
SCARPET: site-specific quantification of methylated and nonmethylated adenosines reveals m6A stoichiometry
- 1Department of Pharmacology, Weill Cornell Medicine, Cornell University, New York, New York 10065, USA
- 2Division of Gastroenterology and Hepatology, Department of Medicine, Weill Cornell Medicine, Cornell University, New York, New York 10065, USA
- 3Department of Physiology Biophysics and Systems Biology, Weill Cornell Medicine, Cornell University, New York, New York 10065, USA
- Corresponding author: srj2003{at}med.cornell.edu
-
Handling editor: Eric Phizicky
Abstract
m6A has different stoichiometry at different positions in different mRNAs. However, the exact stoichiometry of m6A is difficult to measure. Here, we describe SCARPET (site-specific cleavage and radioactive-labeling followed by purification, exonuclease digestion, and thin-layer chromatography), a simple and streamlined biochemical assay for quantifying m6A at any specific site in any mRNA. SCARPET involves a site-specific cleavage of mRNA immediately 5′ of an adenosine site in an mRNA. This site is radiolabeled with 32P, and after a series of steps to purify the RNA and to remove nonspecific signals, the nucleotide is resolved by TLC to visualize A and m6A at this site. Quantification of these spots reveals the m6A stoichiometry at the site of interest. SCARPET can be applied to poly(A)-enriched RNA, or preferably purified mRNA, which produces more accurate m6A stoichiometry measurements. We show that sample processing steps of SCARPET can be performed in a single day, and results in a specific and accurate measurement of m6A stoichiometry at specific sites in mRNA. Using SCARPET, we measure exact m6A stoichiometries in specific mRNAs and show that Zika genomic RNA lacks m6A at previously mapped sites. SCARPET will be useful for testing specific sites for their m6A stoichiometry and to assess how m6A stoichiometry changes in different conditions and cellular contexts.
Keywords
INTRODUCTION
m6A is the most abundant nucleotide modification in mRNA, and has important roles in determining mRNA stability (Sommer et al. 1978; Wang et al. 2015; Zaccara and Jaffrey 2020). m6A is installed in mRNA during or shortly after transcription (Sommer et al. 1978; Ke et al. 2017) by the heterodimeric methyltransferase METTL3–METTL14 (Wang et al. 2018). Abnormalities in methylation have been linked to diverse diseases (Jiang et al. 2021). The advent of m6A mapping technologies (Dominissini et al. 2012; Meyer et al. 2012; Linder et al. 2015) has resulted in a plethora of transcriptome-wide m6A maps in different tissues and cell and disease contexts, providing insights into how m6A can affect cellular functions.
Some m6A mapping studies have reported m6A “dynamics.” A dynamic m6A site can be defined as a nucleotide position in which the fraction of methylated adenosine at a specific site in a specific mRNA changes. The change in methylation can be elicited by a specific signal, or can be seen when comparing methylation in different cellular contexts, such as different tissues or disease states. Dynamic m6A sites have been identified when m6A “peaks” are higher in one condition than another. A major problem is that m6A peak heights can vary substantially even between biological replicates (McIntyre et al. 2020). Thus, peak height changes might not reflect m6A dynamics. It is therefore important to biochemically validate the stoichiometry changes in m6A at specific sites using antibody-independent methods (Grozhik and Jaffrey 2018).
Several methods have been developed for quantitative site-specific modification analysis (Yu et al. 1997; Zhao and Yu 2004; Dai et al. 2007; Hengesbach et al. 2008; Sednev et al. 2018), with SCARPET (site-specific cleavage and radioactive-labeling followed by ligation-assisted extraction and thin-layer chromatography) being the most prominent method for quantifying m6A in mRNA (Liu et al. 2013). SCARLET involves site-specific cleavage of mRNA exactly 5′ to the methylation site using a modified “SCARLET” oligonucleotide. The SCARLET oligonucleotide, referred to henceforth as the SCARPET oligonucleotide in the context of SCARPET experiments, hybridizes in a sequence-specific manner to a specific target mRNA. The SCARPET oligonucleotide contains deoxynucleotides at specific positions such that ribonuclease H (RNase H) will recognize the DNA–RNA hybrid region, and will cleave the mRNA at the 5′ side of the site of interest. After cleavage at a suspected methylated adenosine, the m6A or A at this site can be 5′ radiolabeled, and through a series of purification steps, the m6A and A are resolved by thin-layer chromatography (TLC), which also allows quantification of the amount of both m6A and A (Liu et al. 2013).
SCARLET is problematic since the protocol requires 3 d prior to TLC and involves a low-efficiency ligation step and gel purification steps that cause sample loss. These steps make the overall protocol difficult, which likely accounts for its infrequent use in the literature despite the importance of quantifying m6A stoichiometry at specific sites in specific mRNAs.
Here, we describe SCARPET (site-specific cleavage and radioactive-labeling followed by purification, exonuclease digestion, and thin-layer chromatography), a simple and streamlined biochemical assay for quantifying m6A at any specific site in any mRNA. SCARPET uses the oligonucleotide approach of SCARLET to cleave mRNA at the m6A site for radiolabeling and ultimately for TLC. However, SCARPET uses a series of new steps to remove nonspecifically labeled RNAs and an optimized hairpin oligonucleotide method to selectively capture the radiolabeled RNA of interest. The resulting protocol does not require gel purification or splint ligation steps, and therefore is efficient and rapid. SCARPET requires only a single day for sample processing, and additional time for TLC and for mRNA purification. SCARPET thus provides a biochemical method to confirm m6A dynamics by allowing the stoichiometry of m6A to be determined at specific locations in specific mammalian mRNAs.
RESULTS
Design of the SCARPET assay
SCARLET is particularly powerful for quantifying m6A stoichiometry because both A and m6A at a specific nucleotide position can be seen as separate spots on a TLC plate and thus can be quantified and compared (Liu et al. 2013). The ability to see both A and m6A as spots gives considerable reassurance that the measured signals are likely to be accurate.
Although SCARLET is very powerful, it requires a multiday protocol that utilizes splint ligation steps and gel extraction steps that are time consuming and associated with considerable RNA losses. For this reason, the clearest SCARLET TLC images have been produced when measuring m6A in highly abundant RNAs such as ribosomal RNA (rRNA) and the noncoding RNA MALAT1 (Liu et al. 2013). Therefore, we developed SCARPET, a protocol that diverges from the SCARLET protocol in order to provide more rapid and robust detection of m6A stoichiometry in mRNA (Fig. 1A).
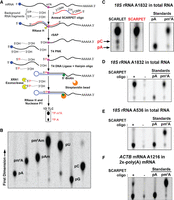
SCARPET reveals m6A stoichiometry at specific sites in RNA. (A) Schematic outline of SCARPET assay. (B) Diagram of one-dimensional thin-layer chromatography (1D-TLC) mobility maps of 5′-monophosphate nucleoside standards of adenosine (A) and its methylated derivates, uridine (U), cytosine (C), and guanosine (G) derived from synthetic RNA oligonucleotides after 32P-labeling, followed by nuclease P1 digestion. (C) Comparison of SCARLET and SCARPET results for determining the m6A stoichiometry at human 18S rRNA A1832 site in 1 µg HEK293T total RNA. The m6A stoichiometry measured by SCARPET and SCARLET assay was the same (∼98%), except the SCARLET assay showed an additional background spot corresponding to cytosine, which was not present with SCARPET. (D) The m6A signal is SCARPET oligonucleotide dependent. When the SCARPET oligonucleotide for human 18S rRNA A1832 is omitted, the m6A signal is completely lost. (E) SCARPET results for human 18S rRNA A536 site, which is not methylated but is found in a DRACH sequence context. SCARPET accurately revealed the m6A stoichiometry at the A536 site to be 0%. (F) SCARPET results for ACTB mRNA A1216 site in 2×-poly(A) HEK293T mRNA. The m6A stoichiometry at A1216 was ∼29%, which is consistent with previous measurements with SCARLET (Liu et al 2013). 32P-labeled m6A and A nucleosides were run as TLC standards in C–F.
SCARLET has several steps which result in considerable loss of material. The first of these is a splint ligation step that is used to ligate a DNA oligonucleotide to the desired mRNA after cleavage of the RNA and phosphorylation with [γ-32P]-ATP. This step uses a splint oligonucleotide to join the primer and the cleaved mRNA. However, formation of a ternary complex has low efficiency (Kurschat et al. 2005; Maroney et al. 2008).
Therefore, in SCARPET we modified this step to make it a binary interaction, using a “self-splinting” hairpin oligonucleotide based on previously used design parameters (Bordoni et al. 2002; Dittmar et al. 2006). The 5′-biotinylated self-splinting DNA hairpin (5Bio-ssHP) oligonucleotide acts as both the splint and also provides a 3′-OH ligation site in a single DNA (Fig. 1A). SCARPET is a one-pot assay which reduces the loss of material at each step in contrast to SCARLET.
The next step of SCARLET involves digestion of the RNA with RNases T1 and A. This causes the RNA to be fully digested at the 3′ side of every ribonucleotide. At the end of this digestion, the DNA oligonucleotide is attached to the radiophosphorylated m6A or A residue. Next, the DNA oligonucleotide linked to the m6A or A is purified by polyacrylamide gel electrophoresis followed by purification by gel extraction. This gel extraction is time consuming and is associated with loss of material, so we eliminated the step by incorporating a biotin on the hairpin oligonucleotide. Thus, in SCARPET the 32P-labeled m6A or A-containing RNA fragment is ligated to the 5′-biotinylated self-splinting hairpin oligonucleotide and is rapidly purified on streptavidin-coated magnetic beads (Fig. 1A). This step improves efficiency and removes the two most time-consuming steps of SCARLET.
To lower background signals, we incorporated a step to remove the 5′ radiolabeled phosphates that are not on the site of interest. To do this, we treat the RNA with Terminator, a 5′-phosphate-dependent exonuclease. This 5′ → 3′ exonuclease removes any 5′ [32P]-labeled phosphate that is not ligated to the hairpin oligonucleotide, thus reducing background signals and facilitating the detection of specific signals deriving from the mRNA site of interest.
Lastly, after DNA and RNA digestion, TLC is used as in the original SCARLET protocol to visualize and quantify both the unmethylated and methylated adenosine.
SCARPET selectively detects m6A in rRNA and in ACTB mRNA
We first tested SCARPET on the m6A site (A1832) in the 18S rRNA (Choi and Busch 1978; Maden 1986). The SCARPET oligo contains four deoxyribonucleotides, with the 3′-most deoxyribonucleotide complementary to the nucleotide upstream of the adenosine of interest in the mRNA. Similar to the SCARLET oligonucleotide, eight 2′-O-methylated nucleotides complementary to the target mRNA are added to either side of the four-deoxynucleotide region. Thus, the overall length of the SCARPET oligo is 20 nt. The SCARPET oligo was annealed to total cellular RNA (1 µg) from HEK293T cells. RNase H was used to site-specifically cleave the target RNA.
After the RNase H cleavage and dephosphorylation step, the 5′ ends of RNA were radiolabeled by phosphorylation with [γ-32P]-ATP and T4 polynucleotide kinase (T4 PNK). We next ligated the 5′’-biotinylated self-splinting DNA hairpin oligonucleotide. This hairpin protects the radioactive phosphate on A1832 during the subsequent Terminator exonuclease step, and also provides a biotin handle for subsequent purification. We designed the hairpin nucleotide based on previous DNA hairpin designs (Bordoni et al. 2002; Dittmar et al. 2006). The DNA hairpin oligonucleotide was complementary to the 18S RNA beginning at A1832 and the subsequent 29 nt 3′ of A1832. The hairpin comprises a 12-bp stem and a 6-nt loop, similar to a previously described hairpin oligonucleotide (Bordoni et al. 2002; Dittmar et al. 2006).
After ligating the DNA hairpin, the DNA–RNA complex was purified on streptavidin magnetic beads. Any contaminating RNA that lacks a hairpin oligo is degraded by incubation with Terminator exonuclease. After washing the beads, the radioactive A1832 is released by incubating the beads with RNase H (1 U) and followed by nuclease P1 (2 U). The hydrolysate was directly analyzed by TLC. We first confirmed that TLC could resolve each of the major nucleotides in RNA: A, G, C, and U, as well as m6A and other methylated forms of adenosine found in RNA (Am and m6Am) (Fig. 1B). We found that the solvent conditions allowed all of these nucleotides to be readily resolved from each other.
We next compared SCARLET and SCARPET for measuring m6A and A levels at A1832 in the 18S rRNA (Fig. 1C). Using both methods, we found a major spot on the TLC plate that co-migrated with an m6A standard, with a substantially lower intensity spot migrating with the adenosine standard (Fig. 1C). Thus, A1832 was ∼98% methylated using either method, consistent with previous results using SCARLET (Liu et al. 2013).
We noticed major differences in SCARPET versus SCARLET. First, the SCARLET assay required 3 d before the TLC step, while the SCARPET protocol was completed in 1 d prior to the TLC step. Second, the amount of recovered RNA was less with the SCARLET assay as evidenced by the longer exposure time needed to image the spots. These data are consistent with the idea that the steps in SCARPET are completed more quickly, result in fewer nonspecific signals, and increase the specific RNA recovery and signal.
We next confirmed that the TLC spots obtained with SCARPET were specific. To test this, we performed a parallel SCARPET experiment in which we left out the SCARPET oligonucleotide, which resulted in a complete loss of the m6A signal (Fig. 1D). Additionally, leaving out the self-splinting hairpin oligo caused a complete loss of the m6A signal (Supplemental Fig. 1A). As an additional control, leaving out the input RNA (1 µg) resulted in a loss of both A and m6A spots. These controls suggest that the signal is highly specific and show relatively low background for this site. As an additional control, we used SCARPET to measure m6A stoichiometry at position A536, an adenosine residue in the 18S RNA that is not methylated but is found in a DRACH (D = A, G, U; R = A, G; H = A, C, U) sequence context typical of m6A sites in mRNA (Linder et al. 2015). Here we saw only the spot corresponding to A (Fig. 1E). Taken together, these results suggest that SCARPET specifically detects m6A and A in the 18S RNA.
SCARPET detects m6A in mRNA
rRNA is highly abundant. We therefore wanted to determine if m6A can be detected using lower amounts of total RNA. For these experiments, we performed SCARPET using 0.1, 0.25, 0.5, and 1 µg total RNA. The TLC was imaged after exposing the plate for 12 h. Using this exposure time, we readily detected m6A in all conditions and found increased signal intensity for the m6A and A spots with increasing input RNA (Supplemental Fig. 1B). This increase corresponded linearly with the input RNA (Supplemental Fig. 1C).
We next asked if SCARPET can quantify m6A at specific sites in an mRNA. For these experiments, we prepared twice-purified poly(A) RNA [2×-poly(A)] from HEK293T cells and used 1 µg in these assays. We selected A1216 in ACTB mRNA. This site has been shown to be ∼21% methylated in HeLa (Liu et al. 2013) and ∼28% methylated in MDA-MB-231 cells by SCARLET (Liu et al. 2013). Indeed, SCARPET was able to detect the methylation levels at A1216 in ACTB mRNA (Fig. 1F). We found the methylation levels were ∼29%, which is consistent with the results using SCARLET (Liu et al. 2013). We also applied SCARPET to TUBB4B, TUBA1B, RACK1, and ACTG1, and detected m6A at each tested site (Supplemental Fig. 2A,B). The abundance of these mRNAs is at similar or lower levels compared to the ACTB in HEK293T cells (Supplemental Fig. 2C; Lachmann et al. 2018). Overall, these data suggest that SCARPET can quantify m6A and A levels at specific sites in mRNA.
Optimization of the steps of SCARPET
In order for SCARPET to provide reliable results, it is important to ensure that each of the enzymatic steps goes to completion. These steps include: SCARPET oligo cleavage of target RNA, 5′ end phosphorylation, and ligation of the hairpin oligonucleotide. If these steps do not go to completion, there could be sequence-dependent biases that might affect the measurement of m6A and A. For example, RNase H cleavage efficiency may be altered when m6A is present, or T4 PNK may have a higher efficiency of 5′ phosphorylation when the 5′ nucleotide is m6A compared to A. We therefore optimized each of these steps of SCARPET using both A1832 in 18S rRNA and A1216 in ACTB mRNA. Importantly, SCARPET oligonucleotide hybridization and the PNK phosphorylation steps were previously optimized in SCARLET (Liu et al. 2013). Nevertheless, we performed optimization steps to confirm the efficiency of each step in the SCARPET protocol. Each step was optimized as follows.
Site-specific cleavage using RNase H and the SCARPET oligonucleotide
The SCARPET oligonucleotide is identical to the oligonucleotide previously used in SCARLET. This oligonucleotide is 20 nt in length, comprising eight 2′-O-methyl-modified nucleotides, followed by four deoxy nucleotides, followed by another eight 2′-O-methyl-modified nucleotides. All 20 nt hybridize to the target mRNA, and the 3′-most deoxynucleotide is complementary to the mRNA nucleotide immediately upstream of the m6A, thus allowing the RNA to be cleaved on the 5′ side of the m6A. The length and design were previously optimized in studies of using RNase H to mediate sequence-specific cleavage of mRNA (Bordoni et al. 2002; Kurschat et al. 2005; Dittmar et al. 2006; Maroney et al. 2008).
We therefore optimized the time required for RNase H-mediated cleavage of target RNA using the SCARPET oligonucleotide. In these experiments, we asked if the reaction conditions (1 µg RNA, 20 ng [3 pmol] SCARPET oligonucleotide, 1 U RNase H, 1 h at 44°C) were sufficient for the reaction to go to completion. To test this, we compared the 1 h digestion protocol to a protocol comprising the same amount of RNase H, but with further digestion for 2 h, as well as a second protocol in which we added additional RNase H after 1 h and continued incubation for an additional 1 h. We reasoned that if the reaction had not gone to completion, either of these treatments might result in an increased amount of radiolabeled A and m6A. However, neither of these treatments resulted in increased total levels of A and m6A when using SCARPET to assay methylation stoichiometry at A1832 in the 18S rRNA using total RNA from HEK293T cells (Fig. 2A). Similar results were obtained using SCARPET on A1216 in ACTB mRNA with 1 µg of 2×-poly(A) mRNA (Fig. 2B). Thus, we used 1 h at 44°C for the RNase H-mediated cleavage step using the SCARPET oligonucleotide.
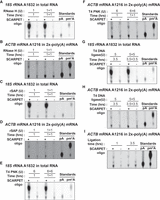
Optimization of SCARPET assay steps. (A,B) RNase H step (A) and (B) optimization of the RNase H-assisted cleavage step in SCARPET by either doubling the amount of RNase H enzyme or the time of incubation compared to standard RNase H reaction conditions (1 U RNase H, 1 h incubation at 44°C). Neither increasing the amount of enzyme nor time of incubation increased total levels of A and m6A at either of these sites. (C,D) rSAP step (C) and (D) Optimization of dephosphorylation step by rSAP enzyme in SCARPET by either doubling the amount of rSAP enzyme or the time of incubation. Compared to standard rSAP reaction conditions (1 U rSAP, 1 h incubation at 44°C), neither increasing the amount of enzyme nor time of incubation resulted in increased levels of A and m6A at either of these sites. (E,F) PNK radiolabeling step (E) and (F) Optimization of radiolabeling by T4 PNK enzyme in SCARPET by either doubling the amount of T4 PNK enzyme or the time of incubation. Compared to standard radiolabeling reaction conditions (6 U T4 PNK, 10 µCi [γ-32P]-ATP, 1 h incubation at 37°C), neither increasing the amount of enzyme nor time of incubation resulted in increased levels of A and m6A at either of these sites. (G,H) Self-splinting hairpin-assisted ligation step (G) and (H) Optimization of ligation step by T4 DNA ligase enzyme in SCARPET by either doubling the amount of T4 DNA ligase enzyme or the time of incubation. Compared to standard ligation reaction conditions (5 U T4 DNA ligase, 3.5 h incubation at 37°C), neither increasing the amount of enzyme nor time of incubation resulted in increased levels of A and m6A at either of these sites. Notably, prolonged ligation seemed to increase the amount of the A spot, which likely reflects increased nonspecific background ligation. (I) Shortening the ligation time to only 1 h leads to a decrease in the background level of A suggesting that 1 h was sufficient for ligation, and prolonged ligation times could increase background signals. The RNA input for all these experiments was 1 µg total RNA for 18S rRNA A1832 site and 1 µg 2×-poly(A) mRNA for ACTB mRNA A1216 site from HEK293T cells and 20 ng of SCARPET oligo was used in all experiments except in the controls where it was omitted. 32P-labeled m6A and A nucleosides were run as TLC standards in A–I.
Dephosphorylation step
After RNase H-mediated cleavage of the target RNA, the 5′ end is dephosphorylated using rSAP to facilitate subsequent phosphorylation by T4 PNK. This reaction (1 U rSAP, 44°C) is performed for 1 h. We compared the yield of radiolabeled A and m6A of the standard protocol with a protocol involving 2 h rSAP treatment, or a protocol involving the initial 1 h of rSAP treatment followed by additional rSAP (1 U) and an additional 1 h treatment. Again, neither of these treatments resulted in an increased level of A and m6A at either A1832 in the 18S rRNA or A1216 in ACTB mRNA (Fig. 2C,D).
PNK radiolabeling step
We next optimized the radiolabeling of the 5′ end, which first requires heat inactivation of rSAP using a standard protocol (Liu et al. 2013). After this step, we add T4 PNK and [γ-32P]-ATP. In this 1 h reaction (6 U T4 PNK, 10 µCi [γ-32P]-ATP, 37°C), we saw no further increase in labeling with further reaction time or further reaction time and enzyme (Fig. 2E,F).
Self-splinting hairpin oligonucleotide-assisted ligation step
We also optimized the ligation step by either increasing the amount of T4 DNA ligase or increasing the incubation time of the ligation reaction. Here we compared the initial reaction conditions (3.5 h ligation, 5 U T4 DNA) to a protocol in which we added an additional 5 U T4 DNA ligase after 3.5 h and continued incubation for an additional 3.5 h. However, these prolonged treatments did not increase the total amount of m6A at A1832 in the 18S rRNA and A1216 in ACTB mRNA (Fig. 2G,H). However, we noticed that prolonged ligation instead seemed to increase the amount of the A spot, which would reflect increased nonspecific background ligation. We therefore tested shortening the ligation time to only 1 h (Fig. 2I). With this shortened time, the background level of A was reduced, suggesting that 1 h was sufficient for ligation, and nonspecific ligation could increase background signals (Fig. 2I). Therefore, 1 h is preferred for this step.
We also tested other ligases besides T4 DNA ligase. However, SplintR and 9°N DNA ligases each failed to produce a ligated product (Supplemental Fig. 1D), suggesting that they cannot ligate the RNA to the DNA hairpin oligonucleotide.
Optimization of the hairpin oligonucleotide
We next wanted to understand the sequence requirements for the hairpin oligo, which comprises a stem–loop structure and a splint sequence that is complementary to the target RNA. We first asked if the stem was stable enough to ensure efficient ligation. To test this, we swapped an A•T base pair near the base of the stem with a C•G base pair (5′Bio-ssHP1). This hairpin oligo did not result in a noticeable increase in m6A (Supplemental Fig. 3A,B), suggesting that the stem stability was likely sufficient for ligation.
The hairpin oligo contains a T at the position opposite to the m6A or A in the RNA. We tested if base-pair complementarity between the splint and the target RNA at the ligation junction was necessary for efficient ligation. To test this, we introduced three mismatches in the splint region near the ligation junction 5′ → 3′ [TGT → ACA] (5Bio-ssHP2). This oligo (5′Bio-ssHP2) produced a markedly lower m6A spot (50% reduction relative to the control oligo [5′Bio-ssHP]) (Supplemental Fig. 3A–C). Thus, correct base-pairing at the ligation junction is needed for efficient ligation.
The hairpin oligo uses 29 nt splint to hybridize to the target mRNA. We tested shorter hybridization domains. We noticed markedly lower m6A levels after shortening the hairpin to 24 nt (5′Bio-ssHP3) and 14 nt (5′Bio-ssHP4) (Supplemental Fig. 3D,E). Thus, even a loss of 5 nt markedly reduces ligation efficiency. Overall, these experiments suggest that the current hairpin oligo design is the best oligo compared to the other tested sequences.
SCARPET accurately predicts m6A stoichiometry using methylated RNA standards
We next assessed the quantitative accuracy of SCARPET. For these experiments, we prepared mixtures of a synthetic 42-nt long RNA oligonucleotide containing a single m6A (m6A-RNA42) or an A (A-RNA42). In these RNAs, the m6A is in the common GGACU consensus site for m6A (A13). The RNA oligonucleotides were then mixed so that the m6A stoichiometry ranged from 0% to 100%. The amount of synthetic RNA mixtures ranged from 0 to 2.7 ng, with 2.7 ng corresponding to 200 fmol total of the synthetic RNA amount. The synthetic RNA mixture was then added to 1 µg of 2×-poly(A) HepG2 mRNA. The SCARPET-predicted m6A stoichiometries were similar to the actual stoichiometries (Fig. 3A,B). Thus, SCARPET shows linear and accurate detection of m6A stoichiometry.
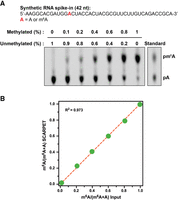
SCARPET accurately predicts m6A stoichiometry using methylated RNA standards. (A,B) SCARPET accurately predicts the stoichiometry of a methylation site in a mixture of two 42-nt long synthetic RNA oligonucleotides—one unmethylated and the other containing a defined m6A modification in a common GGACU consensus site for m6A. The two synthetic RNAs were mixed such that the m6A stoichiometry ranged from 0% to 100%. The mixture was added to 1 µg 2×-poly(A) HepG2 mRNA to simulate the conditions for biological m6A site determination in an mRNA. SCARPET predicted the m6A stoichiometry with high accuracy and showed excellent linearity (R2 = 0.973) to the input m6A fraction (B). 32P-labeled m6A and A nucleosides were run as TLC standards in A.
SCARPET oligonucleotides can have nonspecific cleavage that increases the background signal
We noticed a discrepancy between measurements of m6A stoichiometry at A1216 in ACTB by SCARLET and SCARPET (ranging from 21% to 29%) and the m6A stoichiometry predicted by GLORI (65%), a deamination-based method (Liu et al. 2023). We wondered if SCARPET and SCARLET have a background levels of the A spot due to the SCARPET oligonucleotide binding and cleaving near-cognate binding sites in the transcriptome. If these sites are then ligated by the hairpin oligonucleotide, background A signals would be seen, which would incorrectly reduce the overall measured m6A fraction. Although noncognate cleavage and ligation would have low efficiency due to the lack of complete sequence complementarity, this could still occur at an appreciable level if there are a large number of near-cognate sites in the transcriptome.
We next measured the A and m6A background signals that can originate due to partial hybridization of a SCARPET oligonucleotide to near-cognate GGACU sites in the transcriptome. To measure the background, we designed a SCARPET oligonucleotide against a m6A site in a synthetic 42-nt long RNA oligonucleotide that either contained the A (A-RNA42) or m6A (m6A-RNA42) at nucleotide position 13. This SCARPET oligonucleotide has no exact complementarity to any human mRNA. The A/m6A was in a GGACU sequence context. In these experiments, 200 fmol (2.7 ng) of A-RNA42 or m6A-RNA42 were mixed with 1 µg of 2×-poly(A) HepG2 mRNA. A control sample was prepared with no added oligonucleotide (Supplemental Fig. 4A,B). SCARPET analysis readily detected the A in the sample with A-containing oligonucleotide or the m6A in the sample with the m6A-containing oligonucleotide. However, when no oligonucleotide was added, A was still detected from the cellular RNA. This A signal comprises ∼13.5% of the total adenosine signal (Supplemental Fig. 4A). However, there was no detectable background m6A signal in the cellular RNA sample (Supplemental Fig. 4B). Together these results suggest that background A signals, but not m6A signals, can occur with SCARPET oligonucleotides.
To further assess background levels, we prepared a 1:1 mixture of a methylated and nonmethylated oligonucleotide standard used in Figure 3. We added this mixture at increasing concentrations (0–40 fmol of each RNA) in 1 µg 2×-poly(A) HepG2 mRNA, and measured m6A stoichiometry by SCARPET (Fig. 4A). We found that low concentrations of the standard produced underestimated m6A stoichiometries. Only at the highest levels (40 fmol of each RNA) did the estimated m6A (46.3%) get close to the actual stoichiometry of 50% (Fig. 4A). Notably, the amount of RNA used in Figure 3 was 200 fmol, which accounts for the relatively low m6A estimation in that figure.
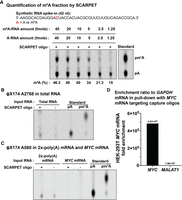
SCARPET oligonucleotides can have nonspecific cleavages that increase background signal. (A) SCARPET underestimates m6A stoichiometries when the concentration of the target RNA is low. SCARPET results are shown for a site in a 42-nt long synthetic RNA standard comprising a 1:1 mixture of methylated and nonmethylated RNA oligonucleotide standards, with amounts ranging from 1.25 to 40 fmol added to 1 µg 2×-poly(A) HepG2 mRNA. At low concentrations of the RNA standard, SCARPET underestimated m6A stoichiometries. Only at the highest levels (40 fmol of each RNA standard) did the estimated m6A (46.3%) get close to the actual stoichiometry of 50%. (B) SCARPET results using a SCARPET oligonucleotide specific for ΦX174 A2768, an arbitrary site, using 1 µg total RNA from HEK293T cells as the input. In principle, no nucleotide should be detected since the ΦX174 RNA is not present in HEK293T RNA. However, a SCARPET oligonucleotide-dependent A spot was detected, demonstrating nonspecific cleavage of cellular RNA. (C,D) SCARPET result for another arbitrary site in ΦX174 (A580). A SCARPET oligonucleotide-dependent A spot was detected. A weak A spot in the absence of the SCARPET oligonucleotide was detected, which likely reflects some ligation of the hairpin oligonucleotide to spurious 5′ ends with weak complementarity to the hairpin oligonucleotide. One microgram of 2×-poly(A) HEK293T mRNA and 1 ng of purified MYC HEK293T mRNA were used as input. (C). The A spot completely disappeared when using purified MYC mRNA as the input which suggests that using purified mRNA can lower the background, and therefore provide more accurate quantification in SCARPET. The RT-qPCR-based fold enrichment of purified MYC mRNA relative to GAPDH mRNA in the pull-down with MYC mRNA targeting capture oligonucleotides compared to MALAT1 lncRNA which was used as a nonspecific control (D). Note: Both ΦX174 A2768 and A580 sites are present in the common GGACU consensus site for m6A. 32P-labeled m6A and A nucleosides were run as TLC standards in A–C.
We further assessed the background using two SCARPET oligonucleotides designed to cleave A580 and A2768 in the ΦX174 sequence. These sites were chosen since they are in a DRACH sequence context. The SCARPET oligonucleotides have no exact complementarity to any RNA in HEK293T cells. Using these oligonucleotides, SCARPET resulted in an A spot (Fig. 4B,C). The A spot was due to the SCARPET oligonucleotide since leaving out the SCARPET oligonucleotide resulted in a loss of the A spot. These data further support the idea that the SCARPET oligonucleotides can cleave near-cognate sites and introduce background levels of A, thus resulting in underestimation of the m6A stoichiometry.
We also detected a weak A spot that was SCARPET oligonucleotide independent (Fig. 4B,C). This likely reflects some ligation of the hairpin oligonucleotide to spurious 5′ ends with weak complementarity to the hairpin oligonucleotide.
To overcome the background problem, we considered using purified mRNAs for SCARPET. To test this, we purified the MYC mRNA from HEK293T cells. The mRNA was purified using the custom RNA affinity capture kit from ElementZero Biolabs (see Materials and Methods). In brief, oligonucleotide probes are used to enrich for the MYC mRNA, which simultaneously de-enriches for other cellular mRNAs such as MALAT1. Affinity enrichment of MYC mRNA was assessed by RT-qPCR and was 4.83 × 105-fold enriched (relative to GAPDH mRNA) compared to MALAT1 lncRNA which was used as a nonspecific control (Fig. 4D). Overall, this protocol leads to a highly enriched MYC RNA fraction.
We next retested the background signal due to the SCARPET oligonucleotide for A580 in ΦX174. This probe (and the hairpin oligonucleotide) should not cleave and label any A in MYC mRNA. Indeed, we observed an A spot when using 2×-poly(A) mRNA (1 µg), but not when using purified MYC mRNA (1 ng) (Fig. 4C).
We next wanted to explore the idea that background signals come from near-cognate sites in the transcriptome. We therefore asked if a SCARPET oligonucleotide for detecting an m6A site at A1361 in the MYC mRNA has near-cognate sites in the transcriptome. Using a simple BLAST analysis, we could readily detect other near-cognate sites that could potentially cause background signals due to cleavage and then ligation to the hairpin oligonucleotide (Supplemental Fig. 4C). Although this analysis was performed on a single site, we suspect that most SCARPET oligonucleotides would have some near-cognate sites and these sites account for the background signal. The use of purified mRNA reduces the likelihood of having these off-target sites.
Overall, these data suggest that using purified mRNA can lower the background, and therefore provide more accurate quantification in SCARPET.
m6A stoichiometry at MYC m6A sites is largely similar in HEK293T and MOLM-13 cells
An important m6A-containing mRNA is MYC. Changes in m6A levels in MYC have been proposed to contribute to the ability of m6A to promote leukemogenesis (Vu et al. 2017; Weng et al. 2018). We therefore wanted to measure m6A stoichiometry at A1361 and A1716 (Fig. 5A), which exhibits a prominent miCLIP in m6A maps (Vu et al. 2017) and may have functional roles (Su et al. 2018). Initially, we tested the A1716 site in MYC mRNA using 1 µg of 2×-poly(A) mRNA as the input. We found m6A stoichiometry at A1716 to be ∼20% (Fig. 5B). The m6A stoichiometry at A1716 is remarkably lower than the stoichiometry that was recently measured by the transcriptome-wide deamination-based methods, GLORI and eTAM-seq (Fig. 5F; Liu et al. 2023; Xiao et al. 2023). The underestimation of m6A stoichiometry at the A1716 site by SCARPET again highlights the importance of using enriched target mRNA as the input.
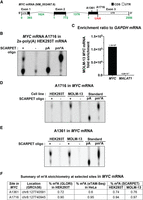
m6A stoichiometry at MYC mRNA m6A sites is largely similar in HEK293T and MOLM-13 cells. (A) Structure of MYC mRNA depicting A1361 and A1716 sites. Both sites have been previously detected by miCLIP m6A mapping (Linder et al. 2015). (B) SCARPET result for A1716 site in MYC mRNA using 2×-poly(A) HEK293T mRNA as the input. The m6A stoichiometry was found to be ∼20%. (C) The RT-qPCR-based fold enrichment of purified MYC mRNA from MOLM-13 cells relative to GAPDH mRNA in the pull-down with MYC mRNA targeting capture oligonucleotides compared to MALAT1 lncRNA which was used as a nonspecific control. (D,E) SCARPET results for MYC mRNA A1361 and A1716 sites using 1 ng of purified MYC mRNA from HEK293T and MOLM-13 cells as input for each site. For both A1361 and A1716, m6A stoichiometry was high (∼75% for A1361 and ∼95% for A1716) and similar between HEK293T and MOLM-13 cells. (F) Comparison of m6A stoichiometries at A1361 and A1716 sites measured by SCARPET to the levels measured by recent transcriptome-wide deamination-based methods, GLORI and eTAM-seq (Liu et al. 2023; Xiao et al. 2023). SCARPET results were remarkably similar to the results measured using these methods. 32P-labeled m6A and A nucleosides were run as TLC standards in B, D, and E.
We therefore remeasured m6A stoichiometry at the A1716 and A1361 in MYC mRNA using purified MYC mRNA as the input. For these experiments, the MYC mRNA was purified from HEK293T cells and MOLM-13 human leukemia cells. We used RT-qPCR to verify that the MYC mRNA was highly enriched relative to rRNAs, GAPDH, and ACTB (Fig. 5C; Supplemental Fig. 5A–F). For both A1716 and A1361, m6A stoichiometry was high (∼95% for A1716 and ∼75% for A1361) and similar between HEK293T and MOLM-13 cells (Fig. 5D,E). These stoichiometries were similar to stoichiometries measured by recent transcriptome-wide deamination-based methods, GLORI and eTAM-seq (Fig. 5E; Liu et al. 2023; Xiao et al. 2023). For these experiments, 1 ng of purified MYC mRNA was used, which required 6 h of exposure time to develop the TLC images. We expect that smaller input amounts would require proportional increases in exposure times. Overall, these results indicate that SCARPET analysis using purified mRNA produces accurate m6A measurements with low background levels from A.
m6A not detectable at selected sites in Zika RNA
We next examined the stoichiometry of m6A sites in the Zika virus. m6A was initially mapped in the Zika virus RNA genome in two studies (Lichinchi et al. 2016; Gokhale et al. 2020). The finding that m6A is present in this genome was unexpected because m6A is thought to be formed during or after transcription in the nucleus (Sommer et al. 1978; Ke et al. 2017). Additionally, METTL3 and METTL14 are usually detected in the nucleus (Ping et al. 2014). Since the RNA genome of Zika virus is thought to remain exclusively in the cytoplasm (Giraldo et al. 2023), the presence of m6A in Zika genomic RNA indicates that new mechanisms for RNA methylation are required to methylate Zika RNA.
We therefore wanted to examine m6A stoichiometry at specific sites in the Zika RNA genome. The previous studies were not performed at single-nucleotide resolution—instead, they used peak-based mapping methods which reveal an m6A-containing region. We selected three peaks identified in both studies. m6A sites are typically predicted within peaks by picking DRACH sites that appear in the center of these peaks, which in this case were A4824, A5811, and A9199 (Fig. 6A).
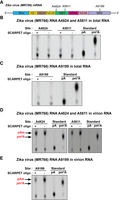
m6A not detectable at selected sites in Zika RNA. (A) Structure of Zika virus (MR766) mRNA depicting A4824, A5811, and A9199 sites. All these sites are in common GGACU consensus site for m6A and are located within m6A peaks that were previously detected using MeRIP-seq m6A mapping (Lichinchi et al. 2016; Gokhale et al. 2020). (B,C) SCARPET results for A4824, A5811, and A9199 sites using DNase I-treated and rRNA-depleted RNA isolated from Huh-7.5 hepatocellular adenocarcinoma cells infected with MR766 Zika virus 72 h postinfection. SCARPET showed a lack of detectable m6A at all three sites. (D,E) SCARPET results for A4824, A5811, and A9199 sites using DNase I-treated and rRNA-depleted RNA isolated from virion particles collected from Zika virus MR766-infected Vero E6 (African green monkey kidney) cells 72 h postinfection. SCARPET showed a prominent Am spot that was independent of the SCARPET oligonucleotide but no m6A spot. The Am spot likely reflects low but measurable ligation of the hairpin oligonucleotide to the 5′ end of the Zika genomic RNA which begins with Am (Coutard et al. 2017). 32P-labeled m6A and A nucleosides were run as TLC standards in B–D.
To prepare Zika RNA fractions, Huh-7.5 (hepatocellular adenocarcinoma) and Vero E6 (African green monkey kidney) cells were infected with Zika virus (African strain, MR766), and total cellular RNA was harvested 72 h postinfection, as described previously (Zhou et al. 2017; Tan et al. 2019; Gokhale et al. 2020). Since Zika genomic RNA becomes highly abundant in the cell and is present at ∼107 RNA copies per µL of viral supernatants after 48 h of infection (Tiwari et al. 2017), we directly tested m6A stoichiometry in total cellular RNA fractions after ribosome depletion, without purification of Zika RNA. SCARPET analysis showed a lack of detectable m6A at all three sites (Fig. 6B,C).
We considered the possibility that m6A could not be detected because the Zika viral RNA was not pure enough. We therefore used the released virion particles from Vero E6 cells, which contain essentially pure viral genomic RNA (Tiwari et al. 2017). The virion particles were prepared from the cell culture supernatants after 72 h postinfection by centrifugation and filtration, as previously described (Lichinchi et al. 2016). The harvested virion RNA was DNase I-treated and rRNA depleted. We next performed SCARPET using the samples. Again, we saw no m6A spot. Instead, we saw a prominent Am spot, that was independent of the SCARPET oligonucleotide (Fig. 6D,E). Thus, the Am spot is not due to cleavage of the viral RNA. The Am spot therefore likely reflects ligation of the hairpin oligonucleotide to the 5′ end of the Zika viral RNA, which begins with Am (Coutard et al. 2017). The high concentration of this viral RNA in the sample, and a low but measurable level of ligation, likely accounts for the large amount of Am in the sample and the resulting Am signal. Overall, the SCARPET data suggest that the selected sites do not contain m6A in Zika RNA.
We may have failed to detect m6A because we did not accurately predict the correct m6A site. The previous studies generated m6A peaks, and we identified a m6A site based on the m6A consensus motif. However, the methylation mechanism in the cytosol may be different from the mechanism in the nucleus for endogenous mRNAs. Nevertheless, the data presented here highlight the importance of testing specific candidate m6A sites rather than assuming the site of methylation. Additionally, further analysis of the Zika RNA genome using methods like GLORI or eTAM-seq (Liu et al. 2023; Xiao et al. 2023) will be useful to discover the exact m6A sites and their methylation stoichiometry.
DISCUSSION
m6A has different stoichiometry at different positions in different mRNAs. However, exact stoichiometries of m6A are often not reported in most studies that have reported m6A dynamics. This is due to the difficulty in making these measurements. The previous SCARLET assay is difficult and requires a large amount of input to detect m6A signals. In this study, we present SCARPET, which involves a site-specific cleavage of mRNA immediately 5′ of an adenosine site in an mRNA. This site is radiolabeled with 32P, and after a series of steps to purify the RNA and to remove nonspecific signals, the nucleotide is resolved by TLC to visualize A and m6A at this site. Quantification of these spots reveals the m6A stoichiometry at the site of interest. An mRNA purification step is used to reduce possible nonspecific signals that cause underestimation of m6A stoichiometry. Overall, SCARPET is streamlined to make it more robust, simpler, and more specific than previous assays. SCARPET will be useful for testing specific sites for their m6A stoichiometry and to assess how the stoichiometry changes in different conditions and cellular contexts. Although we focused on m6A, SCARPET should be useful for determining the stoichiometry of other RNA modifications such as 5-methylcytosine (5mC) and pseudouridine (ψ).
SCARPET maintains some of the key steps of SCARLET, but includes different steps to simplify and increase the recovery of RNA for TLC analysis. SCARPET and SCARLET share the same oligonucleotide step for RNase H-mediated cleavage of RNA at a specific site in the transcriptome (Liu et al. 2013). However, SCARPET uses a bimolecular ligation reaction with a hairpin “self-splinting” oligonucleotide rather than requiring a ternary complex for splint ligation. Also, rather than gel purification of the ligated product, the hairpin oligonucleotide in SCARPET is biotinylated for rapid and efficient purification prior to digestion for TLC. Overall, these changes allow the sample processing steps of SCARPET to be performed in 1 d, rather than the 3 d for SCARLET. Our side-by-side comparison of these two methods showed that the signals were markedly more robust for SCARPET, which shows that more RNA is recovered for the TLC step in SCARPET than SCARLET. The SCARPET name was chosen to credit the earlier SCARLET assay, yet to also highlight fundamentally different steps which simplify the overall protocol.
SCARPET exhibits linearity and quantitative accuracy, based on the methylated and nonmethylated standard RNAs. We found that the accuracy of SCARPET is increased by using purified mRNA in SCARPET. We initially noticed that the calculated stoichiometry of an m6A site in ACTB mRNA using either SCARPET or SCARLET was lower than the stoichiometry recently reported in GLORI, a transcriptome-wide assay for measuring m6A stoichiometry (Liu et al. 2023). Our data suggest that low-level cleavage by the SCARPET oligonucleotide can produce a background signal. Thus, even if cleavage rates or ligation reactions are very low at noncognate sites, the large size of the transcriptome can allow for enough of these sites to contribute to the background. Thus, SCARPET and SCARLET systematically underestimate m6A stoichiometries. For this reason, any m6A measurement should be considered a lower bound for the m6A stoichiometry. Indeed, when we monitored the same m6A site in MYC mRNA using poly(A) RNA compared to the purified MYC mRNA, we noticed a marked increase in stoichiometry when the purified mRNA was used in SCARPET.
Although the background may be lower if the SCARPET oligonucleotide has few off-target sites based on BLAST, it is difficult to remove the background. BLAST analysis typically shows many near-cognate sites in the transcriptome. Although an m6A site can be chosen which has a SCARPET oligonucleotide with fewer off-target sites, the main solution is to use purified mRNA for SCARPET. mRNA purification kits are available, and protocols for purifying specific transcripts from cells have been described (McHugh et al. 2015). We therefore recommend mRNA purification to obtain the highest accuracy.
One of the main advantages of SCARPET is that both the A and m6A can be seen as spots on a TLC plate. In this way, both the modified and unmodified forms are measured. The relative speed of this assay, as well as the steps that reduce the background labeling including exonuclease digestion, sequence-specific RNA cleavage, and sequence-specific ligation, also improve the specificity of this assay. Notably, we do not see C, G, or U at any of the tested sites, which supports the idea that only the intended nucleotide, which is A or m6A, is being measured. The specificity of the assay is also supported by the similar results obtained using SCARLET as well as GLORI or eTAM-seq. Comparison to these orthogonal high-throughput sequencing methods supports the accuracy of m6A stoichiometry measurements using SCARPET.
SCARPET can be highly useful to study an individual site and to monitor its m6A stoichiometry under different experimental conditions. SCARPET can be used to validate widely accepted concepts related to m6A, such as cancer-specific m6A sites (Zhong et al. 2019; Cho et al. 2021), the ability of m6A to be regulated by m6A demethylases (Jia et al. 2011), as well as stimulus or stress-induced m6A (Dominissini et al. 2012). In our study, we examined m6A stoichiometry at adenosines in Zika RNA, which is synthesized in the cytoplasm (Giraldo et al. 2023). In our study, we found no evidence for methylation at any of the three adenosine residues which appeared to be the methylation sites underneath m6A peaks mapped in prior studies. The lack of m6A may indicate that other adenosines in the peaks could have been the true methylated nucleotide. Thus, it will be important to identify the exact methylated nucleotide using transcriptome-wide methods such as GLORI (Liu et al. 2023). Then, SCARPET can be used to study the dynamics of a selected site in multiple different contexts where repeated next-generation sequencing GLORI experiments may not be practical or cost-effective.
The most commonly used assay to determine changes in m6A levels is MeRIP-qPCR. In this assay, m6A-containing RNA is immunoprecipitated in two or more conditions. A specific target mRNA or mRNA region is quantified by reverse transcription and PCR of the immunoprecipitated RNA. There are several problems with this assay. Most importantly, the amount of any mRNA that is immunoprecipitated can be affected by changes in the level of rRNA, a difficult-to-remove contaminant in poly(A) RNA fractions (Cui et al. 2010; Herbert et al. 2018). rRNA is highly abundant in cells and contains m6A (Choi and Busch 1978; Maden 1986) and therefore can influence mRNA immunoprecipitation with m6A antibodies. Another problem is that m6A antibodies also immunoprecipitated m6Am, which is present in ∼40% of cellular mRNAs. Thus, immunoprecipitation cannot be exclusively linked to m6A. The SCARPET assay bypasses these problems and directly measures m6A and A at any specific site in mRNA.
Other methods have also been developed to quantify m6A stoichiometry in mRNA. These include methods such as SELECT (Single-base Elongation- and Ligation-based qPCR amplification) (Xiao et al. 2018). SELECT exploits the fact that m6A hinders the elongation of DNA polymerase and the efficiency of ligases in nick ligation reactions. In this way, m6A impairs the formation of complete cDNA sequences, which can be detected by qPCR. A control sample is prepared by FTO-mediated m6A demethylation. A problem with these assays is that cDNA synthesis efficiency could be affected by sequence contexts or if the amount of input RNA varies between samples. SCARPET has the advantage of detecting both A and m6A regardless of input levels or other conditions. Because both nucleotides are detected in SCARPET, the m6A fraction can be readily measured with high confidence.
MATERIALS AND METHODS
Cell and Zika virus culture
HEK293T, HepG2, and MOLM-13 cells were grown as previously described (Dominissini et al. 2012; Mauer et al. 2017; Vu et al. 2017). Cells were cultured in 15 cm2 plates and allowed to grow until reaching ∼80% confluency before harvesting. The ZIKV MR766 virus was expanded in Huh-7.5 (hepatocellular adenocarcinoma) and Vero E6 (African green monkey kidney) cells according to previously described protocols (Zhou et al. 2017; Tan et al. 2019; Gokhale et al. 2020).
RNA isolation and purification
All steps for RNA purifications were according to the manufacturer's instructions unless otherwise specified. Cells were harvested for total RNA isolation after reaching ∼80% confluency. In experiments using HEK293T and HepG2 cells, the media was removed, and 6 mL TRIzol LS reagent (Thermo Fisher) was directly added to the plates. A cell scraper was used to spread the TRIzol LS reagent evenly. Next, 2 mL of 1× PBS was added to each plate to maintain a 3:1 (TRIzol LS: sample) ratio. In the case of experiments with MOLM-13 cells, which grow in suspension, cells were harvested by centrifugation at 300g for 5 min at 25°C and the media was removed. Next, cells were resuspended in 2 mL 1× PBS and 6 mL TRIzol LS reagent was added to the cell suspension. Samples were mixed up and down with a pipette. Samples were left at 25°C for 10 min to denature nucleic acid–protein complexes. Next, 1.6 mL of chloroform (Thermo Fisher) was added to each sample. After mixing, the samples were transferred into 15 mL precentrifuged (5000g for 5 min at 25°C) MaXtract High-Density tubes (Qiagen). The remaining steps were performed as described in the TRIzol LS protocol. The purified total RNA was treated with DNase I (Thermo Fisher) and the enzymatic reaction was purified with an RNA Clean & Concentrator kit (Zymo Research). Finally, the DNase I-treated total RNA was dissolved in elution buffer (10 mM Tris-HCl pH 7.5), quantified using a NanoDrop (Thermo Fisher), and stored at −80°C until use.
To prepare twice poly(A)-purified mRNA [2×-poly(A)], DNase I-treated total RNA was used as the input, and poly-dT(25) beads (Thermo Fisher) were used to purify the mRNA according to the manufacturer's protocol. A second round of poly(A) purification was done reusing the same oligo-dT(25) beads. Finally, the purified mRNA was concentrated using the RNA Clean & Concentrator kit (Zymo Research) and eluted with 10 mM Tris-HCl pH 7.5, quantified using a NanoDrop (Thermo Fisher), and stored at −80°C until use.
RNA from ZIKV MR766-infected Huh-7.5 and Vero E6 cells was harvested according to the same procedure as above. In addition, the RNA was also extracted from the Zika virion particles secreted into the media by the virus during culture. The supernatant was collected 72 h postinfection, and samples were centrifuged at 800g for 10 min at 25°C to pellet cell debris. Next, the supernatant containing the virion particles was concentrated as described previously (Tan et al. 2019). RNA was extracted using TRIzol LS reagent followed by DNase I treatment to remove residual contaminating DNA. The purified RNA was further subjected to rRNA depletion using the RiboMinus kit (Thermo Fisher) and cleaned and concentrated using the RNA Clean & Concentrator kit (Zymo Research). Finally, the RNA was eluted in the elution buffer, pH 7.5 and stored at −80°C until use.
Oligonucleotide sequences
All the oligonucleotide sequences used in the SCARPET assays and inventoried RT-qPCR assays were ordered from Integrated DNA Technologies (IDT). SCARPET oligonucleotides were ordered with standard desalting purification. The 5′-biotinylated self-splinting hairpin oligonucleotides were all ordered with PAGE purification. The RNA oligonucleotide TLC standards, that is, A, m6A, Am, C, G, and U, were ordered with RNase-free HPLC purification. The m6Am oligonucleotide was synthesized as described previously (Mauer et al. 2017). All the oligonucleotide sequences are listed in Supplemental Table S1.
Preparation of thin-layer chromatography standards
All TLC standards were prepared by radiolabeling with T4 PNK and [γ-32P] ATP, to label the 5′ end of each RNA oligonucleotide standard. Briefly, 250 ng (50 pmol) of each synthetic RNA oligonucleotide standard was aliquoted into a separate tube, and 5 µL (50 pmol) of 10 mCi/mL [γ-32P] ATP (PerkinElmer), 2 µL (20 U) T4 PNK (NEB), was added into each tube. Finally, the volume was brought up to 50 µL with nuclease-free water (IDT). Next, samples were incubated at 37°C for 1 h with intermittent pulse shaking (800 rpm, 30 sec ON/3 min OFF) in an Eppendorf ThermoMixer C with a ThermoTop heated lid. Next, labeled RNA samples were purified with phenol:chloroform:isoamyl alcohol (25:24:1, v/v) (Thermo Fisher) following the manufacturer's instructions, except 5PRIME Phase Lock Gel, Heavy tubes (Quantabio) were used to separate the labeled RNA containing aqueous phase from the organic phase. The labeled RNA pellet was resuspended in 16 µL of nuclease-free water. Next, 2 µL (2 U) of nuclease P1 (NEB) and 2 µL of 10× nuclease P1 buffer (NEB) were added, and the samples were incubated at 37°C for 2 h with intermittent pulse shaking (800 rpm, 30 sec ON/3 min OFF) in an Eppendorf ThermoMixer C with a ThermoTop heated lid. Finally, the volume of each hydrolysate was brought up to 500 µL with nuclease-free water, and the standards were stored at −20°C until use. For the m6Am TLC standard, the RNA oligonucleotide contained a triphosphate at the 5′ end [5′-pppm6Am(N)23-3′], which was removed with rSAP treatment [2 µL (2 U) rSAP, 2 µL 10× T4 PNK buffer, nuclease-free water up to 20 µL] by incubating at 37°C for 1 h. The dephosphorylated RNA oligonucleotide standard was purified with an RNA Clean & Concentrator kit (Zymo Research), labeled with [γ-32P] ATP, and subsequently digested with nuclease P1 as described vide supra.
MYC mRNA capture and RT-qPCR
MYC mRNA was enriched using DNase I-treated total RNA from HEK293T or MOLM-13 cells as the input. We used a Magnetic Instant Capture (MagIC) Beads kit customized for human MYC mRNA (ElementZero Biolabs) following the manufacturer's instructions scaled up accordingly to accommodate 80 µg of input RNA. In this kit, multiple nonoverlapping DNA oligonucleotide probes are covalently immobilized through their 5′ ends on the surface of paramagnetic MagIC beads. The highly efficient capture of the target RNA is achieved through specific binding of these probes under stringent hybridization (high salt, denaturant, and reducing agent) and wash buffer conditions performed at 60°C. These stringent conditions enhance the overall enrichment of the target RNA and lower the nonspecific hybridization to unintended RNA targets. The purified MYC mRNA was eluted with elution buffer, quantified by NanoDrop, and stored at −80°C until use. Typically, 8 ng of MYC mRNA was purified per MagIC bead reaction, and 16 reactions were provided per kit.
The efficiency of MYC mRNA enrichment was assessed with a two-step RT-qPCR. First, cDNA was synthesized with the SuperScript IV First-Strand Synthesis System kit (Thermo Fisher) following the manufacturer's instructions, except both oligo d(T)20 (1 µL of 50 µM) and random hexamer (50 ng) primers were used together for the first-strand cDNA synthesis. For cDNA synthesis, 5 µg of total RNA and 5 ng of captured MYC mRNA were used as input. The cDNA was diluted 200-fold with nuclease-free water, and 3 µL of diluted cDNA was used as the input for quantitative real-time PCR (qPCR) using Power SYBR Green PCR Master Mix (Thermo Fisher) following the manufacturer's instructions. We used inventoried intercalating-dye-based qPCR assays from Integrated DNA Technologies for MYC, ACTB, GAPDH, MALAT1, 18S, and 28S RNAs. The reactions were run on the QuantStudio 5 Real-Time PCR System (Thermo Fisher). All the qPCR reactions were run in triplicate.
To assess the enrichment efficiency of MYC mRNA capture, RT-qPCR data were processed as follows. First, mean quantification cycle (Cq) values from each technical replicate for each RNA were transformed by the 2−Cq formula. Next, transformed values obtained for MYC and MALAT1 transcripts in every sample were divided by the values obtained for ACTB, GAPDH, 18S, and 28S transcripts, thus giving a ratio of the measured transcripts in the input and enriched sample. Finally, the calculated ratios in the enriched sample were divided by the corresponding ratios obtained for the input sample, resulting in the fold enrichment value over input.
Optimization of the steps of SCARPET
We optimized the following steps in SCARPET: (i) RNase H-mediated site-specific cleavage, (ii) dephosphorylation by rSAP, (iii) radiolabeling by T4 PNK, (iv) self-splinting hairpin-assisted ligation, (v) optimization of the hairpin oligonucleotide, and (vi) testing of T4 DNA, SplintR, and 9°N DNA ligases. For optimizing steps (i)–(iv), we selected A1832 in 18S rRNA and A1216 in ACTB mRNA and used 1 µg of DNase I-treated total RNA and 1 µg of 2×-poly(A) mRNA as input from HEK293T cells, respectively. For each of these steps, we optimized the time and the amount of enzyme required by either doubling the amount of enzyme or doubling the reaction time. In the case of steps (v) and (vi) experiments, we only tested the A1832 site in 18S rRNA. In all conditions, we used 20 ng of SCARPET oligonucleotide. Of note, SCARPET oligonucleotide hybridization and the PNK phosphorylation steps were previously optimized in SCARLET (Liu et al. 2013). All the steps were carried out at their specific temperatures in an Eppendorf ThermoMixer C lid with intermittent shaking (800 rpm, 30 sec ON/3 min OFF) with ThermoTop heating. (i) RNase H-mediated site-specific cleavage of target RNA: 1 U RNase H, 1 h at 44°C versus 2 U (1 + 1) RNase H, 2 h (1 + 1) at 44°C. (ii) Dephosphorylation by rSAP: 1 U rSAP, 1 h at 44°C versus 2 U (1 + 1) rSAP, 2 h (1 + 1) at 44°C. (iii) Radiolabeling by T4 PNK: 6 U T4 PNK, 1 h, at 37°C versus 12 U (6 + 6) T4 PNK, 2 (1 + 1) h, at 37°C. (iv) Self-splinting hairpin oligonucleotide-assisted ligation: 5 U T4 DNA ligase, 3.5 h, at 37°C versus 10 U (5 + 5) T4 DNA ligase, 7.5 (3.5 + 3.5) h, at 37°C. (v) Optimization of hairpin oligonucleotide: Stability of the stem of hairpin oligo (5′Bio-ssHP) was tested by swapping an A•T base pair near the base of the stem with a C•G base pair (5′Bio-ssHP1). Requirement of base-pair complementarity between the splint and the target RNA at the ligation junction for efficient ligation was tested by introducing three mismatches in the splint region near the ligation junction (5′→3′ [TGT→ACA] [5′Bio-ssHP2]). Splint length requirement was tested by comparing standard splint length (29 nt) to 24 nt (5′Bio-ssHP3) and 14 nt (5′Bio-ssHP4) splints. (vi) Testing of T4 DNA, SplintR, and 9°N DNA ligases: We compared ligation efficiencies of T4 DNA, SplintR, and 9°N DNA ligases to ligate a self-splinting hairpin oligonucleotide to the radiolabeled target RNA. All these ligases were purchased from New England Biolabs. For this, we selected A1832 in 18S rRNA and used 1 µg of DNase I-treated total RNA as input from HEK293T cells and 20 ng of SCARPET oligonucleotide. We used 5 U of T4 DNA ligase and 25 U of SplintR ligase and incubated samples at 37°C for 3.5 h. In the case of 9°N DNA ligase, we used 40 U of the enzyme and incubated the sample at 45°C for 3.5 h.
SCARPET assay
SCARPET assays were performed to determine the stoichiometry of m6A at a given site in RNA of interest using the optimized protocol. All incubation steps were performed at their specified temperatures using Eppendorf ThermoMixer C with a ThermoTop heated lid with intermittent pulse shaking (800 rpm, 30 sec ON/3 min OFF). For each site, 1 µg of DNase I-treated total RNA or 1 µg of twice poly(A)-purified [2×-poly(A)] mRNA was mixed with 20 ng (∼3 pmol) of SCARPET oligonucleotide in a total volume of 3 µL of 30 mM Tris-HCl, pH 7.5 in 1.5 mL DNA LoBind tubes (Eppendorf). In addition, for each site, a control sample was also set up in which SACRPET oligonucleotide was not added. To anneal the SCARPET oligonucleotide to the target RNA the sample was heated at 95°C for 1 min, followed by incubation at room temperature (RT) for 3 min before putting it on ice for the next step. RNase H-mediated site-specific cleavage and dephosphorylation step: For site-specific cleavage of target RNA and dephosphorylation at the 5′ end, 1 µL RNase H reaction mixture (containing 2× T4 PNK buffer [140 mM Tris-HCl {pH 7.6 at 25°C}, 20 mM MgCl2, and 10 mM DTT, New England Biolabs], 1 U RNase H [New England Biolabs]), and 1 U (1 µL) shrimp alkaline phosphatase (rSAP, New England Biolabs) was added to each sample. The samples were incubated at 44°C for 1 h. The RNase H and rSAP were heat inactivated by incubating the samples at 75°C for 5 min and immediately put on ice. Radiolabeling step: Next, 1 µL of T4 PNK reaction mixture (containing 1× T4 PNK buffer, 6 U of T4 PNK [New England Biolabs]) and 1 µL of 10 µCi/µL [γ-32P]-ATP was added to each sample. The samples were incubated at 37°C for 1 h to radiolabel the cleaved target RNA at its 5′ end. T4 PNK was heat inactivated by incubating the samples at 75°C for 5 min and immediately put on ice. Self-splinting hairpin-assisted ligation: For self-splinting hairpin oligonucleotide-assisted ligation, 2.5 µL of 10 µM (25 pmol) of a 5′-biotinylated self-splinting hairpin oligonucleotide (5′Bio-ssHP) was added to each sample, and samples were mixed using a pipette. For proper folding of 5′Bio-ssHP and splint annealing to the radiolabeled target RNA, samples were heated at 75°C for 3 min followed by incubation at RT for 3 min, and then samples were put on ice. Next, 10 µL of T4 DNA ligation mixture (containing 2.8× T4 PNK buffer, 2 mM ATP, 30% DMSO, 5 U T4 DNA ligase [Thermo Fisher]) was added to each sample, and samples were incubated at 37°C for 3.5 h. The target RNA ligated to the 5′Bio-ssHP was captured by coupling to MyOne Streptavidin C1 magnetic beads (Thermo Fisher). The beads were prepared according to the manufacturer's instructions for RNA applications. To each sample, 5.5 µL of nuclease-free water was added to bring up the final volume to 25 µL. Next, 25 µL of MyOne Streptavidin C1 beads (75 µg beads in 2× B&W buffer [10 mM Tris-HCl {pH 7.5}, 1 mM EDTA, and 2 M NaCl]) was added to each sample bringing the total volume to 50 µL. Samples were incubated at RT on the HulaMixer sample mixer (Thermo Fisher) for 15 min for efficient coupling. Next, samples were put back on a magnet rack (Thermo Fisher) for 1–2 min, and liquid was carefully pipetted out into radioactive liquid waste. Samples were removed from the magnetic rack and beads were washed first with 50 µL of 1× B&W buffer and tubes were placed back on the magnetic rack for 1–2 min, and supernatant was carefully removed. The second wash was performed like the first one with 50 µL of 10 mM Tris-HCl pH 7.5. Terminator step: Next, 20 µL of Terminator 5′-phosphate-dependent exonuclease (Biosearch Technologies) reaction mixture (containing 2 µL of Buffer A, 1 U Terminator exonuclease, and 17 µL of nuclease-free water) was added to each sample and incubated at 30°C for 1 h. Samples were put back on a magnetic rack for 1–2 min, and supernatant was carefully removed. Next, samples were subjected to two rounds of washes as described before. RNase H step: For the RNase H step, 10 µL of RNase H reaction mixture (containing 1 µL 10× T4 PNK buffer, 5 U [1 µL] RNase H, 8 µL of nuclease-free water) was added to each sample, mixed with a pipette, and incubated at 37°C for 1 h. Nuclease P1 step: Finally, samples were digested to 5′-monophosphate nucleotides by adding 3.5 U (3.5 µL) of nuclease P1 (New England Biolabs) and 1.5 µL of 10× nuclease P1 buffer (500 mM sodium acetate [pH 5.5 at 25°C] [New England Biolabs]) and incubated at 50°C for 1 h. Next, samples were placed on a magnetic rack for 1–2 min, and the hydrolysates were transferred to fresh tubes. Thin-layer chromatography (TLC) step: To reveal the m6A stoichiometry, the nucleotide hydrolysate from each sample was separated by 1D TLC on precoated PEI Cellulose F TLC plates (EMD Millipore) as described previously (Grosjean et al. 2004). Briefly, 1 µL RNA hydrolysate from each sample was spotted alongside with 1 µL of m6A and 1 µL of A TLC standard. After the sample spots dried, the TLC plate was developed in a glass tank with 100 mL of running buffer (Isopropanol:HCl:Water [70:15:15, v/v/v]) and allowed to run overnight (∼14 h). Next, TLC plates were dried at RT for 1 h, wrapped with SARAN wrap, and exposed to BAS storage phosphor screens (Cytiva Life Sciences) for 6–12 h. The phosphor screens were imaged using Amersham Typhoon 5 (Cytiva Life Sciences). The phosphor screen images were analyzed using ImageJ 1.53k software (Rasband 1997–2018).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
S.R.J. is the cofounder, advisor, and/or has equity in Chimerna Therapeutics, 858 Therapeutics, and Lucerna Technologies. The other authors have no conflicts to report.
ACKNOWLEDGMENTS
We thank all members of the Jaffrey laboratory for comments and suggestions. This work was supported by the National Institutes of Health grants R35NS111631, S10 OD030335, and R01CA186702 (S.R.J.) and T32 CA062948 (A.H.M.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079776.123.
- Received July 18, 2023.
- Accepted December 4, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








