CPSF30, and WDR33 are involved in the recognition of the AAUAAA PAS, while CPSF100 and CPSF73 participate in endonucleolytic cleavage of the pre-mRNA. FIP1 facilitates the assembly of the polyadenylation and cleavage machinery. Two auxiliary factors, Symplekin and RBBP6, are also required for the cleavage reaction. Lightning bolt: endonucleolytic cleavage.
CPSF has been extensively studied for over three decades, and consists of six core subunits, namely, CPSF160, CPSF100, CPSF73, CPSF30, FIP1, and WDR33 (Fig. 1). In this review, we will first discuss the general functions of these CPSF subunits, followed by discussion of their roles in various biological processes/conditions. Symplekin, which tightly associates with CPSF and is often considered a seventh subunit (Boreikaitė and Passmore 2023), will also be discussed. In addition, RBBP6, which is not a stable component of CPSF, plays a direct role in activating endonucleolytic cleavage (Boreikaitė and Passmore 2023) and will, therefore, also be briefly discussed. On the other hand, CPSF5 (CFIm25 or NUDT21), CPSF6 (CFIm68), and CPSF7 (CFIm59), despite the misleading names, are in fact subunits of CFI , not CPSF, and therefore will not be included in this review.
GENERAL FUNCTIONS OF CPSF SUBUNITS
CPSF160, also known as CPSF1, is the largest CPSF subunit, with a molecular mass of ∼160 kDa. It has three β-propellers in a triangular arrangement, each containing seven WD repeats (Clerici et al. 2017; Sun et al. 2018). Its cDNA was cloned in 1995 and the protein was immediately found to coordinate interactions within the CPA machinery (Jenny and Keller 1995; Murthy and Manley 1995). Indeed, structural studies revealed that CPSF160 is a scaffold protein (Clerici et al. 2017, 2018; Sun et al. 2018), reflecting the function of its WD domains, which generally facilitate protein–protein interactions. Early RNA cross-linking/binding and immunodepletion assays also suggested that CPSF160 interacts directly with the PAS (Keller et al. 1991; Jenny and Keller 1995; Murthy and Manley 1995), and this view persisted for some time. However, later experiments demonstrated that the PAS-interacting sub-units are actually CPSF30 and WDR33 (Chan et al. 2014; Schönemann et al. 2014). CPSF160 was misidentified likely because of its similar molecular mass with WDR33 (∼146 kDa) and its function of positioning CPSF30 and WDR33 for proper PAS recognition (Clerici et al. 2018; Sun et al. 2018).
CPSF100, or CPSF2, was the first cloned CPSF subunit (Jenny et al. 1994). Its calculated molecular mass is ∼88 kDa, although it appears as a ∼100 kDa protein on SDS-PAGE (Jenny et al. 1994). Together with CPSF73, CPSF100 belongs to the metallo-β-lactamase (MβL) protein family (Aravind 1999; Callebaut et al. 2002). In addition to the MβL domain, CPSF100 and CPSF73 both contain aβ-CASP (MβL-associated CPSF Artemis SNM1/PSO2) domain, which confers RNA/DNA processing ability (Callebaut et al. 2002). These properties suggested that one or both of these proteins could possess endonuclease activity. However, CPSF100 is catalytically inactive, due to the absence of evolutionary conserved amino acids in its MβL domain critical for binding two catalytic zinc ions (Callebaut et al. 2002: Mandel et al. 2006). The exact function of CPSF100 is still not well-defined, but it has been shown to be required for cleavage of histone mRNAs, which do not contain a PAS (and thus are not polyadenylated) (Kolev et al. 2008; Sullivan et al. 2009; for a recent review on histone 3′ end formation, see Dominski and Tong 2021). In addition, a more recent structural study identified a CPSF100 segment, named polyadenylation specificity factor interaction motif (PIM), within its highly hydrophilic region near the C-terminus, that interacts with CPSF160 and WDR33 (Zhang et al. 2020a). CPSF100 also interacts with CPSF73, thus bridging the cleavage subunits with the PAS-recognizing subunits (Zhang et al. 2020a).
Given that CPSF100 is catalytically inactive, CPSF73 (or CPSF3), with a molecular mass of ∼73 kDa, became the most probable endonuclease candidate. Evidence supporting this began to emerge in the mid-2000s (Ryan et al. 2004; Dominski et al. 2005; Kolev and Steitz 2005), culminating in its structure determination in 2006 (Mandel et al. 2006), 10 years after its cloning (Jenny et al. 1996). According to the structure, CPSF73 contains two zinc ions at the active site in its MβL domain, and since CPSF100 lacks residues necessary to bind zinc ions, CPSF73 is indeed the 3′ processing endonuclease (Mandel et al. 2006). In addition to cleaving polyadenylated mRNA precursors, CPSF73 is also required for histone pre-mRNA cleavage, along with CPSF100 and Symplekin (Dominski et al. 2005; Kolev and Steitz 2005). More recently, the anti-inflammatory drug JTE-607 (Kakutani et al. 1999) was found to be an inhibitor of CPSF73 (Kakegawa et al. 2019; Ross et al. 2020), revealing CPSF73 as a potential drug target (for review, see Liu and Moore 2021; see also below). Purified CPSF73 displays only inefficient and nonspecific endonuclease activity (Mandel et al. 2006), consistent with the idea that the enzyme must be properly positioned at its site of action before its nuclease activity is unleashed.
An important goal has thus been to elucidate how CPSF73 becomes activated at the cleavage site in a physiological context. Purified CPSF73 activity (but not specificity) was shown to be enhanced by preincubation with calcium ions, and while there is no evidence supporting the involvement of calcium in pre-mRNA cleavage, it was suggested that another protein might fulfill this activating function in vivo (Mandel et al. 2006). In efforts to reconstitute a minimal CPA complex active in cleavage in vitro, Schmidt et al. (2022) and Boreikaite et al. (2022) identified RBBP6 as an indispensable factor for activating cleavage. RBBP6 was initially identified as a protein interacting with the tumor suppressors p53 and Rb (Sakai et al. 1995; Simons et al. 1997). RBBP6 is a large protein (∼202 kDa) containing an N-terminal ubiquitin-like (UBL) domain, a zinc knuckle, a RING finger, an RS domain, Rb- and p53-binding domains, and was postulated to function in UBL pathways and/or mRNA processing (Pugh et al. 2006). RBBP6 was indeed identified as a component of mammalian CPA machinery (Shi et al. 2009), and subsequently shown to function in CPA both in vitro and in vivo (Di Giammartino et al. 2014). RBBP6 activates cleavage by interacting with CPSF73 via its UBL domain, perhaps leading to a conformational change that opens the catalytic MβL domain (Boreikaite et al. 2022; Schmidt et al. 2022). Interestingly, a truncated RBBP6 isoform (iso3) produced by intronic polyadenylation (IPA) and containing only the UBL domain was found to compete with the canonical protein for binding with the CPA machinery, thereby inhibiting 3′ end formation (Di Giammartino et al. 2014).
CPSF30 (CPSF4), the smallest subunit (∼30 kDa), was cloned in 1997 (Barabino et al. 1997). CPSF30 contains five zinc fingers and a putative zinc knuckle, and was initially shown to bind to poly(U) RNA (Barabino et al. 1997). In 2014, however, it was demonstrated to bind AAUAAA (Chan et al. 2014; Schönemann et al. 2014). According to its structure, the second zinc finger recognizes the first two A's in AAUAAA, while the third zinc finger recognizes the two A's immediately downstream from the U (Clerici et al. 2018; Sun et al. 2018). However, CPSF30 by itself binds the PAS poorly, and requires both CPSF160 and WDR33 for proper PAS recognition (Sun et al. 2018).
The last cloned mammalian CPSF subunit was FIP1, or FIP1L1, which was identified based on sequence similarity with the yeast polyadenylation factor Fip1p in 2004 (Kaufmann et al. 2004). FIP1 was found to recognize U-rich upstream sequence elements (upstream of the PAS) (Kaufmann et al. 2004), which had been known to enhance polyadenylation efficiency (Gilmartin et al. 1995; Graveley et al. 1996; Brackenridge and Proudfoot 2000). Consistent with this, FIP1 can interact with and stimulate the activity of PAP (Kaufmann et al. 2004). Having a molecular mass of ∼66 kDa, FIP1 was recently found to be intrinsically disordered, and to function as a scaffold facilitating the assembly of the CPA machinery (Clerici et al. 2017; Hamilton and Tong 2020; Kumar et al. 2021; Muckenfuss et al 2022). Interestingly, 62 FIP1 isoforms appear to be created by alternative splicing (AS), but their significance is unknown (O'Leary et al. 2016).
The last core CPSF subunit to be described was WDR33, which was identified in 2009 (Shi et al. 2009). Its cloning, however, was reported much earlier, in 2001, when it was named WDC146, reflecting the presence of WD repeats, a collagen-like domain, and its molecular mass of ∼146 kDa (Ito et al. 2001). A rat noncanonical isoform (∼38 kDa for the human homolog), named NET14 (nuclear envelope transmembrane 14), was separately identified as an inner nuclear membrane protein (Schirmer et al. 2003; see below for further discussion). Canonical WDR33 is a WD protein with seven repeats, which together form a β-propeller near the N-terminus. Phe153 from its first WD repeat was found to directly contact the PAS (Chan et al. 2014; Schönemann et al. 2014; Clerici et al. 2018; Sun et al. 2018). Similar to CPSF30, WDR33 also binds RNA weakly by itself and requires CPSF160 and CPSF30 for stable RNA interaction (Sun et al. 2018). WDR33's β-propeller, while only occupying the first half of the amino acid sequence, is sufficient to reconstitute the PAS recognition function (Clerici et al. 2018; Sun et al. 2018). The functional significance of the second half, which includes the collagen-like domain and appears to be specific to vertebrates, is still unknown.
The six proteins discussed above are core subunits of CPSF. As mentioned above, a seventh protein, Symplekin, strongly associates with CPSF subunits (Takagaki and Manley 2000; Shi et al. 2009; Chan et al. 2014; Zhang et al. 2020a). Symplekin was first identified as a putative tight junction protein, but localization to the nucleoplasm was also detected (Keon et al. 1996). Symplekin was later uncovered as a ∼135 kDa nuclear protein that interacts with both CstF and CPSF (Takagaki and Manley 2000). Symplekin contains a HEAT domain with seven pairs of antiparallel α-helices near its N-terminal region (Xiang et al. 2010), consistent with it being a scaffold protein during assembly of the CPA complex, as originally suggested (Takagaki and Manley 2000). The protein interacts with CPSF100 and CPSF73 in histone pre-mRNA cleavage (Kolev and Steitz 2005; Sullivan et al. 2009; see also below). Symplekin also functions to modulate transcription-coupled polyadenylation by recruiting and activating the CTD phosphatase SSU72, which in turn may facilitate the association of PAP with the transcription/processing complex (Xiang et al. 2010). Symplekin was also recently found, by proximity labeling, to interact with a newly described Pol II termination complex, called the Restrictor, that plays a role in cleavage-independent termination of many naturally unstable long noncoding RNAs (lncRNAs) (Russo et al. 2023). Symplekin thus appears to have distinct roles in 3′ processing/termination of different types of Pol II transcripts. Finally, Symplekin also functions, likely as part of CPSF, in cytoplasmic polyadenylation, which results in the extension of poly(A) tails in the cytoplasm (Barnard et al. 2004). Cytoplasmic polyadenylation was initially described in oocytes but also has functions in the brain, for example, in synaptic plasticity (for review, see Huang et al. 2023).
Subsequent studies revealed that CPSF exists as two separable subcomplexes, or modules, PSF and CF. PSF consists of WDR33, FIP1, CPSF160, and CPSF30, while CF contains CPSF73, CPSF100, and Symplekin (Chan et al. 2014; Schönemann et al. 2014; Clerici et al. 2017; Zhang et al. 2020a). These two modules can be reconstituted in vitro, with PSF responsible for AAUAAA recognition and recruitment of PAP, and CF for the endonucleolytic cleavage. Although the existence of the CF and PSF modules facilitates the elucidation of functional and structural properties of the CPSF subunits, they likely exist, at least in most instances, as a single complex, CPSF, in vivo. Table 1 summarizes the features and functions of all the CPSF subunits, including RBBP6 and Symplekin.
Summary of the protein features and functions of CPSF subunits
OVERVIEW OF CPSF FUNCTIONS IN MULTIPLE BIOLOGICAL PROCESSES
In addition to the general, central role of CPSF in mRNA 3′ end formation, the complex affects multiple cellular processes beyond this core function. As will be discussed below, its function in many of these events involves the phenomenon of alternative polyadenylation (APA). APA, which occurs with transcripts from ∼70% of mammalian genes (Derti et al. 2012; Hoque et al. 2012), involves the usage of distinct CPA sites within mRNA precursors, giving rise to mRNAs with different 3′-untranslated region (UTR) lengths or, in ∼40% of genes, altered coding potential (for reviews, see Tian and Manley 2013, 2017). The process is subject to regulation, which can occur by multiple mechanisms. Among these, changes in the cellular concentration of individual CPA factors can drive the selection of distinct CPA sites, reflecting a cis-competition between these sites for CPA factors. This mechanism was originally described in the IgM pre-mRNA, where changes in levels of CstF were found to drive the production of mRNAs encoding membrane-bound vs secreted IgM (Takagaki et al. 1996; Takagaki and Manley 1998). Subsequently, it has been found that changes in many core CPA factors, including CPSF, can bring about changes in APA (e.g., Li et al. 2015). Through APA regulation, and other noncanonical functions, CPSF subunits can modulate a diverse collection of biological processes and conditions, which are discussed in the following sections.
TRANSCRIPTION TERMINATION
CPSF, and specifically the cleavage module, is directly linked with transcription termination by Pol II. Perhaps reflecting the immense size of some eukaryotic protein-coding genes, terminating Pol II is a complex, multistep process (see Boreikaitė and Passmore 2023; Rodríguez-Molina and Turtola 2023 for recent reviews). An initial step involves change of the posttranslational modification status of the CTD, which in vertebrates consists of 52 repeats of the consensus sequence YSPTSPS. The phosphatase SSU72 is known to facilitate Pol II termination by dephosphorylating the CTD (Krishnamurthy et al. 2004; St-Pierre et al. 2005; Zhang et al. 2012), and such phosphatase activity is stimulated by direct interaction with Symplekin (Fig. 2A; Xiang et al. 2010). Interestingly, SSU72 can also interact with TFIIB, a general transcription factor involved in transcription initiation, and Symplekin, having a stronger affinity for SSU72, disrupts this interaction (Bratkowski et al. 2018), suggesting that Symplekin's transcription regulation function extends beyond termination.
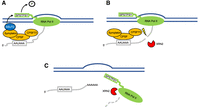
Symplekin and CPSF73 are directly involved in Pol II termination. (A) The first step in Pol II termination involves dephosphorylation of the CTD (YSPTSPS). This is achieved by the phosphatase SSU72, whose activation requires Symplekin, which is associated with CPSF. P in circle: phosphate. (B) The second step involves the endonucleolytic cleavage by the endonuclease CPSF73, which exposes the unprotected 5′ phosphate of the downstream cleavage product, providing a substrate for the 5′ to 3′ exonuclease XRN2. Lightning bolt: endonucleolytic cleavage. (C) Finally, XRN2 degrades the downstream cleavage product (dashed line) and facilitates the displacement of Pol II from the DNA template. The upstream cleavage product is polyadenylated.
The next step in Pol II termination is the cleavage reaction itself. Cleavage of nascent mRNAs by CPSF73 releases the upstream product for polyadenylation by PAP, exposing the unprotected 5′ phosphate of the downstream product (Fig. 2B). This provides a substrate for the 5′ to 3′ exoribonuclease XRN2, which rapidly degrades the remaining RNA, resulting in the eventual “collision” between XRN2 and the continuously transcribing Pol II, thereby, by mechanisms not fully understood, causing displacement of Pol II from the DNA template (Fig. 2C; Eaton et al. 2020). The importance of CPSF73 in this process is highlighted by the observations that its depletion, which eliminates access of XRN2 to the nascent transcript, causes global transcriptional readthrough, i.e., a termination defect (Nojima et al. 2015; Eaton et al. 2018, 2020). Consistent with this, the loss-of-function mutation of CPSF100 in Arabidopsis thaliana also causes transcription readthrough (Lin et al. 2017). For histone transcription termination, however, XRN2 does not appear to play a role (Eaton et al. 2018), and degradation of the downstream cleavage product is achieved by CPSF73 itself, providing evidence that CPSF73 also possesses 5′ to 3′ exoribonuclease activity (Yang et al. 2009, 2020). Whether CPSF73 can also exert this activity in the context of polyadenylated mRNAs is unknown.
In addition to ensuring proper transcription termination at the end of genes, CPSF is also involved in premature/early termination. For example, Cugusi et al. (2022) reported that heat shock increases Pol II elongation rates, but at the same time decreases processivity, reflecting a significant increase in premature transcription termination at cryptic IPA sites. This is in part mediated by the recognition of upstream PASs by CPSF, and CPSF73 depletion indeed suppressed the IPA. This stress-induced early termination may function to promote cell survival by increasing Pol II recycling and by reducing chaperone loads or accumulation of misfolded proteins (Cugusi et al. 2022). Yamazaki et al. (2021) also reported premature transcription termination during oxidative stress, due to dephosphorylation of Ser2 on the CTD. While the underlying mechanism is unclear, an interesting possibility is that CPSF also plays a role, as Lin et al. (2017) reported that in A. thaliana CPSF100 interacts with the CTD to ensure proper Ser2 dephosphorylation during transcription termination.
The function of CPSF in termination control can be dependent on other proteins. For example, FUS, an RNA-binding protein associated with neurodegenerative diseases such as amyotrophic lateral sclerosis (Piol et al. 2023), forms clusters on numerous nascent Pol II transcripts. There it pauses Pol II and recruits CPSF, via interaction with CPSF160, to an upstream PAS if present, thereby facilitating early polyadenylation and transcription termination. Through this CPSF-dependent regulation, FUS controls the expression of genes involved in synaptic activities in neuronal cells (Masuda et al. 2015). Also linking CPSF with transcription, CPSF was shown to be recruited to promoters by interaction with the general transcription factor TFIID, and then transferred to Pol II, perhaps via association with the CTD (McCracken et al. 1997), during initiation (Dantonel et al. 1997). Additionally, FUS also associates with the CTD, preventing hyperphosphorylation and facilitating elongation (Schwartz et al. 2012). How and if all these interactions are related is unclear.
CPSF FUNCTION IN SMALL RNA PROCESSING
CPSF73 function is not limited to mRNA cleavage, and the enzyme can, for example, play a role in the processing of certain microRNAs (miRNAs). miRNAs are processed from larger primary transcripts, pri-miRNAs, transcribed by Pol II and processed at their 3′ end by the canonical CPA machinery (for reviews, see Shang et al. 2023). Pri-mRNAs can contain a single miRNA precursor (pre-miRNA) or a cluster of multiple pre-miRNAs, and the first step in processing occurs in the nucleus and typically involves cleavage of the pri-miRNA by the Microprocessor complex. However, in the case of pri-miR-17–92, which contains a cluster of six pre-miRNAs, transcript cleavage occurs just 5′ to the first pre-miRNA, generating an intermediate species termed a progenitor (pro)-miRNA, and cleavage is carried out by CPSF73 (Fig. 3A; Du et al. 2015). This pro-miRNA is subsequently processed by the Microprocessor and finally by DICER to produce six mature miRNAs, five of which are dependent on CPSF73 cleavage (Fig. 3B; Du et al. 2015). Interestingly, processing of pri-miR-17–92 by CPSF73 was shown to be independent of CPSF100 and FIP1 but to require the little-studied splicing factor ISY1 and components of the U2 small nuclear ribonucleoprotein (snRNP). It is unclear if ISY1 or U2 snRNP can activate the endonuclease activity of CPSF73, or if RBBP6 is also part of this complex.
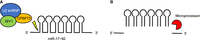
CPSF73 participates in the processing of miR-17–92. (A) The primary miR-17–92 transcript contains six immature miRNAs. Their maturation first involves cleavage by CPSF73, which is in complex with U2 snRNP and ISY1, at the 5′ end of the first miRNA precursor. This generates pro-miR-17–92. Lightning bolt: endonucleolytic cleavage. (B) Pro-miR-17–92 is then further processed by the Microprocessor complex to generate six pre-miRNAs.
Although it is unknown how widespread CPSF73's role in miRNA processing is, the above findings indicate that CPSF73 can function in protein complexes other than CPSF. Indeed, while most human small nuclear RNAs (snRNAs) are processed by the Integrator complex (Baillat et al. 2005), A. thaliana snRNAs are processed by the DSP1 (defective in snRNA processing) complex, in which CPSF73 is an indispensable component (Liu et al 2016; Wang et al. 2020a). This likely reflects the similarity between CPSF73 and INTS11, the endonuclease in the Integrator complex (Fianu et al. 2024).
Endogenous small interfering RNAs (esiRNAs) are derived from double-stranded RNA, such as from retrotransposons to suppress transposable element mobility or from hairpin loci to downregulate mRNA levels (Kawamura et al. 2008). In Drosophila melanogaster somatic cells, retrotransposon-derived esiRNAs are processed in the nucleus by CPSF73 in an apparently CPSF-dependent manner (Harrington et al. 2017). Proper processing requires an interaction between the cleavage enzyme Dicer2 and Symplekin, which recruits CPSF100 and CPSF73, and depletion of Symplekin or CPSF73 affects esiRNA abundance and causes nuclear retention of esiRNA precursors. While the authors suggest processing is catalyzed by the CF module of CPSF, it is unclear whether intact CPSF is in fact responsible (Harrington et al. 2017). Additionally, although esi RNAs are widespread in nature (Okamura and Lai 2008), it is also unclear how universal the role of CPSF is in their generation.
These small RNA processing functions of CPSF subunits suggest a possible regulatory loop involving 3′-UTR APA. Changes in 3′-UTR length brought about by APA dictate the ability of si/miRNAs to target the affected mRNAs. It would thus be interesting if APA targets sensitive to intracellular levels of CPSF subunits contain binding sites for small RNAs that require CPSF for their maturation.
R-LOOP PREVENTION/RESOLUTION AND CPSF
While transcription is of course an essential cellular process, it can expose the genome to potential dangers. R loops, which are three-stranded nucleic acid structures formed by the hybridization of the nascent RNA to the template DNA strand, displacing the nontemplated DNA strand, constitute one such danger. Persistent R loops not only render the DNA unprotected, e.g., from nucleases, but can also block replication fork progression, leading to DNA mutations or even chromosomal fragmentation and rearrangements (Li and Manley 2005; Gan et al. 2011; for reviews, see Sollier and Cimprich 2015; Gaillard and Aguilera 2016; Petermann et al. 2022). Accumulation of such damage can contribute to disease, including neurodegeneration and cancer (for review, see Crossley et al. 2019). In Saccharomyces cerevisiae, mutations affecting the CPA machinery were found to increase chromosomal instability in an R-loop-dependent manner, suggesting that 3′ end processing factors contribute to R-loop resolution/prevention (Stirling et al. 2012). Extending this finding to humans, Stirling et al. (2012) further reported that knockdown (KD) of FIP1 resulted in increased DNA damage and chromosomal breakage, pointing to the importance of CPSF in maintaining genome integrity. This likely reflects the propensity of nascent transcripts downstream from 3′ CPA sites, which would tend to accumulate in the absence of efficient 3′ cleavage and subsequent termination, to form R loops (Skourti-Stathaki et al. 2011).
More recently, a targeted siRNA-mediated screen of potential cancer driver genes identified WDR33 as a key factor in conferring resistance to replication stress-induced DNA damage and genomic stability. WDR33 KD rendered cells more susceptible to replication stress, resulting in reduced replication fork speed and excessive origin firing, which was linked to increased R-loop formation (Teloni et al. 2019). Indeed, inhibition of origin firing prevented R-loop accumulation in WDR33-depleted cells, suggesting R loops can arise as the result of replication fork-transcription complex collisions, which are increased by the elevated transcription arising from defects in 3′ end formation, as noted above. Although genes encoding other CPSF subunits were not represented in the original group of cancer-relevant targeted genes, KD of CPSF160, CPSF30, and FIP1 had the same effects as WDR33 depletion, supporting the conclusion that it is indeed CPSF function in CPA that is critical for preventing replication stress-induced genomic instability (Teloni et al. 2019).
While unscheduled R-loop formation is typically detrimental, the formation of R loops can also in some instances be an essential aspect of gene expression, with a classic example being immunoglobulin class switch recombination (for review, see Li and Manley 2006). A more recent example, which also involves links to CPSF-mediated 3′ end formation, has been described in plants. Specifically, in A. thaliana, cold temperature induces transcription of an antisense RNA called COOLAIR from the 3′ end of the FLC gene, which encodes a protein that suppresses flowering. COOLAIR hybridizes to the FLC locus during transcription, forming an R loop. This R loop is recognized by an RNA-binding protein called FCA, which interacts with FY, the A. thaliana homolog of WDR33 (Fig. 4A). FY was initially discovered genetically as a regulator of flowering (Simpson et al. 2003), and mutations in FY affect over 50% of APA events in the plant (Yu et al. 2019). Importantly, the FCA–FY interaction recruits CPSF and other CPA factors, resulting in COOLAIR CPA at a weaker PAS not used in the absence of FLC. This in turn leads to COOLAIR transcription termination and resolution of the R loop, which ultimately results in epigenetic silencing of the gene, thereby relieving flowering suppression (Fig. 4B; Baxter et al. 2021; Xu et al. 2021). FCA/FY-mediated R-loop resolution also occurs at other loci, indicating that it is a general mechanism in plants, and can function to prevent R-loop-induced DNA damage (Xu et al. 2021). Whether a similar mechanism exists in animals is unclear, although it is known that the formation of R loops can frequently promote antisense transcription in mammals (Tan-Wong et al. 2019).

Arabidopsis thaliana FY (WDR33) regulates FLC gene expression by resolving the COOLAIR R loop. (A) In A. thaliana, low temperature induces antisense transcription at the FLC locus, producing the antisense transcript COOLAIR. COOLAIR hybridizes to the template DNA strand, forming an R loop, which is recognized by the FCA/FY (WDR33) complex. (B) FY (WDR33), as part of CPSF, activates polyadenylation of COOLAIR at a weak PAS, and the R loop is then resolved. This ultimately leads to epigenetic silencing of the FLC gene.
CPSF AND CANCER
Changes in APA, most notably widespread 3′-UTR shortening, are well established to occur in a wide variety of cancers (Mayr and Bartel 2009; Xia et al. 2014; Xiang et al. 2018). These changes frequently reflect alterations in the concentrations of multiple core CPA factors, with factors such as the CstF subunit CstF64 suggested to be “master regulators” (Xia et al. 2014). Consistent with both their central role in CPA and their functions in maintaining genome stability/integrity, multiple CPSF subunits have been implicated in cancer development and progression. In the following paragraphs, we detail a variety of examples of how this can occur.
Perhaps the best characterized and direct example of a CPSF subunit displaying oncogenic activity results from the fusion of the 5′ end of FIP1 with the 3′ ends of the platelet-derived growth factor receptor α (PDGFRA) or retinoic acid receptor α (RARA) genes, which occurs in some leukemia patients (Cools et al. 2003; Buijs and Bruin 2007). The resultant FIP1 fusion proteins can contribute to tumori genesis in two ways. First, these fusion events can be considered gain-of-function mutations. FIP1-PDGFRA is a constitutively active kinase that activates a variety of signaling molecules that lead to cell proliferation and differentiation. Among its targets, STAT5 and AKT are activated dependent on the N-terminal portion of FIP1 (Buitenhuis et al. 2007). In addition, FIP1 sequences are also responsible for the nuclear localization of FIP1-PDGFRA (Iwasaki et al. 2014). For FIP1-RARA, the fusion protein was found to suppress transcription in response to retinoic acid, but this function appears to be independent of FIP1. Homodimerization, on the other hand, requires the N-terminal portion of FIP1 (Kondo et al. 2008; Iwasaki et al. 2014), possibly reflecting FIP1's function as a scaffold protein. How these contribute to leukemogenesis is still poorly understood.
The second way that FIP1 fusion proteins may contribute to cancer is via altered APA. Stirling et al. (2012) reported that removal of the FIP1 C-terminus, as commonly occurs in FIP1-PDGFRA, eliminates the FIP1 function, resulting in FIP1 loss-of-heterozygosity. This, as alluded to above, could lead to enhanced R-loop formation and thus increased DNA mutations and chromosomal instability, perhaps contributing to cancer development. Even though FIP1-PDGFRA and FIP1-RARA likely lack canonical FIP1 function, it is also possible that the fusion proteins themselves could influence APA. It would therefore be interesting to study if and how these proteins alter APA, and whether such changes might contribute to tumorigenesis. Given that alterations in FIP1 levels have well-documented roles in APA regulation (Lackford et al. 2014; Li et al. 2015; Huang et al. 2017; Schwich et al. 2021), this seems an interesting possibility. Notably, two of these studies reveal that FIP1 activity can be modulated in ways other than by changes in expression. Huang et al. (2017) found that a small nucleolar RNA can bind and sequester FIP1, thereby altering APA, while Schwich et al. (2021) showed that an SR protein splicing factor, SRSF7, can bind near certain PASs and directly recruit FIP1.
In contrast to FIP1, which appears not to be overexpressed in cancer, CPSF160 is highly expressed in several cancer types (Mayr and Bartel 2009; Kiehl et al. 2017; Xiong et al. 2022). High CPSF160 mRNA expression is linked to aberrant APA (Wang et al. 2020b; Chen et al. 2021a; Caggiano et al. 2022). Wang et al. (2020b) reported that triple-negative breast cancer (TNBC) can be categorized into four distinct subtypes based on different APA profiles. These aberrant profiles can be reversed by CPSF160 depletion, suggesting that CPSF160 likely functions with different proteins to induce different APA patterns. SQSTM1, or p62, is one of CPSF160's targets in breast cancer. Elevated CPSF160 levels result in the shortening of its mRNA's 3′ UTR, removing the binding site for miRNA 124-3p (Guo et al. 2022). This causes an accumulation of p62 proteins, which can contribute to tumorigenesis (Mathew et al. 2009).
Elevated CPSF160 expression also plays an important role in castration-resistant (CR) prostate cancer (CRPC), an advanced form of the disease (Karantanos et al. 2013). Increased CPSF160 levels were shown to promote the usage of a cryptic PAS within intron 3 of the androgen receptor (AR) pre-mRNA, producing the AR-V7 isoform (Fig. 5A; Van Etten et al. 2017). The AR plays a central role in prostate cancer progression, and AR-V7 is a constitutively active isoform that confers CR in prostate cancer cells. Notably, in CRPC the elevated levels of CPSF160 arise not from changes in CPSF160 mRNA, but by protein stabilization. Specifically, the expression of SIAH1, an E3 ubiquitin ligase that interacts with and targets CPSF160 for degradation, is suppressed in CRPC, resulting in the accumulation of excess CPSF160 (Xia et al. 2022). CPSF160 can also contribute to CRPC via global APA changes (Caggiano et al. 2022).
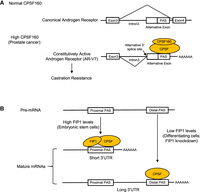
CPSF160 and FIP1 control APA in cancer and differentiation, respectively. (A) Intron 3 of the AR pre-mRNA contains an alternative exon with a cryptic PAS. When CPSF160 is not overexpressed, this intron is removed, producing the canonical transcript (top panel). However, in prostate cancer, CPSF160 is highly expressed. It activates the cryptic PAS, leading to the inclusion of the alterative exon in the mature transcript (bottom panel). This transcript encodes a constitutively active AR isoform termed AR-V7, which confers castration resistance. (B) Many pre-mRNAs contain a proximal and a distal PASs (top panel). In embryonic stem cells, expression levels of FIP1 are high, causing the proximal PAS to be used. Thus, embryonic stem cells tend to have mature mRNAs with short 3′ UTRs (middle panel). In differentiating cells or when FIP1 is knocked down, low FIP1 levels cause CPSF to use the distal PAS, leading to widespread 3′-UTR lengthening (bottom panel).
Similar to CPSF160, CPSF73 expression is also dysregulated in cancer at both mRNA and protein levels. In bladder cancer, CPSF73 mRNA is highly expressed, and such expression is positively correlated with the expression of CD276, which is implicated in immune evasion (Xiong et al. 2022). In breast cancer, the E3 ubiquitin ligase UBE3D stabilizes CPSF73 by preventing its degradation by the proteasome (Liu et al. 2022). KD of UBE3D reduced CPSF73 levels, leading to widespread 3′ cleavage defects, and decreased cell migration, invasion, and cancer stemness in TNBC cells. These phenotypes could be partially recapitulated by CPSF73 KD and partially rescued by CPSF73 overexpression (Liu et al. 2022). Specific CPSF73 targets that contribute to cancer development have also been identified and studied. The proto-oncogene c-FOS is frequently overexpressed in cancers. KD of cyclin-dependent kinase 12 (CDK12) reduced both Pol II Ser2 phosphorylation as well as CPSF73 levels on c-FOS pre-mRNAs, thereby suppressing its expression (Eifler et al. 2015). KD of the exon junction complex (EJC) subunit eIF4A3 also reduced CPSF73 levels on c-FOS transcripts, perhaps reflecting a role for the EJC in recruiting CDK12 to elongating Pol II (Eifler et al. 2015). Furthermore, the translation/nuclear export factor eIF4E was found unexpectedly to enhance 3′ cleavage of pre-mRNAs encoding all seven CPSF subunits, and likely other transcripts, reflecting interaction with specific sequences in their 3′ UTRs as well as with CPSF73, and CPSF160, perhaps contributing to eIF4E oncogenic potential (Davis et al. 2019).
Several recent studies have suggested that CPSF73 may constitute an important target for cancer therapeutics. As mentioned above, the small molecule JTE-607 is a specific CPSF73 inhibitor, binding to its active site and inhibiting cleavage activity in vitro (Ross et al. 2020). This results in widespread transcriptional readthrough accompanied by elevated R-loop formation in vivo, and reduced expression of genes required for proliferation and viability of certain cancer cells, notably acute myeloid leukemia and Ewing's sarcoma. Similarly, JTE-607 blocks pancreatic cancer cell proliferation and growth, while having no effect on nontransformed pancreatic cells (Alahmari et al. 2024). This may reflect the transcriptional readthrough of replication-dependent histone genes, which reduces core histone expression and arrests cells in the S-phase (Alahmari et al. 2024). In keeping with the effects of CPA factor levels on APA (see above), JTE-607 was found to affect APA in cancer cells, consistent with the higher levels of such factors, including CPSF subunits, in these cells (Cui et al. 2023). Finally, the above studies indicated that JTE-607 has widespread but not universal effects on 3′ cleavage, and Liu et al. (2023) showed recently that this reflects specific sequences flanking the cleavage site, which results in different affinities of the CPA machinery for the PAS and thus different, and predictable, sensitivity to JTE-607.
Consistent with the roles of CPSF73 in cancer, RBBP6, which as described above is required for CPSF73 cleavage activity, is also implicated in cancer by regulating CPA. For example, RBBP6 iso3 was found to be downregulated in multiple tumor tissues, such as those from the breast, liver, and cervix (Mbita et al. 2012). As described above, iso3, which contains only the UBL domain and is produced by IPA, is an inhibitor of CPA. As such, lower iso3 levels in these tumors could contribute to more efficient CPA, which could lead to increased expression of mRNAs with AU-rich 3′ UTRs, such as the proto-oncogenes c-Jun and c-Fos (Di Giammartino et al. 2014). Additionally, a CRISPR knockout (KO) screen of ubiquitin E3 ligases reported that RBBP6 is required for glioblastoma stem cell proliferation and tumor initiation. RBBP6, via its RING domain, was found to catalyze K63-linked polyubiquitination of CPSF73, thereby stabilizing it and altering APA (Lin et al. 2024). Among the APA targets are endogenous RNAs that compete with MYC for binding the miRNA miR-590-3p. KD of RBBP6 increased the availability of free miR-590-3p by shortening the 3′ UTRs of such completing endogenous RNAs (ceRNAs), and miR-590-3p thus increasingly targeted MYC, reducing its expression. KO of RBBP6 or CPSF73 in glioblastoma cells or JTE-607 treatment in a mouse model reduced tumor growth and prolonged survival (Lin et al. 2024). Lastly, mutations of RBBP6 have also been reported to associate with familial myeloproliferative neoplasms (MPN), which are blood cancers characterized by the overproduction of blood cells (Harutyunyan et al. 2016). It is unclear whether RBBP6's role in MPN involves CPA, however, as mutations lie in or around the p53-binding domain, which is dispensable for RBBP6 function in CPA.
CPSF30 has also been found to be overexpressed in a wide variety of tumors. While this leads to a variety of consequences, the underlying mechanisms remain for the most part unclear. For example, CPSF30 is highly expressed in oral squamous cell carcinoma, and this correlates with increased proliferation and invasion via activation of the PI3K-AKT pathway (Zhang et al. 2021). In lung adenocarcinoma, in addition to PI3K and AKT, elevated CPSF30 levels are associated with enhanced phosphorylation of the ERK1/2 and JNK proteins and with activation of the NF-κB pathway (Chen et al. 2013; Yi et al. 2016). In hepatocellular carcinoma (HCC), high CPSF30 expression has been suggested to play an unusual role, contributing to a reduction in circular RNAs (circRNAs) (Wang et al. 2021). Many circRNAs, which are typically created by so-called “back-splicing” and can act as “sponges” to sequester miRNAs, also possess one or more PASs. Elevated CPSF30 levels appear to facilitate the linearization of this class of circRNAs by CPA, which would then make the RNAs susceptible to degradation by exonucleases. Reduction of circRNA levels was suggested to lead in turn to degradation of the bound miRNAs, thereby elevating expression of target genes, some of which contribute to tumorigenesis (Wang et al. 2021).
CPSF30 has also been reported to bind to promoters and suggested to modulate transcription. In both lung and colon cancers, CPSF30 appears to bind the promoter region of the TERT gene, as judged by DNA binding and ChIP assays, and facilitates its expression by interacting with the CREB-binding protein or cooperating with NF-κB (Chen et al. 2014; Tang et al. 2016a; Yang et al. 2019). Overexpression of COX-2, a gene implicated in several cancers, also requires CPSF30 binding at its promoter in an NF-κB-dependent manner (Yi et al. 2016). These findings likely reflect the recruitment of CPSF to promoters by interaction with TFIID (Dantonel et al. 1997; see above). The observed increases in expression of target genes may arise from enhanced CPA resulting from more efficient recruitment of CPSF, although it is conceivable that CPSF30 may increase transcription, as suggested by these authors.
The above studies dealing with CPSF30 tend to attribute the various effects observed to elevated levels of only CPSF30. However, it is very unlikely that the protein acts alone, but instead functions as part of CPSF. This though raises an interesting point: Is a single subunit, e.g., CPSF30, limiting for assembly of active CPSF complexes, such that increasing expression of this one subunit alone is sufficient to increase levels of active CPSF? Or do levels of multiple CPSF subunits change in concert? A precedent for the first possibility comes from the initial discovery that altered levels of a CPA factor could affect APA, where it was shown that increased expression of one subunit of the heterotrimeric CstF complex, CstF64, was sufficient to modulate APA of IgM transcripts (Takagaki et al. 1996; see above). On the other hand, Wang et al. (2021) found, by analysis of The Cancer Genome Atlas data, that expression of multiple CPSF subunits was significantly upregulated in HCC.
In this section, we have discussed how CPSF subunits, through a variety of distinct mechanisms, contribute to cancer development and progression. Given the overexpression of many CPSF subunits in cancers, many of the studies mentioned above performed siRNA KD and reported decreased proliferation and invasiveness, and increased apoptosis. We would like to point out that one must be cautious when interpreting results from such experiments, which may involve indirect effects. For example, as discussed earlier in this review, CPSF is required for proper transcription termination. When CPSF is depleted, transcription termination defects will likely occur, and this will induce stress conditions (e.g., DNA damage) that can affect the overall health of normal as well as cancer cells. In addition, the depletion of CPSF subunits likely also affects APA in multiple ways that alter cellular homeostasis. Thus it is important that where necessary, appropriate controls are included to ensure that observed phenotypes are indeed cancer-specific.
DIFFERENTIATION AND DEVELOPMENT
Studies discussed in the above section suggest that the short 3′ UTRs in many cancers are at least in part due to the deregulation of CPSF. 3′ UTRs tend to be lengthened during cellular differentiation and development (Ji et al. 2009; Shepard et al. 2011), and CPSF might also play a role in these processes. Indeed, expression of CPSF100, CPSF73, FIP1, and WDR33 is decreased when embryonic stem cells are cultured under differentiation conditions (Lackford et al. 2014). Among these subunits, KD of FIP1 induces mouse embryonic stem cell differentiation by lengthening the 3′ UTRs of many genes involved in self-renewal and maintenance of pluripotency (Fig. 5B). Consistent with this, FIP1 was also found to be required for the generation of induced pluripotent stem cells, as its depletion strongly reduced reprogramming efficiency (Lackford et al. 2014). Downregulation of CPSF has also been observed during adult tissue-specific stem cell differentiation. For example, expression of all CPSF subunits was found to be decreased during keratinocyte differentiation (Chen et al. 2021b). This results in decreased IPA of transcripts from many genes, including the differentiation activator GRHL3. In this case, IPA serves as a negative regulator, and decreased IPA leads to GRHL3 activation. Supporting the significance of CPSF-mediated regulation, CPSF160 KD was shown to be sufficient to induce premature differentiation (Chen et al. 2021b).
Finally, CPSF can also influence organismal development. A FIP1 A. thaliana mutant was reported to exhibit reduced primary root growth and significantly fewer lateral roots. FIP1 loss causes these root development phenotypes by altered APA, including interestingly in coding sequences and 5′ UTRs (Téllez-Robledo et al. 2019). In Caenorhabditis elegans, differentiation and proliferation of the germline is controlled through fine-tuning of gene expression by miRNAs and 3′-UTR length regulation in a spatiotemporal manner. Mutations affecting this regulation revealed that it is mediated at least in part by FIP1 and CPSF30 (Diag et al. 2018), likely as a part of CPSF. CPSF's involvement in the germline has also been observed in mice. WDR33, before being identified as a polyadenylation factor, was found to be highly expressed during spermatogenesis in mouse testis (Ito et al. 2001). Around the same time, CPSF160 was also reported to be overexpressed in mouse germ cells (Dass et al. 2001). Since miRNAs are also required for mouse spermato genesis and germ cell development (Hayashi et al. 2008), WDR33 and CPSF160 might regulate APA in mammalian germlines. Symplekin is also highly expressed in male germ cells, and germ cell-specific KO of Sympk in mouse leads to male infertility. Spermatocytes with Sympk KO displayed defects in homologous chromosome synapsis, DNA double-strand break repair, and meiotic recombination (Wu et al. 2021). While these defects were correlated with changes in splicing, it is unclear if this reflects a direct role of Symplekin in splicing or indirect effects resulting from Symplekin's canonical function in CPA.
Based on the findings from the above studies, the expression of multiple, perhaps all, CPSF subunits is regulated during differentiation. Among these, though, FIP1 appears to be a central regulator of APA during differentiation and development. This might reflect several properties of FIP1, as discussed above. First, the U-rich region it recognizes is quite variable in different transcripts, especially compared to the more highly conserved AAUAAA. U “richness” could easily be incorporated into the differentiation programs, which could in turn respond to changes in FIP1 expression to regulate PAS usage. Second, FIP1 serves a central scaffolding function, making important contacts with RNA and multiple CPA factors, including PAP, highlighting its potential function as a key regulator. Finally, the potential to produce literally dozens of isoforms by AS (see above) further enhances the regulatory possibilities of FIP1, as these could significantly affect protein/RNA interactions. Notably, the apparent centrality of FIP1 also supports our argument from the previous section that the FIP1-PDGFRA and FIP1-RARA fusion proteins might contribute to tumorigenesis by APA regulation, given that cancer cells resemble stem cells in many aspects.
VIRUS INFECTION AND IMMUNITY
Viruses are known to hijack the infected cell's molecular machineries for their own reproduction. In many cases, this involves the CPA machinery, and CPSF subunits again play important roles. For example, CPSF30 is frequently targeted by viruses to control replication and gene expression, and to suppress the host immune response. The E2 protein from the human papillomavirus is expressed during the early phase of the virus's life cycle, and it was reported to regulate the expression of late genes by interacting with CPSF30 (Johansson et al. 2012). This interaction prevents the assembly of the CPA machinery at the early, but not the late, polyadenylation site, so transcription continues into the late genomic region, producing a transcript that contains the open reading frames of the L1 and L2 proteins. This gene regulation by phases was suggested to contribute to the virus's immune evasion by preventing premature expression of late viral proteins that could expose the infected cell to the host immune system (Johansson et al. 2012).
Influenza A viruses (IAVs) also possess mechanisms to modulate host gene expression via interaction with CPSF30. Specifically, binding of the nonstructural protein NS1 to CPSF30 was found over 25 years ago to inhibit the CPA of cellular mRNAs, thereby preventing the export of host mRNAs from the nucleus (Nemeroff et al. 1998), and the structural basis for the interaction was defined a decade later (Das et al. 2008). Subsequent studies have confirmed the significance of these results with respect to IAV infection. For example, the CPSF30-NS1 inhibition of CPA was found to block the induction of interferon-stimulated genes (ISGs) (von Recum-Knepper et al. 2015). ISGs are needed to confer the antiviral state of cells, and their inhibition is important for efficient viral replication and further infection. Interestingly, different IAV strains were shown to be more or less effective at repressing ISGs, and this was found to reflect polymorphisms in NS1 that allow or prevent binding to CPSF30 (Hale et al. 2010; von Recum-Knepper et al. 2015). More recently, IAV was found to modulate APA in ways that reduce ISG expression and enhance virulence, and this was shown to result from the NS1–CPSF30 inhibition of CPA (Bergant et al. 2023). While this phenomenon was observed in all strains tested, in one the APA changes were observed in the absence of effects on host mRNA expression. This was found to reflect a polymorphism that weakened but did not eliminate the CPSF30–NS1 interaction. In yet another IAV strain, NS1 was found unable to bind CPSF30, but instead to interact, as judged by coIP, with CPSF100 (and CFIm59), and this interaction was also shown to interfere with CPA and prevent activation of ISGs (Kuo et al. 2018). Together, these findings attest to the importance of NS1/CPSF30-mediated inhibition of CPA to the IAV life cycle.
The NS1–CPSF30 interaction has also been reported to modulate splicing. For example, upon infection with IAV, NS1, together with CPSF30, alters the splicing of p53 transcripts, promoting the production of the β and γ isoforms (Dubois et al. 2019). These two isoforms are proviral factors, as their silencing significantly decreases virus production (Dubois et al. 2019). In addition, virus infection can induce endoplasmic reticulum (ER) stress and trigger the unfolded protein response (UPR) that leads to global translation inhibition. UPR activation requires the removal of 26 nt from the XBP1 pre-mRNA to produce the XBP1s protein, an active transcription factor (Lee et al. 2002). NS1 binding to CPSF30 inhibits XBP1 splicing, thus preventing the activation of URP and its inhibitory effect on viral replication (Mazel-Sanchez et al. 2021). The mechanism by which NS1-mediated inhibition of CPSF30 inhibits splicing is unknown, but a likely possibility is that it reflects secondary effects from its effects on CPA/APA. In any event, inhibition of CPSF30/CPSF function by NS1 is advantageous to the virus not only to suppress immune responses but also to facilitate the utilization of host machineries for its reproduction.
The herpes simplex virus ICP27 protein has also been shown to interact with CPSF to modulate 3′ end processing. Different from NS1, however, ICP27 interacts with all six CPSF subunits and can inhibit or activate CPA depending on the sequences upstream of the PAS (Wang et al. 2020c). ICP27's interactions with CPSF appeared to prevent the CPSF-Symplekin association. Given that Symplekin bridges CPSF with CstF and is required for cleavage, as discussed above, ICP27 exerts its inhibitory effect by disrupting CPA machinery assembly and the cleavage reaction. For mRNAs containing GC-rich upstream sequences, which are bound by ICP27, however, efficient CPA was observed. The GC content in upstream sequences is higher in viral mRNAs than in host mRNAs (Corbin-Lickfett et al. 2009), suggesting that ICP27 preferentially activates CPA on viral mRNAs by recruiting CPSF. ICP27's inhibition of CPSF is possibly released as ICP27 is transferred from CPSF to the GC-rich element, but the exact mechanism is unclear. Thus, ICP27 can promote viral gene expression while suppressing host gene expression, a function consistent with its role as an early viral protein. These findings also explain an earlier observation that ICP27 induced IPA at cryptic PASs in host mRNAs containing GC-rich sequences near the 5′ splice sites, producing many transcripts containing novel open reading frames (Tang et al. 2016b). It remains unclear whether such transcripts play a role in viral infection.
Consistent with the targeting of CPSF by NS1 and ICP27 and their roles in immune suppression, CPSF has been found to play additional roles in the proper functioning of the immune system. In macrophages, upon infection by the vesicular stomatitis virus, expression of multiple CPA factors, including CPSF160 and CPSF73, is downregulated. Decreased expression of these proteins induces 3′-UTR APA and modulates mRNA abundance of many genes involved in immune signaling pathways (Jia et al. 2017). In addition to regulating APA during the immune response, CPSF160 is also involved in AS of the Interleukin 7 receptor (IL7R) (Fig. 6A), which is required for T cell maintenance and development (Evsyukova et al. 2013). IL7R exon 6 inclusion generates membrane-bound isoforms, while its exclusion produces the soluble form. A PAS was identified in intron 6, near the 5′ splice site. Based on RNA affinity, CPSF160 binds this PAS, potentially interfering sterically with spliceosome assembly at the 5′ splice site, causing exon 6 to be skipped and generating the soluble IL7R isoform (Fig. 6A; Evsyukova et al. 2013). This model is favored by the fact that CPA could not be detected at this site, arguing against a competition between splicing and CPA. Given that CPSF160 does not directly bind to the PAS, perhaps intact CPSF, or only the PSF module, binds this site.
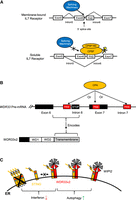
CPSF and WDR33v2 play important roles in innate immunity. (A) Intron 6 of the IL7 receptor pre-mRNA contains a PAS near the 5′ splice site. Removal of intron 6 by the splicing machinery produces the membrane-bound IL7 receptor (top panel). If the PAS is recognized by CPSF, the intron 6 5′ splice site is blocked. Intron 5 5′ splice site is instead used by the splicing machinery together with intron 6 3′ splice site, causing exon 6 to be skipped. This produces the soluble form of the IL7 receptor (bottom panel). CPA do not occur. Lightning bolt: endonucleolytic cleavage. (B) Intron 6 of the human WDR33 pre-mRNA contains an in-frame stop codon, which is associated with multiple functional intronic/exonic PASs (top panel). Usage of either of several of these PASs by the CPA machinery cause intron 6 to be retained, producing the WDR33v2 isoform. WDR33v2 harbors two N-terminal WD repeats and a C-terminal transmembrane domain, which is encoded by the retained intron 6 (bottom panel). (C) WDR33v2 is functionally completely unrelated to the canonical isoform. It localizes to the ER with the intron-encoded transmembrane domain and regulates the immune factor STING. WDR33v2 lowers interferon induction by decreasing STING oligomerization (left), while promoting autophagy activation by recruiting WIPI2 isoforms (right).
WDR33 also functions in immune responses, although in a novel way unrelated to its role in CPA (Liu and Manley 2024). It was first reported in 2009 that WDR33 possesses antiviral activities, as KD of WDR33, as part of a genome-wide screen, increased IAV infection (Brass et al. 2009). Especially in light of the results discussed above, a straightforward explanation for this would be an effect on CPA/APA. Interestingly, however, a noncanonical mRNA variant of WDR33, but not the canonical full-length transcript, was identified as an IAV infection-regulated transcript in another global screen (Miller et al. 2015). This noncanonical mRNA, formed by IPA-related mechanisms (Liu and Manley 2024), encodes the ∼38 kDa WDR33 isoform, which is the human homolog of the rat protein identified in the 2003 nuclear envelope proteomic study mentioned above (Fig. 6B; Schirmer et al. 2003). This isoform, termed WDR33v2 (V2), is an ER transmembrane protein and does not localize to the nucleus or participate in CPA. Strikingly, V2 interacts with and regulates the activities of STING (STimulator of INterferon Genes), an ER protein that induces immune responses to viruses and cytoplasmic dsDNA (Zhang et al. 2020b; Ritchie et al. 2022). V2 was found to regulate STING functions in two ways (Fig. 6C): (i) by suppressing interferon induction, likely by decreasing STING oligomerization, which is essential for activation; and (ii) by promoting autophagy, likely by recruiting isoforms of WIPI2, an essential autophagy factor. Another WDR33 isoform, WDR33v3 (∼30 kDa), also formed by IPA, appears to increase STING levels, perhaps by inhibiting V2. Notably, V2 was recently found to be upregulated when human primary macrophages were induced into a pro-inflammatory state (Wilton et al. 2023), suggesting that V2 also plays a role in macrophage activation. These findings thus reveal that the human WDR33 gene, in addition to encoding a CPSF subunit, also encodes innate immune factors, indicating that protein isoforms encoded by the same gene can be functionally completely unrelated, revealing that APA is a more powerful mechanism than previously thought.
FUTURE DIRECTIONS
As we have discussed CPSF's involvement in multiple biological processes, we also provide our thoughts on future studies that are worth pursuing. In this section, we briefly describe broader research directions that are of interest to our understanding of CPSF.
In cancer and stem cell differentiation/development, it is now well documented how the expression of CPSF subunits changes, and the associated consequences are also well understood. However, how the expression of these genes is regulated, likely to a significant degree at the transcriptional level, but also by posttranslational modifications, e.g., by sumoylation (Vethantham and Manley 2009), remains largely unknown. In addition, the mRNA features of their APA targets are also not well characterized. Detailed molecular insights into these regulatory mechanisms and pathways may facilitate development of antitumor drugs and therapeutic applications of stem cells. Furthermore, how CPSF contributes to the complex conditions of cancer, differentiation, and the immune response by regulating molecular processes other than APA, such as transcription termination and small RNA processing, has also not been studied extensively and warrants further investigation.
The finding that WDR33 isoforms are functionally unrelated underscores the importance of alternative mRNA processing. CPSF30 is subject to AS that generates two isoforms, which are both CPSF subunits (∼30 and ∼28 kDa) (Chan et al. 2014). The functional differences between these two isoforms have never been examined. It is possible that they have different preferences for RNA substrates. As mentioned above, FIP1 is surprisingly annotated to have 62 mRNA isoforms in the RefSeq database (O'Leary et al. 2016), and many of these isoforms would be predicted to affect FIP1 interactions with other CPA factors, or possibly interaction with other regulatory proteins. This raises the possibility that APA regulation by FIP1 might be in part due to different isoform usage rather than changes in protein levels. Understanding how these isoforms affect CPSF functions, and how their production is regulated, will provide interesting links between AS and APA, the two major mRNA processing mechanisms.
CONCLUDING REMARKS
In this review, we have provided a general overview of the functions of CPSF subunits and a discussion of how CPSF influences a diverse collection of biological processes and conditions. Our analyses interestingly revealed that different biological processes involve different CPSF subunits, such as CPSF73's functions outside of 3′ end processing and FIP1's roles in regulating differentiation and development. Through its canonical functions, CPSF as a complex directly regulates gene expression and prevents/resolves R loops to promote genome stability. It also contributes to gene expression fine-tuning by controlling APA and small RNA processing. By regulating such processes, CPSF contributes to complex conditions such as cancer development, differentiation, and infection/immune response. Thus, CPSF, despite being responsible for only two simple biochemical reactions, has profound impacts on cellular homeostasis.
ACKNOWLEDGMENTS
We thank Ikuko Hotta and Julia Parsley for their work on CPSF30 and FIP1 isoforms. L.L. was supported in part by the US NSF Graduate Research Fellowship (DGE-2036197). This work was supported by US National Institutes of Health (NIH) R35 GM118136 (J.L.M.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080108.124.
-
Freely available online through the RNA Open Access option.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵








