
Analysis of programmed frameshifting during translation of prfB in Flavobacterium johnsoniae
- Fawwaz M. Naeem1,2,
- Bryan T. Gemler2,3,
- Zakkary A. McNutt1,2,
- Ralf Bundschuh2,3,4,5,6 and
- Kurt Fredrick1,2,7
- 1Ohio State Biochemistry Program, The Ohio State University, Columbus, Ohio 43210, USA
- 2Center for RNA Biology, The Ohio State University, Columbus, Ohio 43210, USA
- 3Interdisciplinary Biophysics Graduate Program, The Ohio State University, Columbus, Ohio 43210, USA
- 4Department of Physics, The Ohio State University, Columbus, Ohio 43210, USA
- 5Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, USA
- 6Division of Hematology, Department of Internal Medicine, The Ohio State University, Columbus, Ohio 43210, USA
- 7Department of Microbiology, The Ohio State University, Columbus, Ohio 43210, USA
- Corresponding author: fredrick.5{at}osu.edu
-
Handling editor: Marina Rodnina
Abstract
Ribosomes of Bacteroidia fail to recognize Shine–Dalgarno (SD) sequences due to sequestration of the 3′ tail of the 16S rRNA on the 30S platform. Yet in these organisms, the prfB gene typically contains the programmed +1 frameshift site with its characteristic SD sequence. Here, we investigate prfB autoregulation in Flavobacterium johnsoniae, a member of the Bacteroidia. We find that the efficiency of prfB frameshifting in F. johnsoniae is low (∼7%) relative to that in Escherichia coli (∼50%). Mutation or truncation of bS21 in F. johnsoniae increases frameshifting substantially, suggesting that anti-SD (ASD) sequestration is responsible for the reduced efficiency. The frameshift site of certain Flavobacteriales, such as Winogradskyella psychrotolerans, has no SD. In F. johnsoniae, this W. psychrotolerans sequence supports frameshifting as well as the native sequence, and mutation of bS21 causes no enhancement. These data suggest that prfB frameshifting normally occurs without SD–ASD pairing, at least under optimal laboratory growth conditions. Chromosomal mutations that remove the frameshift or ablate the SD confer subtle growth defects in the presence of paraquat or streptomycin, respectively, indicating that both the autoregulatory mechanism and the SD element contribute to F. johnsoniae cell fitness. Analysis of prfB frameshift sites across 2686 representative bacteria shows loss of the SD sequence in many clades, with no obvious relationship to genome-wide SD usage. These data reveal unexpected variation in the mechanism of frameshifting and identify another group of organisms, the Verrucomicrobiales, that globally lack SD sequences.
Keywords
INTRODUCTION
In many bacteria, initiation of translation often involves pairing between a purine-rich Shine–Dalgarno (SD) sequence, located just upstream of the start codon, and the pyrimidine-rich anti-SD (ASD) sequence of the 3′ tail of 16S rRNA (Shine and Dalgarno 1975; Laursen et al. 2005). However, in certain clades of bacteria, including the Bacteroidia (formerly Bacteroidetes), SD sequences are absent from nearly all genes (Nakagawa et al. 2010, 2017; Accetto and Avguštin 2011). In these organisms, the translation machinery must rely on other determinants for accurate and efficient mRNA selection. In the representative Flavobacterium johnsoniae, important features of the translation initiation region (TIR) include low secondary structure; adenines at positions −3, −6, −12, and −13; and a paucity of AUG trinucleotides in the vicinity of the correct start codon (Baez et al. 2019).
Bacteroidia ribosomes are “blind” to SD sequences, in vivo and in vitro, even though these ribosomes carry an intact ASD (Accetto and Avguštin 2011; Wegmann et al. 2013; Jha et al. 2021). A cryo-EM structure of the F. johnsoniae ribosome provides an explanation for this conundrum. The 3′ tail of the 16S rRNA binds a pocket formed by bS21, bS18, and bS6 on the 30S platform (Fig. 1A; Jha et al. 2021). This occludes the ASD, preventing it from interacting with mRNA. Most contacts to the 3′ tail are formed by amino acids that are uniquely conserved in the Bacteroidia, suggesting that the mechanism is ubiquitous in this group (Jha et al. 2021).
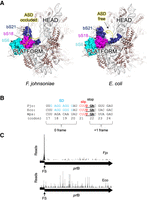
Flavobacterium johnsoniae and Escherichia coli ribosomes differ in structure and function. (A) Comparison of the 30S subunit from F. johnsoniae (left) and E. coli (right), viewed from “above” the platform domain. Proteins bS21, bS18, and bS6 form a pocket that binds the 3′ tail and occludes the ASD in F. johnsoniae. (B) Sequence comparison of the prfB frameshift site in F. johnsoniae (Fjo), E. coli (Eco), and Winogradskyella psychrotolerans (Wps). Codon numbers (based on F. johnsoniae) are indicated below, with the zero and +1 reading frames indicated. The SD, slippery sequence (slip), and UGA stop codon (stop) are shown in blue, red, and bold-underscore, as indicated. (C) Ribosome distribution along the prfB coding region in F. johnsoniae (Fjo) and E. coli (Eco). Reads (arbitrary scale) were mapped with respect to the central nucleotide of the predicted P codon. (FS) Frameshift site.
Remarkably, in F. johnsoniae, and most other Flavobacteriales, the TIR of only one gene—rpsU—contains an obvious SD sequence (Jha et al. 2021). Gene rpsU encodes bS21, one of the three proteins responsible for ASD occlusion. These observations, and the fact that bS21 is one of the last proteins incorporated during 30S biogenesis, led Jha et al. (2021) to propose a straightforward model for bS21 autoregulation. According to this model, ribosomes lacking bS21 contain a liberated ASD and translate rpsU mRNA at an enhanced rate, increasing bS21 production. This promotes bS21 incorporation and full assembly of the subunit. The corresponding replete ribosomes still translate rpsU mRNA but at a reduced rate (due to ASD sequestration), dialing down further bS21 synthesis (Jha et al. 2021). To test this model, McNutt et al. (2023) targeted rpsU in F. johnsoniae. They found that mutation or depletion of bS21 in the cell increases protein synthesis from rpsU′-gfp and other reporters containing a strong SD but has no such effect on SD-less reporters. Purified ribosomes lacking bS21 (or its carboxyl terminus) exhibit enhanced initiation on rpsU mRNA and reduced initiation on other (SD-less) mRNAs. These results provide strong evidence that rpsU is autoregulated via a subpopulation of ribosomes that specifically lack bS21 in F. johnsoniae. Across the Bacteroidota, the rpsU gene is characterized by an unusually strong SD, suggesting widespread autoregulation that requires an extended SD–ASD helix (ΔG < −13 kcal/mol) (McNutt et al. 2023).
In bacteria, two release factors are responsible for translation termination—RF1 and RF2. RF1 recognizes UAA and UAG, while RF2 recognizes UAA and UGA. In ∼80% of bacteria, RF2 is encoded from two open reading frames (ORFs) and its production requires a programmed +1 frameshift event (Craigen et al. 1985; Craigen and Caskey 1986; Curran and Yarus 1988; Baranov et al. 2002; Bekaert et al. 2006). The mechanism of prfB programmed frameshifting has been extensively studied in E. coli and is thought to be similar in other bacteria. The first ORF of prfB is short, encoding the first 25 amino acids of RF2, and terminates with UGA. The second ORF, in the +1 reading frame, encodes the remainder of RF2 (340 residues). During translation elongation, the ribosome pauses when the in-frame UGA enters the A site. One of two competing events then occurs: (i) RF2 catalyzes termination, releasing a small unstable polypeptide, or (ii) peptidyl-tRNALeu shifts on the slippery sequence CUUU, repairing with UUU in the +1 frame, enabling production of full-length (FL) RF2. There is an SD sequence just three nucleotides upstream of the slippery sequence. SD–ASD pairing is thought to act as a compressed spring, pulling the mRNA in the 5′ direction and facilitating the frameshift event (Curran and Yarus 1988; Devaraj and Fredrick 2010). Indeed, base substitutions in the SD sequence nearly eliminate frameshifting, in line with this model (Weiss et al. 1987, 1988; Curran and Yarus 1988). A cytidine just downstream from UGA “weakens” this stop codon, increasing the dwell time of the paused ribosome (Poole et al. 1995, 1998; Major et al. 1996; Mottagui-Tabar and Isaksson 1997). Collectively, these RNA elements make up the frameshift site, which promotes +1 frameshifting at high frequency (∼50%) in E. coli (Craigen and Caskey 1986; Weiss et al. 1987; Curran and Yarus 1988).
Programmed frameshifting in prfB is just as prevalent in Bacteroidia as in other bacterial lineages. In F. johnsoniae, and many other Bacteroidia, the prfB frameshift site contains a “perfect” SD sequence (Fig. 1B). This is puzzling, since Bacteroidia ribosomes fail to recognize SD sequences due to ASD sequestration (Jha et al. 2021). In this work, we show that the efficiency of prfB frameshifting is substantially lower in F. johnsoniae than in E. coli. Mutations of bS21, which compromise ASD sequestration, increase the efficiency of frameshifting in F. johnsoniae substantially. In some members of the Flavobacteriales, such as W. psychrotolerans (Wps), the SD sequence of the frameshift site has degenerated. In F. johnsoniae, the Wps site supports frameshifting at levels comparable to that of the native site, and frameshifting is unaffected by mutations of bS21. These data suggest that prfB frameshifting does not normally involve SD–ASD pairing in F. johnsoniae, at least under standard laboratory conditions. Strains carrying chromosomal mutations that remove the frameshift or ablate the SD grow normally under many conditions but exhibit subtle defects in the presence of paraquat or streptomycin, respectively. These data indicate that both the autoregulatory mechanism and the SD element contribute to cell fitness. Evaluation of frameshift sites across bacteria reveals SD degeneration in certain clades and SD conservation in others, indicating variable use of the element.
RESULTS
Ribo-seq data suggest a large dwell time for ribosomes paused at the frameshift site in F. johnsoniae
In previous work, ribosome profiling was used to elucidate determinants of translation initiation in F. johnsoniae (Baez et al. 2019). Cells were grown in rich media at optimum temperature to mid-log phase and then subjected to RNA-seq and ribo-seq analyses. Each ribo-seq read was mapped to a single genomic position, corresponding to the center of the predicted P codon. A plot of the frequency of reads across the prfB gene reveals a large peak corresponding to ribosomes at the slippery sequence, with CUU in the P site and UGA in the A site (Fig. 1C). The relative height of this peak is larger than that of the average terminating ribosome at either UGA or UAA (Supplemental Fig. S1). Equivalent treatment of ribo-seq data from E. coli, also grown in rich media at optimum temperature, shows a different pattern (Balakrishnan et al. 2014). Ribo-seq reads are distributed more evenly across prfB, with no obvious peak at the frameshift site (Fig. 1C). These observations suggest differences in kinetics between the two systems. In F. johnsoniae, ribosomes appear to spend more time at the frameshift site, as though termination and frameshifting are delayed in some way. This led us to hypothesize the existence of a downstream RNA element that stalls the ribosome, giving enough time for the 3′ tail of 16S rRNA to dissociate from the 30S platform and engage with the SD sequence.
Downstream sequence has no apparent effect on the steady-state level of frameshifting
To determine the region of prfB needed for programmed frameshifting in F. johnsoniae, we designed fusion constructs with variable portions of the prfB gene. In the first set of constructs, a portion of prfB (containing the frameshift site) was cloned between gfp and infA, such that synthesis of GFP-RF2-IF1 requires +1 frameshifting. For each frameshift (FS) construct, a corresponding in-frame control (C) construct, encoding the identical protein product, was also made (Fig. 2A). These C constructs harbor a single base pair deletion which converts the slippery sequence CUUU to CUU. Protein products were detected and quantified by western blot, using commercial anti-GFP antibodies. In cells carrying an FS construct, both the truncated (T) product of termination and the FL product of +1 frameshifting can be quantified, so we calculated frameshift efficiency (FE) as FL/(FL + T)·100% (method A). An advantage to method A is that both products are quantified from the same cells. We also estimated FE as the ratio of FL product encoded by the FS construct to FL product encoded by the corresponding in-frame C construct (FE = FLFS/FLC × 100%; method B). An advantage to method B is that it accounts for any instability of the particular fusion protein, as corresponding FS and C constructs encode the same polypeptide. Typically, both methods gave similar values. Constructs are named based on the 3′-most codon of prfB included. For example, FS-23 contains codons 1–23 of prfB, FS-74 contains codons 1–74 of prfB, and so on. Construct FS-365 carries the entire coding region of prfB. One construct, FS-M (minimal), carries only 24 bp of prfB, containing codons 17–23 (Fig. 1B).
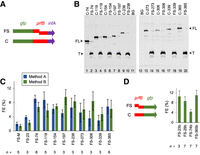
Measuring prfB frameshifting in F. johnsoniae. (A) To quantify frameshifting, DNA of prfB (various lengths) was cloned between gfp and infA, such that the production of FL fusion protein depends on frameshifting. For each frameshift-containing (FS) construct made, a corresponding in-frame control (C) construct was also made. These C constructs carry a single base pair deletion that converts the slippery sequence CUUU to the codon CUU. Numbered constructs include all codons upstream of the frameshift site and are named based on the downstream-most codon of prfB. Construct “FS-M” is the only exception and contains only codons 17–23 of prfB. (B) Examples of western blots using anti-GFP antibodies to detect truncated (T) and FL products of the fusion constructs. Lysate from untransformed F. johnsoniae was loaded in adjacent lanes (11,12) to assess background (BG). (C) The efficiency of frameshifting on mRNA from various FS constructs (as indicated), estimated by two methods, is shown. With method A (blue bars), FE was calculated as FE = FLFS/(FLFS + TFS) × 100%, where FLFS and TFS correspond to the full-length and truncated products from a given FS construct. With method B (green bars), FE = FLFS/FLC × 100%, where FLFS represents the full-length product from the FS construct, and FLC represents the same product from the corresponding in-frame C construct. (D) Another reporter system, involving prfB–gfp fusions, was also used to measure frameshifting, and method B was used for quantification. Data represent the mean ± SEM. The number of biological replicates (n) is indicated below the bars, and the raw data are given in Supplemental Table S1.
Frameshift efficiencies ranged from 2% to 10% across the strains, with most measurements near 6%–9% (Fig. 2B,C). Construct FS-74 supported a level of frameshifting as high as any other, arguing against any stimulatory role for sequences downstream from codon 74 (Fig. 2C). Constructs FS-M and FS-23 supported somewhat lower levels of frameshifting (∼2%–4%), consistent with a modest contribution of RNA between codons 23 and 74. To further investigate this, we designed another reporter system in which prfB lies upstream of gfp (Fig. 2D). Four FS constructs (FS-23b, FS-29b, FS-74b, and FS-365b; nomenclature following that described above) and their corresponding in-frame controls (C) were made, and frameshifting efficiencies were calculated using method B. Levels of frameshifting ranged from 4% to 11%, with FS-74b setting the low end of this range. FS-23b promoted frameshifting as efficiently as FS-29b and FS-365b, effectively ruling out a cis-acting element downstream from codon 23.
Idiosyncratic effects often stem from engineered gene fusions. To estimate the steady-state level of prfB frameshifting in F. johnsoniae, we used all FE measurements, except those from FS-M/C-M (which lack codons 1–17). Mean values from the 14 construct sets were averaged, yielding a value of 7%, which we consider to be a reasonable estimate of prfB FE in F. johnsoniae under these steady-state growth conditions.
The steady-state level of prfB frameshifting is substantially lower in F. johnsoniae than in E. coli
To directly compare programmed frameshifting in F. johnsoniae and E. coli cells, we cloned gfp–prfB–infA fusions FS-M, C-M, FS-23, and C-23 into the expression vector pBAD24. The resulting plasmids were transformed into E. coli, transformants were grown in the presence of arabinose to induce reporter gene transcription, and western blots were performed to quantify GFP-containing protein products (Fig. 3). Analogous F. johnsoniae strains were analyzed in parallel. We found that these constructs, based on F. johnsoniae prfB, support high-level frameshifting in E. coli (40%–60%), akin to analogous E. coli prfB constructs in E. coli (Craigen and Caskey 1986; Weiss et al. 1987; Curran and Yarus 1988). In F. johnsoniae cells, the same constructs support a much lower level of frameshifting (2%–4%) (Fig. 3).
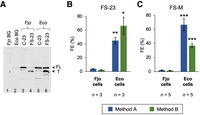
Programmed prfB frameshifting is considerably less efficient in F. johnsoniae than in E. coli. (A) An example western blot, comparing products generated from constructs FS-23 and C-23 in F. johnsoniae (Fjo) and E. coli (Eco) cells, as indicated. Annotations are as described in the legend to Figure 2. (B,C) Quantification of FE on mRNA from constructs FS-M and FS-23 (as indicated), using method A (blue) and method B (green). Data represent the mean ± SEM. The number of biological replicates (n) is indicated below the bars, and the raw data are given in Supplemental Table S1. A two-tailed t-test was used to evaluate differences (Eco vs. Fjo). Uncorrected P-values: (*) P < 0.05; (**) P < 0.005; (***) P < 0.0005.
Mutation of bS21 increases prfB frameshifting in F. johnsoniae
In E. coli, SD–ASD pairing is known to promote prfB programmed frameshifting. In F. johnsoniae, SD–ASD pairing is inhibited due to ASD sequestration on the 30S platform (Jha et al. 2021), which could explain the lower levels of frameshifting in this organism. To investigate this possibility, we moved several reporters into each of two mutant rpsU (bS21) strains of F. johnsoniae. Protein bS21 is one of the three proteins responsible for ASD sequestration, and ribosomes lacking bS21 (or its carboxyl terminus) exhibit enhanced initiation on mRNAs with strong SD sequences (including rpsU mRNA) (McNutt et al. 2023). One strain encodes the carboxy-terminally truncated variant ΔC, and the other strain encodes the singly substituted variant Y54A. In these strain backgrounds, the efficiency of frameshifting increased substantially (Fig. 4A–D). On FS-M, FS-23, and FS-74 templates, mutation ΔC increased frameshifting by approximately eightfold, approximately ninefold, and approximately fourfold, respectively. The levels of frameshifting seen in the ΔC strain of F. johnsoniae (∼30%) approach those seen in E. coli (40%–60%). Mutation Y54A also increased frameshifting, albeit to a smaller degree (by two- to fourfold). The fact that ΔC confers stronger phenotypes than Y54A is fully in line with the effects of these mutations on initiation (McNutt et al. 2023).
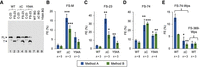
Mutations in rpsU (bS21) increase the efficiency of prfB frameshifting in F. johnsoniae. (A) An example western blot, comparing the products generated from constructs FS-23 and C-23 in wild-type (WT) and mutant (ΔC, Y54A) strains of F. johnsoniae (as indicated). Annotations are as described in the legend to Figure 2. (B–E) Quantification of FE on mRNA from construct FS-M (B), FS-23 (C), FS-74 (D), FS-74-Wps (E), or FS-369-Wps (E), using method A (blue) and method B (green). Data represent the mean ± SEM. The number of biological replicates (n) is indicated below the bars, and the raw data are given in Supplemental Table S1. A two-tailed t-test was used to evaluate differences from WT. Uncorrected P-values: (*) P < 0.05; (**) P < 0.005; (***) P < 0.0005.
The prfB sequence of Winogradskyella psychrotolerans promotes frameshifting at levels comparable to the F. johnsoniae sequence
We noticed degeneracy of the SD sequence in several Flavobacteriales species, including W. psychrotolerans (Wps) (Fig. 1B). Two FS constructs were made in which codons 1–74 or 1–369 of Wps prfB were cloned between gfp and infA. These constructs, termed FS-74-Wps and FS-369-Wps, are analogous to those described above and differ only in the source of prfB. For both, an in-frame control (C) was made, enabling us to quantify FE by both methods. These plasmids were moved into F. johnsoniae, and the efficiency of frameshifting was measured. In WT F. johnsoniae cells, FS-74-Wps and FS-74 promoted frameshifting at comparable levels, 5%–12% (Fig. 4D,E), suggesting that the SD plays no stimulatory role, at least under these conditions. As expected, mutation of the slippery sequence in either context results in no detectable FL product, confirming that product formation depends on programmed frameshifting (Supplemental Fig. S2). FS-369-Wps promoted frameshifting at ∼2% efficiency (Fig. 4E), lower than that seen for FS-365 (∼7%), FS-74 (∼8%), and FS-74-Wps (∼9%) (Figs. 2C and 4D,E). While the basis of this decrease remains unclear, it may stem from altered ribosome traffic downstream from the frameshift site. In the ΔC strain background, no higher level of frameshifting was seen for FS-74-Wps; in fact, a three- to fourfold lower level of frameshifting was observed (Fig. 4E). This suggests that the ability of mutation ΔC to stimulate prfB frameshifting depends on the SD sequence. Collectively, these data provide evidence that, in WT F. johnsoniae, prfB frameshifting does not generally involve SD–ASD pairing. However, when the sequestration mechanism is compromised by mutation, SD–ASD pairing does stimulate frameshifting.
Prevalence of the SD of the frameshifting site across the Bacteroidota
Next, we took a computational approach to assess the degeneracy of the SD in the frameshifting site across the phylum Bacteroidota. Using the program free_scan.pl (Starmer et al. 2006) and nearly the entire 3′ tail of 16S rRNA (nt 1532–1544), we determined the SD strength (i.e., mRNA–rRNA pairing free energy) for each of 256 organisms with a frameshift-containing (FS-containing) prfB (Supplemental Fig. S3A). Most organisms exhibited a strong SD (86%, ΔG < −7 kcal/mol; 71%, ΔG < −10 kcal/mol). Weaker SD sequences (−7 kcal/mol < ΔG < −4 kcal/mol) were seen for six organisms (2%), while degenerate (ΔG > −4 kcal/mol) sequences were seen for 29 organisms (11%). These latter organisms fall into distinct clades within the Flavobacteriales, Cytophagales, and Ignavibacteriales, indicating loss or degeneration of the SD in multiple lineages. Loss of the SD appears to be nonrandom, since all members of other clades, including the entire orders of Chitinophagales, Bacteroidales, and Sphingobacteriales, retain a strong SD. Conservation of the SD sequence at the nucleic acid level was confirmed by comparison of prfB sequence logos from FS-containing and FS-lacking organisms (Supplemental Fig. S3B).
Chromosomal mutations that target the SD or eliminate programmed frameshifting impact cell growth under certain stress conditions
The SD of prfB is conserved across the genus Flavobacterium (Supplemental Fig. S3A), implying that the element provides some fitness benefit. We created a strain of F. johnsoniae, FN226, that has three mutations in the SD of the prfB frameshift site (Supplemental Fig. S4A). Strain ZAM11, constructed previously (McNutt et al. 2021), carries the same SD substitutions and a single base pair deletion within the slippery sequence that eliminates the frameshift. ZAM11 showed no loss of fitness in casitone yeast extract (CYE) media at 30°C, based on growth competition assays in which co-cultures were propagated for 36 d (McNutt et al. 2021).
Growth of WT, FN226, and ZAM11 was compared in liquid CYE media at various temperatures (20°C, 30°C, 32°C, and 34°C) and in the presence of sublethal concentrations of erythromycin, kanamycin, streptomycin, tetracycline, or paraquat (Table 1; Supplemental Fig. S4B). Neither mutant showed a growth defect at suboptimal temperatures or in the presence of erythromycin, kanamycin, or tetracycline. However, in the presence of streptomycin, ZAM11 grew slightly faster than WT while FN226 grew slightly slower. In the presence of paraquat, which causes oxidative stress, ZAM11 grew slower than WT, whereas FN226 showed no difference. These data indicate that autoregulation of RF2 is beneficial under certain conditions and that the SD can be used in WT cells.
Doubling times of the wild-type and mutant strains under various growth conditions
Prevalence of the SD of the frameshifting site across all phyla
To see whether degeneration of the SD has occurred in other groups, we expanded our computational analysis to include 2686 organisms across all phyla of bacteria. Loss of the SD from the frameshift site was evident for multiple clades distributed across many phyla (Fig. 5A; Supplemental Table S1). Degeneracy was common in the Planctomycetota, Firmicutes_A, Firmicutes_B, and Cyanobacteria, as most organisms of these phyla lack the stimulatory element. While most Proteobacteria contain the SD, the genera Francisella and Buchnera show loss or degeneracy of the element. These observations underscore that programmed frameshifting in prfB can function without SD–ASD pairing.
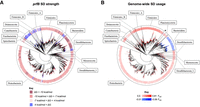
Degeneration of the prfB SD sequence in certain clades of bacteria. (A) Phylogenetic tree of 2763 bacterial species with FS-containing prfB. The leaves are colored according to the free energy of pairing between the ASD and SD of the FS site: dark red, ΔG < −10 kcal/mol; light red, −10 kcal/mol < ΔG < −7 kcal/mol; pink, −7 kcal/mol < ΔG < −4 kcal/mol; blue, −4 kcal/mol < ΔG. (B) Phylogenetic tree of the same bacterial species but with leaves colored by overall SD usage across the respective genomes. Red indicates higher SD usage than expected by chance, with FSD values ranging from 0.0 (lightest tint) to 0.81 (darkest shade). Blue indicates lower SD usage than expected by chance, with FSD values ranging from −0.01 (lightest tint) to −0.09 (darkest shade). Phyla with ≥25 representative organisms are indicated. The Verrucomicrobiales are marked with an asterisk.
Genome-wide prevalence of SD sequences in all representative bacteria
Nakagawa and coworkers compared SD usage in 277 bacteria, by identifying SD sequences within all TIRs of all genomes (Nakagawa et al. 2010). However, their analysis was done 13 yr ago, prior to most metagenomic studies, and hence did not fully capture the diversity
of bacteria. With a set of 4362 representative bacteria, we revisited the question of SD usage in initiation. Using free_scan.pl
(and nt 1532–1544 of 16S rRNA), we identified SD sequences by screening all TIRs in all genomes. As a control, we identified
mock-SD (MSD) sequences by screening an equivalent window too far upstream to contain an authentic SD. To assess SD usage
genome wide, we first calculated the average SD strength 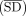 , defined as
, defined as 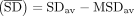 , where SDav corresponds to the mean ΔG value of all SD–ASD interactions and MSDav corresponds to the mean ΔG value of all MSD–ASD interactions (Supplemental Table S1). We also calculated FSD, a parameter analogous to dRSD of Nakagawa, which reflects the fraction of genes that contain an SD (defined by an energy threshold, in our case −4 kcal/mol)
minus the fraction of genes that contain an MSD. Supplemental Figure S5 compares the heatmaps of
, where SDav corresponds to the mean ΔG value of all SD–ASD interactions and MSDav corresponds to the mean ΔG value of all MSD–ASD interactions (Supplemental Table S1). We also calculated FSD, a parameter analogous to dRSD of Nakagawa, which reflects the fraction of genes that contain an SD (defined by an energy threshold, in our case −4 kcal/mol)
minus the fraction of genes that contain an MSD. Supplemental Figure S5 compares the heatmaps of  and FSD across the phylogenetic tree of bacteria, and the two parameters closely reflect one another. The Bacteroidia stand out in
both heatmaps, because frequencies of SD sequences in these organisms are lower than expected by chance (Nakagawa et al. 2010; Jha et al. 2021; McNutt et al. 2023). Low SD usage is also evident in the Cyanobacteria, particularly the Cyanobiaceae, in line with earlier studies (Nakagawa et al. 2010). Interestingly, like the Bacteroidia, Verrucomicrobiales also lack SD sequences, as indicated by negative FSD values (and positive
and FSD across the phylogenetic tree of bacteria, and the two parameters closely reflect one another. The Bacteroidia stand out in
both heatmaps, because frequencies of SD sequences in these organisms are lower than expected by chance (Nakagawa et al. 2010; Jha et al. 2021; McNutt et al. 2023). Low SD usage is also evident in the Cyanobacteria, particularly the Cyanobiaceae, in line with earlier studies (Nakagawa et al. 2010). Interestingly, like the Bacteroidia, Verrucomicrobiales also lack SD sequences, as indicated by negative FSD values (and positive  values) for seven of seven representative species (Supplemental Figs. S5, S6; Supplemental Table S1).
values) for seven of seven representative species (Supplemental Figs. S5, S6; Supplemental Table S1).
One might expect SD usage in programmed frameshifting to relate in some way to SD usage in initiation. However, a comparison
of prfB SD strength versus FSD (or  ) in all FS-containing organisms reveals no obvious relationship (Fig. 5A,B).
) in all FS-containing organisms reveals no obvious relationship (Fig. 5A,B).
DISCUSSION
Programmed frameshifting in prfB has been thoroughly characterized in E. coli. Key mRNA determinants include the slippery-stop sequence CUUUGA and an SD sequence just 3 nt upstream (Weiss et al. 1987). Pairing between this SD and the ASD of the paused ribosome puts tension on mRNA, which destabilizes codon–anticodon interaction in the P site and promotes the +1 shift in frame (Curran and Yarus 1988; Devaraj and Fredrick 2010). Many bacteria were found to contain the prfB frameshifting mechanism, and the SD seemed nearly as conserved as the slippery-stop sequence (Baranov et al. 2002; Bekaert et al. 2006). Thus, it was widely presumed that SD–ASD pairing is an integral part of the mechanism.
In this work, we provide evidence that prfB frameshifting does not generally involve SD–ASD pairing in F. johnsoniae. In this organism, FS constructs with or without an SD support the same level of frameshifting, ∼7%. Growth of FN226, which harbors an SD-less version of prfB, is indistinguishable from WT under eight of nine conditions tested, suggesting that RF2 production is quite similar in the two strains. Mutations of bS21 (Y54A and ΔC) increase the efficiency of frameshifting, specifically when the FS site harbors an SD. These bS21 mutations are predicted to liberate the 3′ tail of 16S rRNA from the 30S platform pocket, enabling the ASD to pair with mRNA (Jha et al. 2021). Initiation on rpsU mRNA (and other SD-containing mRNAs) is specifically stimulated by Y54A and to a greater degree by ΔC (McNutt et al. 2023), findings fully in line with the SD-dependent effects of these mutations on prfB frameshifting reported here. Thus, we infer that ASD sequestration usually prevents SD–ASD pairing during prfB frameshifting, explaining its relatively low steady-state level (∼7%).
Our deletion analysis effectively rules out any downstream stimulatory element. Most constructs, including those with the entire prfB coding region, support a frameshifting level of 6%–9%. In E. coli, certain slippery sequences can by themselves promote frameshifting at similar levels, depending on the sequence context (Gurvich et al. 2003). Amino acids of the nascent chain and nucleotides just downstream from the UGA stop codon can influence the efficiency of frameshifting (Poole et al. 1995, 1998; Major et al. 1996; Mottagui-Tabar and Isaksson 1997). These proximal features may collectively tune the frameshift site in F. johnsoniae, W. psychrotolerans, and other Flavobacteriales, enabling sufficient frameshifting in the absence of SD–ASD pairing. Autoregulation only depends on the ability of intracellular RF2 concentration to influence the fate of ribosomes (i.e., probability of termination) at the slippery-stop sequence CUUUGA. So, a change in the overall steady-state level of frameshifting from ∼50% to ∼7% may be largely inconsequential for RF2 autoregulation and cell physiology.
In E. coli, RF2 is three- to ninefold more abundant than RF1, UGA (0.30) is more than three times as common as UAG (0.09), and compelling genetic evidence suggests that RF2 is primarily responsible for termination at UAA (Mora et al. 2007; Balakrishnan et al. 2014; Baggett et al. 2017). In F. johnsoniae, on the other hand, RF1 appears to play the more prominent role in termination. RNA-seq and ribo-seq coverage on prfB is fourfold and eightfold lower, respectively, than on prfA (Baez et al. 2019). These data suggest that prfA is translated more efficiently than prfB and that RF1 is considerably more abundant than RF2 in the cell. Stop codon usage follows the trend UAA (0.79) > UAG (0.14) > UGA (0.07) (Nakamura 2000), in line with higher RF1 activity in F. johnsoniae. It has been proposed that release factor levels correlate with RF-specific stop codons across organisms and environmental conditions (Korkmaz et al. 2014), an idea that merits further investigation.
Degeneration of the prfB SD is evident in certain clades of Bacteroidota. However, the SD is conserved across the genus Flavobacterium and in orders Bacteroidales, Chitinophagales, and Sphingobacteriales, implying that the element confers some selective advantage to these organisms. Indeed, FN226 shows a subtle growth defect in the presence of 30 µg/mL streptomycin. This likely stems from inadequate RF2 levels, since ZAM11 exhibits the opposite phenotype and contains the constitutive prfB(-FS) allele. We were unable to confirm that the SD can stimulate programmed frameshifting in WT cells exposed to streptomycin (Supplemental Fig. S7); however, a small or transient response to streptomycin might be masked by the time cells reach the mid-log phase. How might streptomycin enable SD–ASD interaction in F. johnsoniae? Streptomycin binds near the 30S A site and alters ribosome structure, dynamics, and function (Shoji et al. 2006; Milon et al. 2008; Qin et al. 2012; Demirci et al. 2013a,b). Biophysical studies have shown that spin label and fluorescent probes attached to C11 of bS18 gain mobility when streptomycin (but not neomycin) binds the E. coli small subunit (Noreau et al. 1980). The corresponding residue of F. johnsoniae, C33, lies within the platform pocket (Jha et al. 2021). It is possible that streptomycin allosterically affects the pocket, destabilizing the 3′ tail and enabling SD engagement. Further studies will be needed to determine when and how the SD of prfB is used in F. johnsoniae.
Strain ZAM11 exhibits two subtle phenotypes—faster growth in the presence of streptomycin and slower growth in the presence of paraquat. To our knowledge, these data are the first to link prfB autoregulation to physiology in any bacterium. The former phenotype hints at dysregulation of growth rate control. In E. coli and other bacteria, guanosine tetra/penta-phosphate [(p)ppGpp] plays a central role in regulating cell growth in response to nutrient changes or stress (Vercruysse et al. 2011; Ma et al. 2019; Horvatek et al. 2020; Wu et al. 2022; Zhao et al. 2023; Zhu and Dai 2023). The (p)ppGpp synthetase, RelA, is activated by binding the A site of stalled ribosomes along with cognate deacyl tRNA (Arenz et al. 2016; Loveland et al. 2016). Increased ppGpp globally shunts transcription in the cell, reducing the expression of ribosome-related genes and increasing the expression of metabolic, transport, and stress-protective genes (Horvatek et al. 2020; Zhu and Dai 2023). RF2 also binds the A site, and the bound factor would occlude RelA and tRNA interactions. It is possible that in ZAM11 cells, excess RF2 acts as a competitive inhibitor of RelA on the ribosome, perturbing growth rate control. This could lessen the cell's growth rate response to streptomycin and compromise the cell's defense against oxidative stress, a hypothesis worth testing in the future.
We observed degeneration of the prfB SD beyond the Bacteroidota, in certain clades of multiple phyla, providing further evidence that the frameshifting mechanism can operate without SD–ASD pairing. Rampant loss of the stimulatory element was seen in the Planctomycetota, Firmicutes_A, Firmicutes_B, and Cyanobacteria. High SD usage is characteristic of the former three groups, indicating that their ribosomes contain a functional (nonsequestered) ASD. Why the intragenic SD of prfB would be lost in these organisms is particularly puzzling, as there is predicted cost (less efficient RF2 production) with no apparent benefit. Future work will be needed to solve this paradox.
Finally, as part of this work, we revisited the question of genome-wide SD usage across bacteria, using a much larger set
of representative organisms. To do so, we identified and calculated pairing free energies of putative SD sequences for all
TIRs of 4362 organisms. The resulting data confirm and extend Nakagawa's finding that SD usage varies widely across bacteria
(Nakagawa et al. 2010, 2017). SD sequences are most prevalent in the Firmicutes, Firmicutes_A–H, Fusobacteriota, Synergistota, and Thermotogota, with
FSD values typically greater than 0.6. More moderate SD usage (0.2 < FSD < 0.5) is characteristic of Acidobacteriota, Actinobacteriota, Aquificota, Bdellovibrionota, Chloroflexota, Deinococcota,
Desulfobacterota, Myxococcota, Planctomycetota, Proteobacteria, and Spirochaetota. SD usage is lowest in three groups—Bacteroidia,
Verrucomicrobiales, and Cyanobiaceae. In many of these organisms, SD sequences are detected at frequencies lower than expected
by chance, as indicated by negative FSD values (and positive  values). Ribosomes of F. johnsoniae sequester the 3′ tail of 16S rRNA in a pocket on the 30S platform, a mechanism conserved across the Bacteroidia (Jha et al. 2021). Mutant ΔC ribosomes, which have a liberated 3′ tail, initiate poorly on SD-less mRNA, consistent with the possibility that
sequestration plays a beneficial role for initiation on most mRNAs (McNutt et al. 2023). It will be of interest to determine the structures of ribosomes from representative Verrucomicrobiales and Cyanobiaceae
species and see if the ASD is functionally occluded in these cases as well.
values). Ribosomes of F. johnsoniae sequester the 3′ tail of 16S rRNA in a pocket on the 30S platform, a mechanism conserved across the Bacteroidia (Jha et al. 2021). Mutant ΔC ribosomes, which have a liberated 3′ tail, initiate poorly on SD-less mRNA, consistent with the possibility that
sequestration plays a beneficial role for initiation on most mRNAs (McNutt et al. 2023). It will be of interest to determine the structures of ribosomes from representative Verrucomicrobiales and Cyanobiaceae
species and see if the ASD is functionally occluded in these cases as well.
MATERIALS AND METHODS
Constructs with gfp–prfB–infA translational fusions
Gibson assembly (Gibson et al. 2009) was used to clone a gfp–prfB–infA gene fusion into the vector portion of pSCH710 (Baez et al. 2019), linearized with Bam HI and Sph I. The gfp fragment was amplified from pSCH710, and the prfB and infA fragments were amplified from the genome of F. johnsoniae UW101. All of the fragments were designed to have appropriate overlapping sequences. Linkers encoding three glycines were introduced at the gfp–prfB and prfB–infA junctions. Restriction sites Nhe I and Sac I were also introduced to facilitate subsequent cloning of prfB (various lengths) between gfp and infA. The resulting plasmid, pFN20, was then modified by replacing the Kpn I–Sal I fragment containing lacI with a DNA cassette containing Pant, a constitutive E. coli promoter. The resulting plasmid, pFN21, has both an E. coli promoter and F. johnsoniae promoter upstream of the gfp–prfB–infA gene fusion. Next, the rrnB T1 T2 terminator sequence was PCR amplified from pBAD18 (Guzman et al. 1995), and infA was PCR amplified from pFN21. The primers were designed to allow for overlap extension PCR, generating a fragment carrying infA with a Pst I site and the terminator sequence downstream. This fragment was then cloned into pFN21 via Sac I and Sph I, replacing the terminator-lacking infA fragment, to generate pFN22.
Various lengths of the prfB gene (excluding the start codon) were amplified from the F. johnsoniae chromosome. In parallel, the same primers were used to amplify corresponding fragments from strain ZAM11 (McNutt et al. 2021), which harbors a single base pair deletion in prfB that removes the frameshift. The amplified fragments were each cloned into the Nhe I and Sac I sites of pFN22, replacing the resident fragment, to create pFN29 to pFN46 (Supplemental Table S1). To generate plasmids pFN98–pFN101, the same approach was used except that the slippery-site mutations were introduced via overlap extension PCR. To create constructs FS-M, C-M, FS-23, and C-23, complementary oligonucleotides were annealed, and the resulting cassettes were cloned into the Nhe I and Sac I sites of pFN22, resulting in pFN25, pFN26, pFN75, and pFN76 (Supplemental Table S1).
To study frameshifting in E. coli, the fusion-containing Xba I–Sph I fragments of pFN25, pFN26, pFN75, and pFN76 were cloned into the same sites of pBAD24 (Guzman et al. 1995). This resulted in plasmids pFN49, pFN50, pFN77, and pFN78 (Supplemental Table S1).
Constructs with prfB–gfp translational fusions
To create the prfB–gfp reporter constructs (FS-23b, FS-29b, FS-74b, and FS-365b), prfB fragments of various lengths (which all include 138 bp of the leader region) were amplified from F. johnsoniae UW101. Each was cloned upstream of gfp in pDW01 (Baez et al. 2019), via Bam HI and Xho I restriction sites. The resulting fusions contain six glycine codons between the prfB and gfp coding regions. The corresponding in-frame control constructs (C-23b, C-29b, C-74b, and C-365b) were made the same way, except that strain ZAM11 (McNutt et al. 2021) DNA was used as the PCR template. All plasmid names and descriptions are listed in Supplemental Table S1.
Constructs with Winogradskyella psychrotolerans DNA
The full prfB sequence of W. psychrotolerans (Wps), flanked by sequences corresponding to the polylinker region of vector pCR2.1-TOPO (Thermo Fisher), was ordered from Integrated DNA Technologies as a gene block (gBlock). Using Gibson assembly, this DNA was cloned into pCR2.1-TOPO, creating pFN65. Site-directed mutagenesis (Hemsley et al. 1989) using Phusion DNA polymerase (New England Biolabs) was then performed to make a variant, pFN66, in which the frameshift is removed (CTTTGAC to CTTGAC). Using pFN65 and pFN66 as templates, fragments carrying fusions of various lengths were amplified and cloned into pFN22 via Nhe I and Sac I, generating pFN69, pFN70, pFN71, and pFN72 (Supplemental Table S1).
Strain construction
Plasmids were moved into F. johnsoniae strains UW101 (WT), ZAM64 (ΔC), and ZAM65 (Y54A) by conjugation, using triparental mating as described (McBride and Kempf 1996; McNutt et al. 2023). Plasmids were moved into E. coli CSH142 (Miller 1992) by transformation.
To make strain FN226, precise allelic replacement was used (Zhu et al. 2017). Mutant alleles were generated by separately amplifying ∼1 kb regions from the F. johnsoniae chromosome both upstream and downstream from the mutagenesis site. The primers contained three mutations that effectively removed the SD sequence (AGGAGG to CGTAGA). The two fragments were then combined via PCR overlap extension, and the combined fragment was cloned into the Bam HI and Sph I sites of suicide vector pYT313 (Zhu et al. 2017). The resulting plasmid, pFN102, was moved into F. johnsoniae, via triparental mating (McBride and Kempf 1996), and erythromycin (Erm, 100 µg/mL) resistant transconjugants were selected. Colonies were then screened for plasmid integration at the appropriate chromosomal locations using PCR. Confirmed recombinants were then grown overnight in the absence of Erm, to allow for loss of the plasmid via a second recombination event, and then cells were plated on 5% (w/v) sucrose, to select against the plasmid. Sucrose-resistant, Erm-sensitive colonies were then screened via PCR for the mutant strain.
Western blotting
Flavobacterium johnsoniae cells were grown in CYE media (McBride and Kempf 1996) at 30°C without or with Erm (100 µg/mL). E. coli strains were grown in LB media at 37°C without or with ampicillin (100 µg/mL). Overnight cultures were diluted 100-fold into fresh media on the day of the experiment. For cells harboring pBAD24-derived or pDW01-derived plasmids, arabinose (1.33 mM) or IPTG (1 mM) was included in the media, respectively, to induce fusion expression. Note, pFN22-derived plasmids lack lacI, hence the fusions are constitutively expressed in F. johnsoniae. For the experiment of Supplemental Figure S7, streptomycin was added at 30 µg/mL. Cells (1 mL) were grown to OD600 ≈ 0.5, pelleted, washed with 1 mL of PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4; pH 7.4), and resuspended in 1 mL of PBS. OD600 measurements were performed, and equivalent amounts of cells were transferred to fresh tubes, pelleted, and stored overnight at −80°C. Pellets were resuspended in 50 µL RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% SDS, 0.5% sodium deoxycholate, 1% Triton-X-100) with freshly added PMSF (1 mM). Samples were heated to 98°C for 6 min, and lysates were clarified by centrifugation (21,130g for 10 min at 4°C). Lysates were mixed 1:1 with 2× Laemmli loading buffer (125 mM Tris-HCl pH 6.8, 4% SDS, 20% glycerol, 0.2% bromophenol blue) containing freshly added β-mercaptoethanol (710 mM). Samples were heated to 98°C for 5 min and clarified by microcentrifugation for 1 min. Samples were then loaded into an SDS polyacrylamide gel (10%–12%) and electrophoresed. Transfer of proteins from the gel to a nitrocellulose membrane was done using a Bio-Rad transfer apparatus (Mini-PROTEAN Tetra System), with constant amperage (250 mA; 40–50 mV) for 2.5 h. The membrane was then incubated at 4°C overnight with 3% BSA in TBS-T buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% Tween 20). The blocking solution was removed, and the primary anti-GFP-HRP antibody (Thermo Fisher Scientific) was diluted 1000-fold into TBS-T and incubated with the membrane for 2 h at room temperature. The membrane was then washed with TBS-T five times (three 30-min washes, two 15-min washes). Clarity Western ECL substrate (Bio-Rad) was then applied to the membrane for 6 min, and images were taken using the Bio-Rad Chemi-Doc imager. Quantification of images was done using Image-Lab software (Bio-Rad).
Growth rate measurements
Flavobacterium johnsoniae strains UW101 (WT), ZAM11, and FN226 were cultured overnight in CYE media at 30°C. Cells were diluted 500-fold into media (without or with various inhibitors), and aliquots (200 µL) were distributed into 96-well plates. Plates were incubated at 30°C, 32°C, or 34°C with continuous double orbital shaking in an Epoch 2 BioTek Microplate Spectrophotometer. Growth was monitored by measuring OD600 every 15 or 30 min. Inhibitors were added at sublethal concentrations (erythromycin, 4 µg/mL; kanamycin, 80 µg/mL; streptomycin, 30 µg/mL; tetracycline, 1 µg/mL; paraquat, 100 µg/mL). MgSO4 was omitted from CYE when kanamycin or tetracycline was present. Growth at 20°C was measured by culturing cells (50 mL) in Erlenmeyer flasks (250 mL) and monitoring OD600 every 60 min. For each experiment, log (OD600) was plotted versus time, and points clearly within the exponential phase were used to calculate doubling time.
Selection of genomes
A list of bacterial organisms designated as “species representative” and with an NCBI assembly level “complete genome” was obtained from the Genome Taxonomy Database (GTDB) (Parks et al. 2022). Genomes and annotations were obtained from NCBI using the RefSeq assembly summary file available on NCBI's file transfer protocol system. A total of 4465 such organisms were identified (Supplemental Table S1).
Detection of prfB frameshifting sites
The ARFA tool was run on all assemblies with default parameters (Bekaert et al. 2006). A prfB gene was identified in 4318 of these assemblies (Supplemental Table S1). An E-value threshold of 0.1 for ORF0 was used to qualify prfB gene annotations with a detected frameshift site (threshold set based on previously reported results; McNutt et al. 2021). This resulted in 2763 assemblies with a frameshift-containing prfB (Supplemental Table S1).
Creation of sequence logos
Organisms belonging to class Bacteroidia and containing a detected prfB gene were split into two groups: those with (n = 246) and without (n = 133) a detected frameshift site. Sequence logos were created using the Python package Logomaker (Tareen and Kinney 2020) for each group. Nucleotide and amino acid sequences used to generate the logos are included in Supplemental Table S1.
Detection of SD sequences
For each organism, ASD sequences were identified at the 3′ ends of the 16S rRNA. In brief, barrnap v0.9 was used with default settings to predict 16S rRNA sequences. For each 16S rRNA prediction, the sequence region from 100 nt upstream of the 3′ end to 10 nt downstream from the 3′ end was extracted. CutAdapt v3.4 (Martin 2011) was used to find a highly conserved 16S rRNA motif with the settings [-g AAGTCGTAACAAGGTAGCCGT –e 0.25 -O 21 –discard-untrimmed], and the sequence positioned 19–32 nt downstream was isolated as the extended ASD (i.e., 16S nucleotides 1532–1544). To ensure high-quality data, ASDs from every rRNA were collected and only organisms with one unique ASD were maintained.
SD sequences were identified using the free_scan.pl program, which enables the calculation of the free energy of pairing between the ASD and mRNA (Starmer et al. 2006). For the prfB frameshifting analysis, we defined the SD region as the window 20 nt upstream to 4 nt downstream from the prfB frameshifting site. For the genome-wide SD analysis, we detected SD and MSD sequences as described previously (McNutt et al. 2023), scanning a 29 nt window just upstream of the start codon (−29 to 0) for SDs and a 29 nt window further upstream (−55 to −26) for MSDs. Organisms were stratified by taxonomy into groups of interest (see Supplemental Table S1).
Annotation of phylogenetic trees
Phylogenetic trees using the GTDB taxonomy system were generated using iTOL (Letunic and Bork 2021). GTDB taxonomy tree distances (e.g., branches) between each node were maintained. For organisms containing a detected prfB frameshifting site, their immediate branch and name were color coded based on their SD pairing free energy (ΔG) via the following schema: dark red (ΔG < −10 kcal/mol), light red (−10 kcal/mol < ΔG < −7 kcal/mol), pink (−7 kcal/mol < ΔG < −4 kcal/mol), blue (−4 kcal/mol < ΔG). Phyla containing at least 25 organisms with a prfB frameshifting site were annotated.
For the genome-wide SD analysis, we defined two metrics to measure a genome's SD usage:  and FSD.
and FSD. 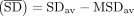 , where SDav corresponds to the mean ΔG value of all SD–ASD interactions, and MSDav corresponds to the mean ΔG value of all MSD–ASD interactions. FSD = SDfract − MSDfract, where SDfract is the fraction of genes with an SD (energy threshold −4 kcal/mol), and MSDfract is the fraction of genes with an MSD (energy threshold −4 kcal/mol). These metrics were painted as heatmaps onto the phylogenetic
trees, as specified in the keys or legends.
, where SDav corresponds to the mean ΔG value of all SD–ASD interactions, and MSDav corresponds to the mean ΔG value of all MSD–ASD interactions. FSD = SDfract − MSDfract, where SDfract is the fraction of genes with an SD (energy threshold −4 kcal/mol), and MSDfract is the fraction of genes with an MSD (energy threshold −4 kcal/mol). These metrics were painted as heatmaps onto the phylogenetic
trees, as specified in the keys or legends.
DATA DEPOSITION
All custom scripts used in data analysis for this manuscript are available on GitHub under https://github.com/bundschuhlab/PublicationScripts/tree/master/prfBFrameshiftingEfficiencyFJO. SD and MSD strengths for each gene for each of the 4362 organisms analyzed are available at Zenodo under doi: 10.5281/zenodo.7930098.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Science Foundation (NSF; MCB-2029502 to K.F.) and by a fellowship from the OSU Center for RNA Biology (to F.M.N.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079721.123.
-
Freely available online through the RNA Open Access option.
- Received May 15, 2023.
- Accepted October 27, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Fawwaz M. Naeem is the first author of this paper, “Analysis of programmed frameshifting during translation of prfB in Flavobacterium johnsoniae.” Fawwaz is a graduate student in the Ohio State Biochemistry Program at The Ohio State University, working in Kurt Fredrick's lab. His research involves translation in Flavobacterium johnsoniae, focusing on prfB and bS21.
What are the major results described in your paper and how do they impact this branch of the field?
Our work shows that translation of prfB is different in Flavobacterium johnsoniae (Fjo) compared to Escherichia coli (Eco). Here, we show that the prfB +1 programmed frameshifting frequency is much lower in Fjo compared to Eco and that frameshifting does not generally involve Shine–Dalgarno (SD)–anti-Shine–Dalgarno (ASD) pairing in Fjo. This is mainly due to the unique ability of Fjo ribosomes to sequester the ASD sequence of the 16S rRNA. We also show degeneration of the SD sequence within the prfB frameshift site in many clades of bacteria. Additionally, this work is the first to link prfB autoregulation to physiology in any bacterium.
What led you to study RNA or this aspect of RNA science?
Ever since I learned about the RNA world hypothesis in my undergraduate career, I became interested in studying RNA as part of my graduate school journey. RNA research is an exciting and emerging field with many possibilities for therapeutics and future discoveries both on the cellular and medicinal level. It was inspiring to see RNA research scientists playing an important role during the COVID-19 pandemic, where their rigorous efforts came together to quickly develop an mRNA vaccine.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
When I first started working on this project, we had an initial hypothesis about an RNA element downstream from the prfB frameshift site; however, our results did not show evidence for such an element and revealed that frameshifting was lower than expected based on the Eco model, which was surprising to us. This led us to consider new hypotheses and change the trajectory of the work, resulting ultimately in this published paper.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
My natural sense of curiosity led me toward the path to become a scientist. While I was an undergraduate student at Kansas State University, I immediately joined a research team studying muscle proteostasis in fruit flies under the direction of Dr. Erika Geisbrecht, which provided me the tools that helped shape me as a scientist today and inspired me to continue my scientific journey into graduate school. During my undergraduate research experience, it felt natural to work in a lab and be part of a research team. I cannot imagine a place better for myself now than in Dr. Kurt Fredrick's lab, where I continue to expand my work and lab skills to become an independent scientist.
If you were able to give one piece of advice to your younger self, what would that be?
My advice to my younger self is to not let worry deter you from pursuing and achieving your goals. Believe in yourself because no matter what barriers and obstacles you face, you will overcome them. And remember that your family will always support your efforts and be there for you.
What are your subsequent near- or long-term career plans?
I would like to continue my journey in academia through a post-doc where I will expand my scientific toolkit. In the future, I hope to either lead a lab or work in industry as part of a research team focusing on RNA therapeutics.








