
Rational design of oligonucleotides for enhanced in vitro transcription of small RNA
- Department of Applied Chemistry, Graduate School of Science and Engineering, Ehime University, Matsuyama, Ehime 790-8577, Japan
- Corresponding authors: yamagami.ryota.bn{at}ehime-u.ac.jp, hori.hiroyuki.my{at}ehime-u.ac.jp
-
Handling editor: Eric Westhof
Abstract
All kinds of RNA molecules can be produced by in vitro transcription using T7 RNA polymerase using DNA templates obtained by solid-phase chemical synthesis, primer extension, PCR, or DNA cloning. The oligonucleotide design, however, is a challenge to nonexperts as this relies on a set of rules that have been established empirically over time. Here, we describe a Python program to facilitate the rational design of oligonucleotides, calculated with kinetic parameters for enhanced in vitro transcription (ROCKET). The Python tool uses thermodynamic parameters, performs folding-energy calculations, and selects oligonucleotides suitable for the polymerase extension reaction. These oligonucleotides improve yields of template DNA. With the oligonucleotides selected by the program, the tRNA transcripts can be prepared by a one-pot reaction of the DNA polymerase extension reaction and the transcription reaction. Also, the ROCKET-selected oligonucleotides provide greater transcription yields than that from oligonucleotides selected by Primerize, a leading software for designing oligonucleotides for in vitro transcription, due to the enhancement of template DNA synthesis. Apart from over 50 tRNA genes tested, an in vitro transcribed self-cleaving ribozyme was found to have catalytic activity. In addition, the program can be applied to the synthesis of mRNA, demonstrating the wide applicability of the ROCKET software.
Keywords
INTRODUCTION
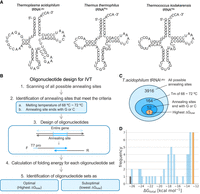
In vitro transcription (IVT) from a template DNA by T7 RNA polymerase can synthesize any class of RNA molecule such as mRNA, rRNA, tRNA, small RNA, or long noncoding RNA. The technique is broadly used in RNA-related research including biochemistry, structural biology, and molecular biology. In addition, based on the recent advancements in RNA-based technologies including the emergence of the SARS-CoV-2 mRNA vaccines and the CRISPR–Cas13 system for RNA targeting, RNA has become widely recognized as a pivotal molecule for next-generation gene therapy, diagnosis, and fundamental research (Pardi et al. 2018; Pickar-Oliver and Gersbach 2019; Palaz et al. 2021). Successful application of various RNA technologies requires large quantities of high-quality RNA. To meet this demand, RNA preparation methods have been extensively studied over the past five decades.
Synthetic RNAs are commonly transcribed from plasmid DNAs or PCR products by IVT with T7 RNA polymerase. The template DNA contains the T7 promotor sequence attached to the 5′ end of the gene of interest. IVT from plasmid DNAs is particularly useful for preparing relatively long RNAs, such as mRNA, rRNA, and long noncoding RNAs. In addition, the IVT method is applicable to small RNAs. In the 1980s, Uhlenbeck and coworkers pioneered the use of IVT to prepare yeast phenylalanine-transfer RNA (tRNAPhe) from a tRNAPhe gene that was cloned in between the T7 promotor and BstNI restriction site of a plasmid. The plasmid, linearized by the BstNI restriction enzyme, was then used for transcription (Fig. 1A; Sampson and Uhlenbeck 1988; Sampson et al. 1989; Behlen et al. 1990). This IVT method has since found application in numerous tRNA-related studies in molecular biology, structural biology, and biochemistry (Himeno et al. 1990; Motorin and Grosjean 1999; Droogmans et al. 2003; Shitivelband and Hou 2005; Tukalo et al. 2005; Byrne et al. 2010; Swinehart et al. 2013; Nakano et al. 2016; Dégut et al. 2019; Hibi et al. 2020; Schultz et al. 2023). While IVT from plasmids accommodates various RNA species, plasmid construction involves molecular cloning and sequencing steps, consuming several days. An additional step of site-directed mutagenesis is required when RNA variants for the characterization of RNA structure or function have to be generated (Hall et al. 1989). Moreover, the use of restriction enzymes can be limited when their cleavage sites are present within the target gene.

Schematic illustrations of the preparation of double-stranded DNA template for in vitro RNA transcription. (A) Preparation of a DNA template from plasmid DNA. A target DNA fragment harboring the T7 promoter sequence (T7 pro) at the 5′ end of the gene is prepared from plasmid DNA by linearization using a restriction enzyme or by PCR amplification. (B) Three distinct strategies to prepare a DNA template from two oligonucleotides. Strategy 1: A double-stranded DNA template is prepared by annealing nontemplate strand (NTS) and template strand (TS) DNA oligonucleotides encoding the T7 pro sequence and the entire target gene. Strategy 2: A hemi-duplexed DNA template is prepared by annealing a NTS oligonucleotide encoding the T7 pro sequence to a TS oligonucleotide encoding the T7 pro and the entire gene. Strategy 3: Two oligonucleotides are designed to possess an overlapping region that facilitates their annealing and are extended by DNA polymerase.
The development of methods for preparing in vitro RNA transcripts has been determined by the quality, cost, and length of chemically synthesized DNA oligonucleotides. With the widespread use of dideoxy-DNA sequencing and PCR, chemically synthesized DNA oligonucleotides became commercially available in the 1980s. At that time, synthetic DNA oligonucleotides were expensive, and short DNA oligonucleotides (30 mer or shorter) were designed and template DNAs for preparing RNA transcripts were generated from plasmids by PCR. In the 1990s, relatively long DNA oligonucleotides (50 mer or longer) became commercially available, and over the past two decades technological innovations in solid-phase DNA oligonucleotide synthesis with phosphoramidite, coupled to automatization of synthesis, processing, and purification, enabled the production of affordable high-quality DNA oligonucleotides with lengths of up to 100–200 nt (Kosuri and Church 2014). Such oligonucleotides can cover a complete gene of a small RNA such as a tRNA gene. Consequently, the use of synthetic DNA oligonucleotides has become an alternative for preparing template DNA for IVT. Oligonucleotide design for IVT has followed three distinct strategies (Fig. 1B). In Strategy 1, PCR products harboring the T7 promoter and the full-length gene are used for IVT after amplification from plasmid DNA/genomic DNA or complete double-stranded DNA consisting of nontemplate strand and template strand. Strategy 2 uses a hemi-duplexed DNA template consisting of a nontemplate strand oligonucleotide corresponding to the T7 promoter and a complete template strand oligonucleotide (Korencic et al. 2002; Beckert and Masquida 2011; Leamy et al. 2019; Kartje et al. 2021; MalagodaPathiranage et al. 2023). For Strategy 3, nontemplate strand/template strand DNA oligonucleotides are designed to have an overlap region that enables annealing with an approximate melting temperature (Tm) of 65°C–70°C. These DNA oligonucleotides are extended with DNA polymerase, and the resulting double-stranded DNA is used as a template (Sherlin et al. 2001; Francklyn et al. 2008; Yamagami et al. 2012, 2018, 2022; Avcilar-Kucukgoze et al. 2020; Albers et al. 2021, 2023; Yamagami and Hori 2023). While Strategies 1 and 2 are straightforward and simplify the design of the DNA oligonucleotides, they often result in a low transcription efficiency because of the relatively long DNA oligonucleotides (∼100 nt). Such long DNA oligonucleotides could form stable secondary structures that might pause the extension reaction of DNA polymerase (LaDuca et al. 1983; Bedinger et al. 1989) which would lead to a reduction in the amount of double-stranded DNA template. In contrast, Strategy 3 requires DNA oligonucleotides that are shorter than those utilized in Strategy 1 or 2. Short oligonucleotides are structurally flexible, which reduces the chance that internal secondary structures are formed. Strategy 3 adheres to a basic concept of the polymerase cycling assembly method (PCA) to synthesize a long double-stranded DNA fragment from multiple short DNA oligonucleotides (Hoose et al. 2023). Although Strategy 3 can be used for the preparation of DNA templates to synthesize various RNA species, the design of the oligonucleotides is somewhat complicated by following empirical rules, which is time-consuming and error-prone. To address this, Das and coworkers developed Primerize, a program that automated the design of oligonucleotides for preparing IVT DNA templates according to Strategy 3 (Tian et al. 2015). The output of Primerize is oligonucleotides that avoid mispriming during the extension reaction with DNA polymerase (Tian et al. 2015). In addition, nearest-neighbor thermodynamic models have been widely used for accurate prediction of DNA/RNA secondary structure and stability (Xia et al. 1998). The nearest-neighbor models assume that the thermostability of a given base pair depends on the identity of neighboring base pairs (SantaLucia 1998). The Primer3 software, one of the most cited software that designs PCR primer, and similar software use the thermodynamic models to calculate melting temperatures of priming regions and predict formation of primer dimers (Rozen and Skaletsky 2000; Untergasser et al. 2012). To our knowledge, while the thermodynamic models have been utilized in the rational design of PCR primers, it has yet to be applied to the oligonucleotide design for IVT.
Here, we report the results obtained with a Python tool for the rational design of oligonucleotides calculated with kinetic parameters for enhanced in vitro transcription, so-called ROCKET. The ROCKET software uses thermodynamic parameters, performs folding energy calculations, and selects oligonucleotides suitable for preparing template DNA for IVT from a pool of candidates. To test the versatility of ROCKET and the specificity of its output, we used the oligonucleotides in the construction of DNA templates for in vitro tRNA transcription. We find that the extension efficiency of template DNA from ROCKET-selected oligonucleotides by the DNA polymerase reaction is significantly improved. Various tRNA species are successfully transcribed from the DNA templates prepared from ROCKET-selected oligonucleotides. We then compare the Primerize software to ROCKET. Also, we show that the software is applicable to other small RNA as well as mRNA. The software can be downloaded from GitHub at https://github.com/TEPPEI-MAT/ROCKET under the MIT license.
RESULTS
ROCKET-selected oligonucleotides increase the extension efficiency of DNA polymerase
To compare oligonucleotides selected by ROCKET with those that have been published, we focused initially on three tRNA genes (Fig. 2A); tRNALeu and tRNAPro genes from Thermoplasma acidophilum HO-62 and Thermus thermophilus HB8 for which template strand/nontemplate strand oligonucleotides had been designed and used for the DNA polymerase reaction (Yamagami et al. 2012; Kawamura et al. 2015). In addition, we tested the tRNAHis gene from Thermococcus kodakarensis KOD1 because tRNAHis has been used for biochemical studies in our laboratory. ROCKET automates the oligonucleotide design strategy that is depicted in Figure 2B. For the T. acidophilum tRNALeu gene, the ROCKET software identified 3916 different annealing sites (Fig. 2C). Of these sites, 164 sites met the Tm criteria of 68°C–72°C (Fig. 2C), which was further refined to 52 sites after selection of those in which the annealing region ended with G or C (Fig. 2C). Then, the software generated 52 sets of template strand/nontemplate strand DNA oligonucleotides, calculated their folding energies (Fig. 2D) and selected the oligonucleotide pair with the highest ΔGtotal of −12.6 (kcal•mol−1). These oligonucleotides are predicted thermodynamically to be less structured, which would help to eliminate internal oligonucleotide folding and increase the efficiency of strand extension by DNA polymerase. We also selected the oligonucleotide set that scored the lowest ΔGtotal of −26.5 (kcal•mol−1) as the suboptimal control (Fig. 2B, step 5). These oligonucleotides are predicted thermodynamically to be more structured, which would decrease the efficiency of the DNA extension reaction.
Overview of the ROCKET software. (A) Cloverleaf structures of Thermoplasma acidophilum tRNALeu, Thermus thermophilus tRNAPro, and T. kodakarensis tRNAHis. (B) Workflow for computational design of DNA oligonucleotides for in vitro transcription. (C) Oligonucleotide selection for T. acidophilum tRNALue. The number of usable oligonucleotides is narrowed down in ROCKET in subsequent steps. (D) The ΔGtotal (ΔGforward oligo + ΔGforward oligo) was calculated for each set of forward/reverse oligonucleotides. The suboptimal oligonucleotide set with the lowest (dark-gray) and the optimal set with the highest ΔGtotal value (orange) are highlighted.
We first compared published oligonucleotides for the three tRNA genes to those selected by the program, which turned out to be based on other annealing sites. For example, in the case of the T. kodakarensis tRNAHis gene, the published oligos anneal in the D-arm region, whereas the computationally selected oligos cover the anticodon arm and T-arm (Fig. 3A). We used Dream-Taq DNA polymerase for DNA extension and in-house prepared T7 RNA polymerase for RNA transcription (Supplemental Fig. S1). We first examined the NTPs concentration in our transcription assay at 37°C for 4 h and found that the transcription yield from the 2.5 mM NTPs condition was comparable to those from 3.75 mM and 5.0 mM NTPs conditions (Supplemental Fig. S2A,B). Thus, we used the final concentration of 2.5 mM NTPs for the following transcription assays. We conducted a one-pot reaction of DNA polymerase extension and RNA transcription. In other words, the reaction mixture of DNA polymerase was directly used for IVT without DNA purification. For T. acidophilum tRNALeu and T. kodakarensis tRNAHis, a significant increase in the transcription yield was observed (Fig. 3B; Supplemental Fig. S3A). In the case of T. thermophilus tRNAPro, the three sets of DNA oligonucleotides supported comparable levels of IVT (Supplemental Fig. S3B). Thus, the oligonucleotides selected by the program as optimal improved transcription yields for two of the three tested tRNAs. In contrast, transcription levels were comparable when we used the same amount of template DNAs from each set of oligonucleotides (Fig. 3C). Therefore, we hypothesized that the transcription yields were enhanced by an increase in the template DNA amounts from ROCKET-selected oligonucleotides in the DNA polymerase extension reaction.
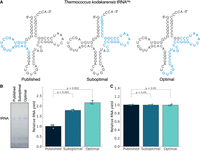
Oligonucleotides selected by ROCKET enhance transcription yields. (A) Secondary structures of T. kodakarensis tRNAHis with nucleotides highlighted in blue that form the annealing region for published forward/reverse oligonucleotides (left) or for oligonucleotide pairs selected by ROCKET as optimal (right) or suboptimal (center). (B) Transcription yields from the one-pot reaction of DNA extension and RNA transcription were monitored by staining of RNA with Toluidine blue after 10% denaturing PAGE (7 M urea) (left) and quantified (right), as described in the Materials and Methods. Errors (x), SD, and P-values are shown. (C) In vitro transcription yields with comparable amounts of template DNA. Errors, SD (n = 3), and P-values are shown.
To address our hypothesis, we separately characterized the DNA extension reaction (Fig. 4) and the transcription reaction (Fig. 5). We first performed an electrophoretic mobility shift assay (EMSA) to monitor the oligonucleotide duplex-formation (Supplemental Fig. S4). From the extent of annealing between oligonucleotides and their input concentration (Fig. 4A), apparent KD values were calculated to be 30 nM, 40 nM, and 70 nM for published, suboptimal, and optimal oligonucleotide pairs, respectively. A larger fraction of optimal/suboptimal oligonucleotides can form duplexes, which would lead to a larger yield of template DNA after strand extension (Fig. 4A). This turned out to be the case; the relative yield of template DNA produced by DNA polymerase from duplexes formed by optimal/suboptimal oligonucleotides was slightly increased compared to the yields obtained with duplexes formed by published oligonucleotides, with a P-value of 0.003 and 0.008, respectively (Fig. 4B). In this experiment, we used ∼250 nM Dream-Taq DNA polymerase (2.5 U Dream-Taq DNA polymerase in the 100 µL reaction) according to the manufacturer's instruction. To gain detailed insights into the reaction efficiency of the polymerase extension, we reduced the polymerase concentration to ∼10 nM (0.01 U Dream-Taq DNA polymerase in the 100 µL reaction) to achieve oligonucleotide-excess conditions. The amounts of synthesized template DNAs were traced PCR cycle-dependently (Fig. 4C). The cycle experiment clearly showed that the optimal DNA oligonucleotides were more efficiently extended by DNA polymerase; the extension reaction of ROCKET-selected oligonucleotides reached a plateau within 10 cycles, whereas the reaction of published oligonucleotides did not finish at 10 cycles and even after 30 cycles (Fig. 4C). At 10 cycles, the polymerase reaction using the ROCKET-selected oligonucleotides yielded ∼2.5-fold amounts of template DNAs as compared with the reaction using the published oligonucleotides (Fig. 4D). The ROCKET-selected oligonucleotides for the other two tRNA genes (T. acidophilum tRNALeu and T. thermophilus tRNAPro genes) consistently gave similar results (Supplemental Fig. S5). Also, time-course experiments showed that ROCKET-selected oligonucleotides were more efficiently extended than published oligonucleotides (Fig. 4E). Importantly, even in the polymerase-diluted condition, the yields of the extended DNA products after 25 cycles were comparable to the yields obtained from the manufacturer's recommended condition when we used ROCKET-selected oligonucleotides (Fig. 4F).
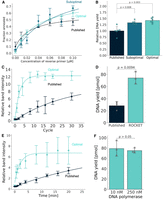
Characterization of the DNA extension reaction. (A) Duplex-formation of published (black), suboptimal (green), and optimal (light green) oligonucleotide pairs. Numbers were obtained by quantification of electrophoretic mobility shift assays (EMSAs) shown in Supplemental Figure S4; SD (n = 3) are shown for each data point. (B) Relative DNA yields obtained in DNA polymerase extension reactions were quantified as described in the Materials and Methods. Errors, SD (n = 6), and P-values are shown. (C) PCR cycle-dependent amounts of extended DNA obtained by DNA polymerase reactions in the presence of 1 µM oligonucleotides and ∼10 nM Dream-Taq DNA polymerase (0.01 units) were quantified. The data were fitted to a single exponential curve. Errors, SD (n = 3) are shown. (D) The DNA template after 10 cycles of the DNA polymerase extension reaction was analyzed. After the DNA purification steps, DNA concentration was quantified by UV absorbance. Errors, SD (n = 3) are shown. (E) Time-dependent amounts of extended DNA obtained by DNA polymerase reactions in the presence of 1 µM oligonucleotides and ∼10 nM Dream-Taq DNA polymerase (0.01 units) were quantified. Errors, SD (n = 3) are shown. (F) DNA yields after 25 cycles of the extension reaction in the presence of ∼10 nM and ∼250 nM Dream-Taq DNA polymerase were analyzed after DNA purification steps. DNA concentration was quantified by UV absorbance. Errors, SD (n = 3) are shown.

Characterization of the transcription reaction. (A) RNA yields obtained by in vitro transcription at specific time points were analyzed by Toluidine blue staining after 10% denaturing PAGE and (B) relative band intensities were quantified as described in the Materials and Methods and fitted to a single exponential curve (Equation 4). Errors, SD (n = 3) are shown.
Next, to test if the DNA templates from ROCKET-selected oligonucleotides affect the T7 RNA polymerase reaction, we performed transcription experiments (Fig. 5). We monitored the transcription reaction in the presence of 500 nM template DNAs and 100 nM T7 RNA polymerase. The RNA transcripts were comparably synthesized from both DNA templates (Fig. 5A,B, Compare ROCKET and published), suggesting that template DNAs from ROCKET-selected oligonucleotides do not change the transcription efficiency.
Based on the results from DNA extension and RNA transcription experiments, we conclude that the use of optimal oligonucleotides leads to an increase in the DNA polymerase extension efficiency, which consequently contributes to an increase in transcription levels in the one-pot reaction (Fig. 3B).
Options available in the ROCKET software
The T7 RNA polymerase reaction is known to be drastically repressed when transcription does not initiate at a G residue (Kuzmine et al. 2003). To eliminate this, the ROCKET software has an option [-p or ‐‐precursor] to attach a leader sequence to the 5′ end of those tRNA genes that start with an A, T, or C residue. The leader sequence can be removed by RNase P posttranscriptionally (Fig. 6A). To test precursor tRNAs synthesized in vitro with ROCKET-selected oligonucleotides, we reconstituted RNase P using the Escherichia coli C5 protein and M1 RNA (Supplemental Fig. S1B,C). As shown in Figure 6B–D, the leader sequence in the tested precursor tRNAs could be completely removed by RNase P cleavage. Note that according to the literature, the RNase P reaction can be combined with the IVT reaction (Fukunaga et al. 2006). Also, it should be mentioned that the RNase P reaction yields transcripts with a 5′ monophosphate (Esakova and Krasilnikov 2010), whereas a regular T7 RNA polymerase reaction produces transcripts with a 5′ triphosphate. Alternatively, tRNAs that lack a G at their 5′ end could be prepared in vitro after introducing a nucleotide substitution at the end of the acceptor stem, which can be made in the sequence for the input in the ROCKET software. For example, replacing the A1-U74 pair of T. kodakarensis tRNAiMet with a G1-C74 pair resulted in good transcription yields (Fig. 7, see iMet-CAT*). When the application of the in vitro synthesized RNA relies on the original sequence, however, we strongly recommend the RNase P strategy for the preparation of tRNAs that do not start with a G.
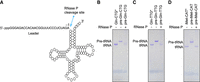
In vitro RNase P reaction on precursor tRNA. (A) A cloverleaf structure of precursor tRNA. (B–D) Cleavage of the leader sequence by RNase P. Based on optimal, ROCKET-selected oligonucleotides precursors for (B) tRNAGln-CTG, (C) tRNAGln-TTG, and (D) tRNAiMet-CAT were in vitro synthesized and subsequently incubated with (+) or without RNase P (−). The RNAs were visualized by Toluidine blue staining after 10% denaturing PAGE (7 M urea). The asterisk marks tRNAs used as a migration control.
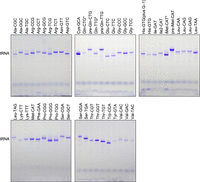
In vitro transcription of all T. kodakarensis tRNA genes. DNA templates were based on tRNA genes retrieved from the genomic tRNA database (Chan and Lowe 2016) and constructed with oligonucleotides selected as optimal by the ROCKET software (Supplemental Table S1). RNAs were visualized by Toluidine blue staining after 10% denaturing PAGE of 5 µL transcription mixture. Nucleotides at positions 1 and 72 in those tRNAs that do not have a G residue as the 5′ end (marked with an asterisk), have been substituted with G1-C72.
In eukaryotes, tRNAHis is known to undergo posttranscriptional processing whereby an extra G residue is enzymatically attached to the 5′ end (G–1) (Gu et al. 2003). In prokaryotes, this G is encoded in the genome. The ROCKET software has an option [-g or ‐‐g_addition] that automatically adds a G residue at the −1 position.
The most well-known role for tRNA is to deliver an amino acid specified by an mRNA codon during translation by ribosomes. For this, tRNAs carry a CCA terminal sequence required for the aminoacylation reaction and peptide-bond formation (Xiong and Steitz 2006). The CCA sequence is not always genetically encoded but posttranscriptionally added by tRNA nucleotidyltransferase (Heinemann et al. 2010). For those tRNA genes that lack the CCA terminus, ROCKET has an option [-c or ‐‐cca_addition] to extend the gene with this triplet.
We note that T7 RNA polymerase often produces some undesired byproducts (Milligan et al. 1987; Cazenave and Uhlenbeck 1994; Pleiss et al. 1998). For example, T7 RNA polymerase is known to synthesize tRNA transcripts having heterogeneous sequences at the 5′ end when target tRNA genes have consecutive G residues at the 5′ end (Pleiss et al. 1998). In theory, this issue can be addressed by attaching a precursor sequence and cleaving it off by RNase P after transcription. The cleavage reaction of RNase P would result in providing tRNA transcripts having homogenous sequences at the 5′ end. Also, T7 RNA polymerase often adds extra nucleotides at the 3′ end of transcripts, which produces longer RNA transcripts than expected RNA sizes. This extension reaction occurs primally due to the loopback extension reaction where the 3′ end of runoff transcripts rebinds to T7 RNA polymerase and anneals with its transcript. The polymerase adds extra nucleotides to the bound RNA using itself as a template (Gholamalipour et al. 2018,2019). This self-primed extension reaction can be inhibited by adding a DNA oligonucleotide that is designed to bind to the 3′ end of runoff transcripts (Gholamalipour et al. 2019). These troubleshooting procedures should be considered when researchers have difficulty in preparing target RNAs.
Transcription of all tRNA genes encoded in T. kodakarensis
Using the optimized IVT reaction as well as the ROCKET software, we transcribed all 46 tRNA genes encoded in T. kodakarensis (Fig. 7). The tRNAHis that was programmatically extended with a G at the 5′ end, was successfully synthesized in the transcription reaction (Fig. 7, see His-GTG [plus G–1]). Furthermore, tRNAs with leader sequences attached in ROCKET were successfully transcribed (Fig. 7, See pre-Gln-CTG, pre-Gln-TTG, pre-iMet-CAT). Note that the G1-C72 mutants of these tRNAs were also transcribed (Fig. 7, see inputs marked with an asterisk). Finally, we found that the transcription efficiency for some tRNAs, like tRNAVal, was apparently linked to their sequence.
ROCKET and Primerize select different oligonucleotide sequences
The Primerize software designs oligonucleotides that can be assembled and polymerase-extended for IVT (Tian et al. 2015). Primerize focuses on finding oligonucleotides that avoid mispriming and have a minimal oligonucleotide length. To test how ROCKET compared to Primerize, we tested oligonucleotide selection by ROCKET and Primerize for 46 tRNA genes of T. kodakarensis. We found that ROCKET-selected oligonucleotides of different sequences and lengths (in other words, with a different region of overlap) than those suggested by Primerize. For example, in the tRNATrp gene, ROCKET selected the oligonucleotides that anneal in the D- and anticodon arms, whereas Primerize selected oligonucleotides that cover the acceptor and D-stems (Fig. 8A). Those outputs varied, related to the folding energy calculation incorporated in ROCKET. Oligonucleotide pairs selected with ROCKET had an average ΔGtotal value of −15.4 ± 4.2 kcal/mol, whereas those picked by Primerize came with an average ΔGtotal value of −18.6 ± 4.8 kcal/mol (Fig. 8B). The difference between these average ΔGtotal values was significant with a P-value of 2 × 10−9, implying that the ROCKET-selected oligonucleotides are predicted to be more structurally flexible. As for oligonucleotide length, the ROCKET-selected oligonucleotide sets have an average length of 128.1 ± 6.8 nt, whereas the Primerize-selected pairs have an average length of 123.8 ± 8.2 nt. These average values were also significantly different with a P-value of 1 × 10−6. Next, we monitored the DNA polymerase extension using both sets of ROCKET- and Primerize-selected oligonucleotides. We found that ROCKET-selected oligonucleotides were more efficiently extended than the Primerize-selected oligonucleotides in a cycle-dependent manner under the DNA polymerase-diluted condition (Fig. 8C), similar to Figure 4. Also, the time course experiment showed the same trend (Fig. 8D). These results reflect the unique features built into each software: selection by ROCKET is guided by structural dynamics of the oligonucleotides, whereas Primerize centers on the overall size of the DNAs and the length of the annealing region. In addition, we examined the one-pot reaction of DNA extension and RNA transcription. We observed that the yields from the ROCKET-selected oligonucleotides were significantly higher than that from the Primerize-selected oligonucleotides in these three tRNAs due to the enhancement of the DNA polymerase extension reaction with the ROCKET-selected oligonucleotides (Fig. 8E–G).
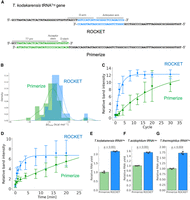
Comparison of oligonucleotide selection by ROCKET and Primerize. (A) Forward and reverse oligonucleotides selected by ROCKET (top) and Primerize (bottom) for T. kodakarensis tRNATrp with the annealing sites in color. (B) Density plot of ΔGtotal values for 46 sets of oligonucleotides for T. kodakarensis tRNA genes, selected by ROCKET (blue) and Primerize (green). (C) PCR cycle-dependent amounts of extended DNA obtained by DNA polymerase reactions in the presence of 1 µM oligonucleotides and ∼10 nM Dream-Taq DNA polymerase (0.01 units) were quantified. The data were fitted to a single exponential curve. Errors, SD (n = 3) are shown. (D) Time-dependent amounts of extended DNA obtained by DNA polymerase reactions in the presence of 1 µM oligonucleotides and ∼10 nM Dream-Taq DNA polymerase (0.01 units) were quantified. Errors, SD (n = 3) are shown. (E–G) Transcription yields of (E) T. kodakarensis tRNAHis, (F) Thermoplasma acidophilum tRNALeu, and (G) Thermus thermophilus tRNAPro from one-pot reactions using Primerize-selected oligonucleotides and ROCKET-selected oligonucleotides were compared. Transcription yields were quantified by 10% denaturing PAGE (7 M urea) as described in the Materials and Methods. Errors (x) from three biological replicates, SD, and P-values are shown.
The ROCKET software is not specific for tRNA genes
To test whether ROCKET can be used for finding oligonucleotides to synthesize small RNAs other than tRNAs, we obtained DNA oligonucleotides for the self-cleaving ribozyme CPEB3 (Fig. 9A; Salehi-Ashtiani et al. 2006; Yamagami et al. 2019). With a high concentration of MgCl2 in the transcription mixture, self-cleavage can occur between the leader and CPEB3 sequence (Fig. 9B). In our assay (see Materials and Methods), we observed that both the full-length transcript and cleaved products accumulated as a function of time (Fig. 9C), indicating that the transcribed CPEB3 ribozyme is active. This result demonstrated that our software package can be applied to various small RNAs. Furthermore, we tested if the ROCKET strategy is applicable to the synthesis of mRNA. We designed 20 DNA oligonucleotides using the modified version of ROCKET. These oligonucleotides selected by the software can be assembled into a template DNA that encodes the EGFP gene by polymerase cycling assembly (Fig. 10A). The software first identified the longest annealing site (ASlong) from all possible annealing sites in the EGFP gene (Fig. 10A, Steps 1 and 2). Because all DNA oligonucleotides except for the first and final oligonucleotides (in this case, O1 and O20 oligonucleotides) have two annealing sites at both 5′ and 3′ ends, they need to have more than 2ASlong lengths. The software, then, selected O1 and O2 oligonucleotides for the first 69 nt of the gene (Fig. 10A, Step 3). Next, the O3 oligonucleotide was designed to have an overlapping site with the O2 oligonucleotide (Fig. 10A, Step 4). This selection step was iterated for the rest of the oligonucleotides. Finally, the O2, O4, ••• O18, and O20 oligonucleotides were reverse complemented (Fig. 10A, Step 5). After two rounds of polymerase cycling assembly (Fig. 10B), we observed the desired band with the expected DNA size (∼819 bp) of the EGFP template (Fig. 10C). Thus, the oligonucleotides were successfully assembled by PCR. We performed IVT and translation experiments using PUREflex 2.0 and confirmed the expression of EGFP by fluorescence detection, and demonstrated that functional EGFP mRNA was prepared from ROCKET-selected oligonucleotides (Fig. 10D). Overall, we could apply the ROCKET strategy to the synthesis of mRNA.
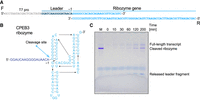
Application of ROCKET in the preparation of catalytically active CPEB3 ribozyme. (A) Oligonucleotide sequences selected by ROCKET with the portions covering T7 promoter (T7 pro, gray), leader (dark blue), and CPEB3 ribozyme (light blue) highlighted. (B) Secondary structure of the produced transcript. The cleavage site of the CPEB3 ribozyme (between A-1 and G1) is indicated by an arrow. (C) Synthesis and self-cleavage of the CPEB3 ribozyme was monitored by 10% denaturing PAGE as described in the Materials and Methods.
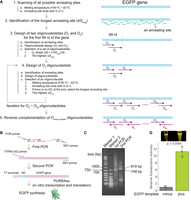
Application of ROCKET to the synthesis of EGFP protein. (A) Computational workflow for design of DNA oligonucleotides for the EGFP gene. (B) Experimental workflow for EGFP synthesis. The experiment was perfomed by two successive PCRs. The resulting template DNA was gel-purified and then used for the protein synthesis. (C) One microliter of the PCR product from each PCR step was analyzed by 2% agarose gel electrophoresis. The gel was stained by SYBR gold. (D) EGFP synthesis was monitored by fluorescence intensities where EGFP was excited by the 488 nm light, and the emission at 510 nm was measured. Errors (x) from three biological replicates, SD, and P-values are shown.
DISCUSSION
Due to the recent technical innovations in RNA biology, the development of a method that can prepare RNA with high quality in a time-effective manner is in great demand (Qin et al. 2022). A DNA template for IVT can be prepared from a set of template strand/nontemplate strand oligonucleotides that have an overlap region. The two oligonucleotides are extended by DNA polymerase, and the resulting double-stranded DNA template can guide RNA polymerase in an IVT reaction (Fig. 1B, Strategy 3). The oligonucleotide extension method is pivotal in this strategy because it contributes to the rapid preparation of RNA and has been widely used in high-throughput approaches to analyze RNA sequence and function (Kladwang et al. 2011; Yamagami et al. 2019). In this study, we developed ROCKET, a Python package that takes thermodynamic parameters into account for the selection of oligonucleotides that are optimal for IVT using this strategy. The software selects oligonucleotides that have the highest ΔG value to eliminate self-priming, primer–dimer formation, and folding of the DNA (Fig. 2A). With the increase in length of oligonucleotides that are economically affordable, it has become a challenge to consider DNA flexibility when designing these oligonucleotides. ROCKET provides a tool that addresses this difficulty and is easy to use. We found that oligonucleotide choice influenced the amount of double-stranded DNA template obtained after the extension reaction. This, in turn, determined the efficiency of the transcription reaction, because this increased with the amount of template. Other parameters of the IVT procedure could be optimized as well. By combining automated selection of DNA oligonucleotides with optimal reaction conditions we observed a significant increase in the transcription yield compared to our previous approach for IVT (Fig. 3B).
The ROCKET software has options that support the design of oligonucleotides that will be used to synthesize tRNA: (i) addition of a leader sequence for those tRNAs that do not start with a G, (ii) addition of an extra G at position −1 for tRNAHis, and (iii) addition of a CCA sequence at the 3′ end for those tRNA genes that do not possess this sequence. Of these options, the addition of a leader sequence can be used for any RNA. The leader sequence can be cleaved by RNase P in the presence of an external guide RNA (Forster and Altman 1990) that can form a double helix with the downstream sequence of the cleavage site in the target RNA. The guide RNA needs to have a 3′-CCA terminus for substrate recognition by RNase P (for more details, see Forster and Altman 1990). In addition, we demonstrated that the CPEB3 ribozyme prepared from DNA oligonucleotides obtained with ROCKET, is catalytically active (Fig. 9). Thus, our result showed that the software can be utilized for the selection of oligonucleotides for IVT of other small RNAs.
Despite the technological advances in solid-phase oligonucleotide synthesis, the length of error-free oligonucleotides remains limited to 100–200 nt (Kosuri and Church 2014). This size restriction also determines the maximum length of DNAs that are output by the ROCKET package. With 100 nt forward/reverse DNA oligonucleotides for synthesizing in vitro transcripts, the maximum length of these transcripts is estimated to be about 150 nt. However, such long DNA oligonucleotides are still not cost-effective and come with the risk of many DNA folding intermediates which would affect duplex formation. In this study, we applied the ROCKET strategy to fully automated oligonucleotide design for polymerase cycling assembly to synthesize the EGFP template DNA. The ROCKET-selected oligonucleotides were successfully assembled. While the EGFP protein could be synthesized by the E. coli cell-free transcription and translation system with the template, the current ROCKET strategy has not been compatible with eukaryotic cell-free translation systems, including wheat-germ and human systems (Sawasaki et al. 2002; Mikami et al. 2008), where the internal ribosome entry site (IRES) must be attached to mRNAs as well as codon-usage must be considered for good yields of target proteins. To address these issues, further development and optimization of the ROCKET package is needed.
In summary, we developed the ROCKET software for the automated selection of DNA oligonucleotides based on thermodynamic parameters of DNA structure. ROCKET is useful for designing DNA oligonucleotides for IVT of tRNA and other small RNAs.
MATERIALS AND METHODS
The ROCKET software
ROCKET was implemented in Python. As model genes for which DNA templates for IVT had to be prepared, we selected three different tRNAs (T. acidophilum tRNALeu, T. thermophilus tRNAPro, and T. kodakarensis tRNAHis, see Fig. 2A). The workflow to retrieve oligonucleotides for producing a DNA template is shown in Figure 2B. Central to the design strategy is seqfold (https://github.com/Lattice-Automation/seqfold), a Python package used for predicting the minimum free energy structure of nucleic acids. With seqfold incorporated in ROCKET, the software automates the selection procedure in the following five steps to find the optimal pair of oligonucleotides for a given sequence:
In Step 1, a list of all possible sites of overlap for forward/reverse oligonucleotides is generated from an input sequence.
The number of all possible annealing sites (Nall) for a DNA sequence of length n is given by Equation 1: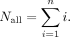 (1)
For example, if the input DNA sequence is 75 nt long, there can be 2850 different annealing sites in total.
(1)
For example, if the input DNA sequence is 75 nt long, there can be 2850 different annealing sites in total.
In Step 2, the melting temperature Tm (°C) is calculated for each annealing site using Equation 2: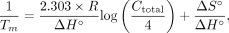 (2)
where R is the molar gas constant 1.9872 (cal•K−1•mol−1), Ctotal is the total concentration of oligonucleotide, and ΔS° and ΔH° are the standard entropy (kcal•mol−1•K−1) and standard enthalpy (kcal•mol−1) calculated from the nearest-neighbor model (SantaLucia and Hicks 2004). In addition, the Gibbs free energy (ΔG) at 37°C is calculated with Equation 3:
(2)
where R is the molar gas constant 1.9872 (cal•K−1•mol−1), Ctotal is the total concentration of oligonucleotide, and ΔS° and ΔH° are the standard entropy (kcal•mol−1•K−1) and standard enthalpy (kcal•mol−1) calculated from the nearest-neighbor model (SantaLucia and Hicks 2004). In addition, the Gibbs free energy (ΔG) at 37°C is calculated with Equation 3: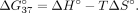 (3)
After calculation of Tm values, the program finds the annealing sites with a Tm value between 68°C and 72°C, only considering annealing sites ending with a G or C that can form more stable double-stranded
DNA according to the terminal-AT penalty rule (SantaLucia and Hicks 2004).
(3)
After calculation of Tm values, the program finds the annealing sites with a Tm value between 68°C and 72°C, only considering annealing sites ending with a G or C that can form more stable double-stranded
DNA according to the terminal-AT penalty rule (SantaLucia and Hicks 2004).
In Step 3, a set of forward/reverse oligonucleotides is designed based on the selected annealing sites. For the forward oligonucleotide, the T7 promoter sequence (5′-GCC TAA TAC GAC TCA CTA TA-3′) is attached to the 5′ end of the gene. The forward oligonucleotide ends at the 3′ end of the annealing site. As for the reverse oligonucleotide, the DNA sequence from the 5′ end of the annealing site to the 3′ end of the gene is reverse complemented.
In Step 4, the folding energy for each set of oligonucleotides is calculated by seqfold, utilizing the nearest-neighbor parameters for a DNA duplex in 1 M NaCl buffer, retrieved from previous reports (Zuker and Stiegler 1981; SantaLucia and Hicks 2004).
In Step 5, the optimal oligonucleotide set is selected; this set scores the highest ΔGtotal value among all sets where ΔGtotal is defined as the sum of ΔGforward oligo and ΔGreverse oligo. The set with the lowest ΔGtotal value is taken as the suboptimal set.
The selected oligonucleotides were purchased from Thermo Fisher Scientific at a synthesis scale of 50 nmol and validated by IVT experiments. The oligonucleotides have been desalted. In the experiments, Rocket-selected oligonucleotides are compared to those that have been used previously and for abbreviation purposes are called “published” (as is the case for the majority of these oligonucleotides; Yamagami et al. 2012; Kawamura et al. 2015). The oligonucleotides used in this research are provided in Supplemental Table S1.
For the transcription of the enhanced GFP gene (Zhang et al. 1996), we modified the ROCKET software so that we could design multiple DNA oligonucleotides. These oligonucleotides can be assembled into one piece by the polymerase cycling assembly method (Hoose et al. 2023). The modified version of the software performed the following selection procedure composed of six steps to find the optimal pair of 20 oligonucleotides (O1–O20) for the EGFP gene (Fig. 10A):
In Step 1, a list of all possible sites of annealing sites was generated from the EGFP sequence, and their melting temperatures were calculated by Equation 2. The software found the annealing sites end with a G or C with a Tm value of between 60°C and 63°C.
In Step 2, the software identified the longest annealing site (ASlong) from all annealing sites found in Step 1.
In Step 3, two oligonucleotides (O1 and O2) for the first 69 nt of the EGFP gene were designed. The software found annealing sites in the region using the same criteria with those used in the regular ROCKET software (Step 3a). Then, the software designed O1 and O2 oligonucleotides for all sets of annealing sites (Step 3b). The optimal oligonucleotide set was selected; this set must satisfy the following two criteria. (i) O2 oligonucleotide length is more than 2ASlong length determined in Step 2 (Step 3c-1). (ii) The oligonucleotide set has the highest ΔGtotal value (Step 3c-2).
In Step 4, an O3 oligonucleotide was designed to have an overlapping region with the O2 oligonucleotide. First, possible overlapping regions ending with the 3′ end of the O2 oligonucleotide with a Tm value of between 60°C and 63°C were identified. The annealing sites ending with G or C were then selected. If there is no G or C residue at the end of the annealing sites, the software selects the longest annealing site. Then, O3 oligonucleotide candidates having the annealing sites at the 5′ end were designed. Finally, the software calculated a ΔGO3 value for each candidate and selected the optimal O3 oligonucleotide that has the highest ΔGO3 value. This step was iterated for O4–O20 oligonucleotides. For O3–O20 oligonucleotides, the oligonucleotide lengths were designed to have 68 nt.
In Step 5, the Oeven number oligonucleotides were reverse complemented.
Preparation of T7 RNA polymerase
The pAR1219 plasmid encoding the T7 RNA polymerase gene was a gift from Dr. Takuya Ueda (Waseda University). A culture of E. coli BL21 (DE3) Rosetta2 transformed with pAR1219 was set up in 250 mL LB medium supplemented with 100 µg/mL ampicillin and grown at 37°C for 16 h, and then diluted with 1 L fresh LB. The E. coli cells were further grown at 37°C until OD600 ∼0.6–0.8, when expression of T7 RNA polymerase was induced by the addition of 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After another 4 h incubation at 37°C, the cells were harvested, and 3 g of cells were resuspended in 30 mL buffer A containing 50 mM potassium phosphate (pH 8.0), 20 mM NaCl, 1 mM EDTA, 10 mM 2-mercaptoethanol, 5% glycerol, and 1× protease inhibitor solution (Nacalai Tesque). Cells were broken by sonication with an ultrasonic disruptor (VCX-500, Sonics and Materials) on ice for 30 min with a 2 sec interval between pulses of 1 sec. After centrifugation at 10,000g at 4°C for 15 min, the cleared extract was loaded onto a HiTrap Q HP (5 mL; Cytiva) column pre-equilibrated with 25 mL buffer A. The unbound proteins were washed out with 25 mL buffer A. The bound T7 RNA polymerase was eluted with a linear gradient of NaCl from 20 to 500 mM in 50 mL buffer A. The fractions containing T7 RNA polymerase were loaded onto a HiTrap Heparin HP column pre-equilibrated with 25 mL buffer B containing 50 mM potassium phosphate (pH 8.0), 200 mM NaCl, 1 mM EDTA, 10 mM 2-mercaptoethanol, 5% glycerol. The unbound proteins were washed out with 25 mL buffer B. The bound T7 RNA polymerase was eluted with a linear gradient of NaCl from 200 mM to 2 M in 50 mL buffer B. The fractions containing T7 RNA polymerase were diluted with buffer A to decrease the concentration of monovalent ions to <500 mM. The diluted sample was loaded onto a HiTrap Blue HP column pre-equilibrated with 25 mL buffer C containing 50 mM potassium phosphate (pH 8.0), 500 mM NaCl, 1 mM EDTA, 10 mM 2-mercaptoethanol, 5% glycerol. The unbound proteins were washed out with 25 mL buffer C. The bound T7 RNA polymerase was eluted with a linear gradient of NaCl from 500 mM to 3 M in 50 mL buffer C. The purity of T7 RNA polymerase was monitored by 15% SDS-PAGE at each purification step. The purified T7 RNA polymerase was concentrated to 1 mg/mL storage buffer containing 50 mM Tris-HCl (pH 7.6), 50 mM KCl, 6 mM 2-mercaptoethanol, and 50% glycerol using Vivaspin 15R centrifugal filter units (Sartorius Stedim Biotech) and stored at −30°C. The purity of 2 µg T7 RNA polymerase was assessed by 10% SDS-PAGE and staining with Coomassie brilliant blue (Supplemental Fig. S1A).
Preparation of template DNAs
Template DNAs were prepared by a DNA polymerase extension reaction in an end-volume of 100 µL containing 1× Dream-Taq buffer (Thermo Fisher, EP0703), 0.2 mM dNTPs (TOYOBO, NTP-101), 1 µM forward oligonucleotide, 1 µM reverse oligonucleotide, and 0.025 U/µL Dream-Taq DNA polymerase (Thermo Fisher, EP0703). The polymerase extension reaction was done in a thermal cycler with the following program: 94°C for 2 min; 25 cycles of 94°C for 30 sec, 68°C for 30 sec, 72°C for 30 sec; 72°C for 5 min; 20°C hold. Purification of the template DNA is not necessary but can be done by phenol extraction followed by ethanol precipitation. In the DNA polymerase experiments performed in Figure 4D and F and RNA transcription experiments performed in Figure 5, the concentration of the DNA template was quantified. For quantification of DNA yields, the extended DNA template formed in the polymerase reaction was separated by 8% nondenaturing PAGE at 180 V. After staining the gel with SYBR gold, bands with double-stranded DNA template were excised. The DNA was eluted from the gel pieces in a buffer containing 500 mM ammonium acetate, 10 mM MgCl2, 1 mM EDTA, and 0.1% SDS at 25°C for 12 h and then recovered by ethanol precipitation. The yield was measured by UV absorbance at 260 nm.
In vitro transcription
In vitro transcription was performed in a 200 µL reaction containing 40 mM HEPES-KOH (pH 7.6), 20 mM MgCl2, 1 mM spermidine, 5 mM dithiothreitol, 50 µg/mL bovine serum albumin, 2.5 mM NTPs, 100 nM T7 RNA polymerase, and 500 nM template
DNA (or 100 µL of the reaction mixture from the DNA extension reaction), at 37°C for 240 min. The transcription yield was
monitored by 10% denaturing PAGE (7 M urea) of a 5 µL reaction mixture combined with 5 µL 2× loading dye containing 7 M urea
and 1× TBE. The RNA was visualized by toluidine blue staining. The relative transcription yield was calculated from band intensities
quantified with ImageJ. A control RNA was run on the same gel, and the band intensity of the control was set as 1.0. Standard
deviations were calculated from three independent experiments; P-values were calculated from the t-test. Data were fit to Equation 4: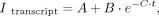 (4)
where Itranscript is the relative band intensity of tRNA transcript, A is the relative intensity of the transcript at completion, B is the amplitude of the observable phase, C is the rate constant for RNA transcription, and t is time. In the case of the CPEB3 IVT reaction, the mixture contained 50 mM MgCl2 to enable self-cleavage of the ribozyme. Samples of 10 µL were taken and mixed with 10 µL 2× loading dye at various time
points (0, 3, 5, 10, 30, 60, 120, and 240 min) for the PAGE analysis.
(4)
where Itranscript is the relative band intensity of tRNA transcript, A is the relative intensity of the transcript at completion, B is the amplitude of the observable phase, C is the rate constant for RNA transcription, and t is time. In the case of the CPEB3 IVT reaction, the mixture contained 50 mM MgCl2 to enable self-cleavage of the ribozyme. Samples of 10 µL were taken and mixed with 10 µL 2× loading dye at various time
points (0, 3, 5, 10, 30, 60, 120, and 240 min) for the PAGE analysis.
Electrophoretic mobility shift assay
Duplex formation between forward and reverse oligonucleotides was tested by EMSA. The concentration of the forward oligonucleotide
was kept the same in each sample (0.1 µM), and that of the reverse oligonucleotide was increased from sample to sample, from
an initial 0.01 µM to a final 0.1 µM (see Fig. 3C or Supplemental Fig. S4). Forward and reverse oligonucleotides were denatured in 1× Dream-Taq buffer at 94°C for 3 min and annealed at 68°C for 5
min. The DNAs were mixed with a nondenaturing loading dye and immediately separated by 8% nondenaturing PAGE. The DNA was
visualized by SYBR gold staining and band intensities were quantified with ImageJ. Standard deviations were calculated from
three independent experiments; P-values were calculated from the t-test. Duplex-formation was quantified by ImageJ. Data were fit to Equation 5: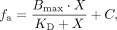 (5)
where fa is the fraction annealed oligonucleotide, Bmax is the maximum annealed, KD is the dissociation constant, X is the concentration of the oligonucleotide, and C is a constant. The fraction of annealed oligonucleotide was calculated from Equation 6:
(5)
where fa is the fraction annealed oligonucleotide, Bmax is the maximum annealed, KD is the dissociation constant, X is the concentration of the oligonucleotide, and C is a constant. The fraction of annealed oligonucleotide was calculated from Equation 6: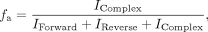 (6)
where IForward, IReverse, and IComplex are the band intensities of forward, reverse, and annealed oligonucleotides, respectively.
(6)
where IForward, IReverse, and IComplex are the band intensities of forward, reverse, and annealed oligonucleotides, respectively.
Oligonucleotide design by Primerize
Forty-six sets of oligonucleotides for T. kodakarensis tRNA genes were designed with Primerize webserver at https://primerize.stanford.edu/. The advanced options (minimum Tm of 68°C, maximum length of 100 nt, and minimum length of 7 nt) were used. Primerize automatically attaches the T7 promoter sequence with 5′ flanking nucleotides of TTC (5′-TTC TAA TAC GAC TCA CTA TA-3′) to the forward oligonucleotides. The flanking nucleotides of TTC were then converted into GCC because ROCKET uses those 5′ flanking nucleotides. Then, the folding energies were calculated by seqfold.
Preparation of RNase P
The C5 protein and M1 RNA are the components of RNase P1 (Fukunaga et al. 2006). RNase P cleaves precursor tRNAs with a reader sequence at their 5′ end. RNase P is useful for the transcription of those tRNA genes that do not start with a G. Plasmids pETC5 and pUCM1 encoding the C5 protein and M1 RNA, respectively, were kind gifts from Dr. Takashi Yokogawa (Gifu University). A detailed protocol for the preparation of RNase P1 has been published (Fukunaga et al. 2006). In short, as described for T7 RNA polymerase, the C5 protein was expressed in the Rosetta2 strain at 37°C for 4 h after the addition of 0.5 mM IPTG. After harvesting, 3g cells were resuspended in 15 mL buffer D containing 50 mM Tris-HCl (pH 7.6), 1 M NaCl, 5 mM EDTA, 10% glycerol, and 1× protease inhibitor solution, and sonicated on ice for 15 min with a 3 sec interval between 2 sec pulses. After centrifugation at 10,000g at 4°C for 15 min, ammonium sulfate was added to the supernatant to 50% saturation. The solution was centrifuged at 10,000g at 4°C for 15 min and ammonium sulfate was added to the supernatant to 80% saturation, and the insoluble fraction was collected by centrifugation at 10,000g at 4°C for 15 min. The precipitate was dissolved in buffer E containing 50 mM sodium acetate (pH 6.5), 5 mM EDTA, and 250 mM NaCl. To remove ammonium sulfate, the sample solution was dialyzed against buffer E. The dialyzed solution was loaded onto a HiTrap SP column pre-equilibrated with 25 mL buffer E. The bound protein was eluted with a linear gradient of NaCl from 250 mM to 1 M in 50 mL buffer E. With Vivaspin 15R centrifugal filter units, the purified C5 protein was concentrated to 1.5 mg/mL storage buffer containing 20 mM Tris-HCl (pH 7.6), 1 mM MgCl2, 40 mM KCl, 6 mM 2-mercaptoethanol, and 50% glycerol. The C5 protein (Supplemental Fig. S1B) was stored at −30°C. The M1 RNA component of RNase P was prepared by IVT using a DNA template generated by PCR using plasmid pUCM1 and primers 5′-GGG GCT GCA GTA ATA CGA CTC ACT ATA-3′ and 5′-AGG TGA AAC TGA CCG ATA AG-3′. The transcribed M1 RNA was purified by 6% denaturing PAGE (7 M urea) (Supplemental Fig. S1C).
RNase P assay
The RNase P reaction was performed in a 100 µL reaction containing 40 mM HEPES-KOH (pH 7.6), 20 mM MgCl2, 1 mM spermidine, 5 mM dithiothreitol, 50 µg/mL bovine serum albumin, 10 µM C5 protein, 10 µM M1 RNA, and 1.7 µM tRNA at 37°C for 60 min. The cleaved tRNA was analyzed by 10% denaturing PAGE (7 M urea).
Preparation of EGFP DNA template
The O1–O20 oligonucleotides for EGFP designed by the software (see the ROCKET software section) were assembled by two rounds of PCR. The first PCR was performed in a 50 µL reaction containing 1×PCR buffer for KOD-Plus-Neo (TOYOBO), 200 µM dNTPs, 1.5 mM MgSO4, 0.3 µM oligonucleotides (FOR primer and REV primer), 2 nM each O1–O20 oligonucleotide, and 1.0 U KOD-Plus-Neo (TOYOBO) with the following program: 98°C for 2 min; 30 cycles of 98°C for 10 sec; 68°C for 60 sec; 68°C for 5 min; 20°C hold. The second PCR was performed in a 50 µL reaction containing 1×PCR buffer for KOD-Plus-Neo (TOYOBO), 200 µM dNTPs, 1.5 mM MgSO4, 0.3 µM oligonucleotides (T7PRO-SD primer and REV primer), 1 µL of the first PCR product, and 1.0 U KOD-Plus-Neo (TOYOBO) with the following program: 98°C for 2 min; 30 cycles of 98°C for 10 sec, 68°C for 60 sec; 68°C for 5 min; 20°C hold. The sequences of FOR, T7PRO-SD, and REV primers are provided in Supplemental Table S1. The final product was purified by agarose gel and used for IVT and translation experiments.
In vitro transcription and translation of the EGFP template DNA
For the IVT and translation experiment, the E. coli pure system (Shimizu et al. 2001) (PUREfrex2.0, PF201-0.25, GeneFrontier) was used according to the manufacturer's instruction. In brief, the reaction was performed in a 20 µL containing 10 µL Solution I, 1 µL Solution II, 2 µL Solution III, and 60 ng gel-purified EGFP template DNA at 37°C for 13 h. The EGFP synthesis was monitored with the GloMax Discover System (GM3000, Promega).
DATA DEPOSITION
The source code of the ROCKET algorithm is available on GitHub (https://github.com/TEPPEI-MAT/ROCKET). All raw data obtained in this study are available upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Dr. Takuya Ueda (Waseda University) for plasmid pAR1219, Dr. Takashi Yokogawa (Gifu University) for plasmids pETC5 and pUCM1, and Professor Kazuyuki Takai for technical suggestions. This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) (22K15035 to R.Y.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079923.123.
- Received December 10, 2023.
- Accepted February 14, 2024.
This article is distributed exclusively by Cold Spring Harbor Laboratory Press for the first six months after the full-issue publication date (see http://genome.cshlp.org/site/misc/terms.xhtml). After six months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Teppei Matsuda is the first author of this paper, “Rational design of oligonucleotides for enhanced in vitro transcription of small RNA.” Teppei is working on his master's degree under the supervision of Dr. Ryota Yamagami and Dr. Hiroyuki Hori at Ehime University in Japan. His research focuses on the development of a Python tool that designs optimal DNA oligonucleotides for in vitro RNA transcription.
What are the major results described in your paper and how do they impact this branch of the field?
This paper describes an easy and efficient pipeline for designing a set of DNA oligonucleotides for in vitro transcription with T7 RNA polymerase, based on thermodynamic parameters and prediction of DNA secondary structure, which was fully automated by Python. We show that our tool outputs a set of DNA oligonucleotides optimized for in vitro transcription in seconds (depending on the computer's performance). These DNA oligonucleotides are extended by DNA polymerase to make double-stranded DNA templates. This DNA extension reaction as well as in vitro transcription can be performed in a one-pot reaction. While we focused on short RNAs such as tRNA and ribozyme in this paper, our tool is broadly applicable to other RNAs.
What led you to study RNA or this aspect of RNA science?
The Hori lab is interested in how RNA modification regulates gene expression. To address this, we especially focus on tRNA and tRNA methylation. The Hori lab has reported many important findings, including the identification of a tRNA methyltransferase that has a novel methyltransferase domain and tRNA modification network (or tRNA modification circuit) in thermophilic bacteria. Since I joined this lab in 2022, I've been studying tRNA methylation and finding myself enjoying working on my projects in the lab. It is fascinating to explore the diverse roles of tRNA modification in many cellular events, including quality control of protein synthesis, regulation of codon–anticodon recognition, and stabilization/destabilization of RNA structure. It has been enough to keep me hooked. In 2022 and 2023, I attended international conferences and meetings of the RNA Society Japan and heard cutting-edge RNA-related talks, where I realized that my enthusiasm for RNA was still growing. I believe that this paper makes a significant contribution to the routine process of preparing RNAs of interest in your lab. It is my hope that this technical advancement will save researchers time and cost in preparing RNA materials, which would eventually allow them to focus more on addressing RNA-related biological questions.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
While our lab works on tRNA, some of the lab members have struggled with designing DNA oligonucleotides for in vitro tRNA transcription. It took them about 20 minutes to design a set of DNA oligonucleotides (for only one tRNA sequence!), and they sometimes made mistakes in the design, resulting in low yields of RNA transcription. In April 2023, I began developing this Python tool to meet the demands of lab members who wanted a fully automated, time-saving, and rational method for designing the oligonucleotides for in vitro transcription. I found that not only does the Python software work great, but also the optimal DNA oligonucleotides designed by the software significantly increase the final RNA yield, due to the improved yield of the DNA template. This conclusion seemed to be very simple, but it took me a long time to prove this (I performed countless in vitro transcription experiments…). Additionally, as an application, I designed multiple DNA oligonucleotides for the EGFP gene (∼10 times longer than tRNA genes) using the software described in this paper. However, there are still many areas for improvement in this regard, which is one of my current research projects.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
First, I would like to express my heartfelt gratitude to the university and the Hori lab for giving me the opportunity to challenge myself and engage in RNA research as a student. I am particularly grateful to my colleagues, who have not only been companions in learning, challenging, discussing, and researching together but also wonderful friends. My supervisors, Drs. Hiroyuki Hori and Ryota Yamagami, have influenced me a lot. Dr. Hori taught me the importance of communicating with many people, broadening perspectives, thinking rationally, formulating hypotheses, and persistently advancing research. These are fundamental and essential skills applicable in any field beyond scientific research. Ryota has been training me since 2022, and he taught me his expertise in RNA-related techniques from biochemistry and molecular biology to bioinformatics. Also, I learned project management skills from him. There is no doubt that their approach to RNA science has contributed to this paper.








