
Identification, characterization, and structure of a tRNA splicing enzyme RNA 5′-OH kinase from the pathogenic fungi Mucorales
- 1Molecular Biology Program, Memorial Sloan Kettering Cancer Center, New York, New York 10065, USA
- 2Microbiology and Immunology Department, Weill Cornell Medical College, New York, New York 10065, USA
- Corresponding author: shumans{at}mskcc.org
-
Handling editor: Eric Phizicky
Abstract
Fungal Trl1 is an essential tRNA splicing enzyme composed of C-terminal cyclic phosphodiesterase and central polynucleotide kinase end-healing domains that convert the 2′,3′-cyclic-PO4 and 5′-OH ends of tRNA exons into the 3′-OH,2′-PO4 and 5′-PO4 termini required for sealing by an N-terminal ATP-dependent ligase domain. Trifunctional Trl1 enzymes are present in most human fungal pathogens and are untapped targets for antifungal drug discovery. Mucorales species, deemed high-priority human pathogens by WHO, elaborate a noncanonical tRNA splicing apparatus in which a stand-alone monofunctional RNA ligase enzyme joins 3′-OH,2′-PO4 and 5′-PO4 termini. Here we identify a stand-alone Mucor circinelloides polynucleotide kinase (MciKIN) and affirm its biological activity in tRNA splicing by genetic complementation in yeast. Recombinant MciKIN catalyzes magnesium-dependent phosphorylation of 5′-OH RNA and DNA ends in vitro. MciKIN displays a strong preference for GTP as the phosphate donor in the kinase reaction, a trait shared with the stand-alone RNA kinase homologs from Mucorales species Rhizopus azygosporus (RazKIN) and Lichtheimia corymbifera (LcoKIN) and with the kinase domains of fungal Trl1 enzymes. We report a 1.65 Å crystal structure of RazKIN in complex with GDP•Mg2+ that illuminates the basis for guanosine nucleotide specificity.
Keywords
INTRODUCTION
Intron-containing tRNAs are present in all Eukarya, whereby the intron is inserted at position 37 within the anticodon loop. Because the intron is inimical to tRNA function, it must be removed by incision at the exon–intron junctions followed by joining of the tRNA exons. Eukaryal tRNA splicing endonuclease breaks the phosphodiester at the exon–intron junctions to generate 2′,3′-cyclic phosphate and 5′-OH termini on the tRNA exons and the excised intron (Hayne et al. 2023). In fungi, tRNA exon joining entails four enzymatic reactions comprising sequential end-healing, end-sealing, and junction-healing phases (Shuman 2023). The fungal tRNA ligase enzyme Trl1 executes three distinct RNA repair reactions: (i) the 2′,3′-cyclic phosphate (>p) end is hydrolyzed to a 3′-OH,2′-PO4 by a cyclic phosphodiesterase (CPD); (ii) the 5′-OH end is phosphorylated by a GTP-dependent polynucleotide kinase; and (iii) the 3′-OH,2′-PO4 and 5′-PO4 ends are sealed by an ATP-dependent RNA ligase to form an unconventional 2′-PO4, 3′-5′ phosphodiester at the splice junction (Fig. 1A; Greer et al. 1983). Trl1 is composed of three separable catalytic domains: a C-terminal CPD module belonging to the 2H phosphoesterase superfamily; a central GTP-dependent kinase module of the P-loop phosphotransferase superfamily; and an N-terminal ATP-dependent RNA ligase domain that belongs to the covalent nucleotidyltransferase superfamily (Fig. 1B; Sawaya et al. 2003; Remus et al. 2017; Banerjee et al. 2019a,b,c; Peschek and Walter 2019). The CPD and kinase reactions are collectively referred to as end-healing (Schwer et al. 2004). The distinctive feature of the ligase domain of Trl1 is that its RNA end-sealing activity is strictly dependent on the 2′-PO4 moiety (Greer et al. 1983; Ghosh and Shuman 2024). The residual 2′-PO4 mark at the splice junction formed by Trl1 is removed by a dedicated 2′-phosphotransferase enzyme, Tpt1, that transfers the tRNA 2′-PO4 to NAD+ to form ADP-ribose-1′,2′-cyclic phosphate and nicotinamide in a two-step pathway that proceeds through a 2′-phospho-ADP-ribosylated tRNA intermediate (McCraith and Phizicky 1991; Culver et al. 1993; Spinelli et al. 1999; Steiger et al. 2005; Munir et al. 2018; Banerjee et al. 2019b; Jacewicz et al. 2023).
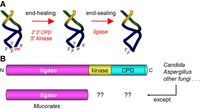
Pathway of fungal tRNA splicing and domain organization of the canonical trifunctional tRNA ligase Trl1 versus the monofunctional Mucorales RNA ligase. (A) Intron removal by tRNA splicing endonuclease leaves 2′,3′-cyclic phosphate and 5′-OH ends on the broken tRNA halves. The tRNA exons are then joined by Trl1—a trifunctional tRNA ligase. Trl1 catalyzes two end-healing reactions, performed by a 5′-OH polynucleotide kinase domain and a polynucleotide 2′,3′-CPD domain, to generate the 5′-PO4 and 3′-OH,2′-PO4 termini required for sealing by an ATP-dependent RNA ligase domain. The 2′-PO4 at the resulting splice junction is subsequently removed by the NAD +-dependent RNA 2′-phosphotransferase enzyme Tpt1. (B) Fungal Trl1 consists of N-terminal ligase, central kinase, and C-terminal CPD catalytic modules. Mucorales species have a stand-alone RNA ligase enzyme homologous to the Trl1 ligase domain. Initial searches of Mucorales proteomes did not identify candidate orthologs of the Trl1 kinase and CPD domains.
Each of the four enzymatic steps catalyzed by Trl1 and Tpt1 is essential for the viability of the budding yeast Saccharomyces cerevisiae (Sawaya et al. 2003; Wang and Shuman 2005; Wang et al. 2006). Metazoa rely on an entirely different series of chemical steps to achieve tRNA exon splicing, catalyzed by the GTP-dependent RNA ligase RtcB (which is structurally and mechanistically unrelated to the fungal RNA ligase domain), in a pathway that does not involve a 5′-OH kinase step and does not generate a 2′-PO4 splice junction (Shuman 2023). This divergence in tRNA splicing along phylogenetic lines highlights the fungal tRNA splicing enzymes as plausible targets for the discovery of novel antifungal agents active against the foremost human fungal pathogens, which has been designated by the World Health Organization as a top priority for research and public health action (World Health Organization 2022).
Nearly all the WHO-prioritized fungal pathogens—including Aspergillus, Candida, Coccidioides, Blastomyces, Histoplasma, Cryptococcus, Talaromyces, Lomentospora, Scedosporium, and Pneumocystis species—encode a trifunctional Trl1 enzyme homologous to S. cerevisiae Trl1. The biological activity of exemplary pathogen tRNA splicing enzymes has been affirmed by genetic complementation in budding yeast (Remus et al. 2016; Dantuluri et al. 2021; Ahammed and van Hoof 2024), and their biochemical activities have been interrogated as purified recombinant enzymes (Remus et al. 2016, 2017; Dantuluri et al. 2021). The exceptions to the rule are the priority pathogens of the order Mucorales, which cause serious (often fatal) infections acquired either by inhalation of fungal spores leading to infection of the sinuses, brain, and/or lungs or by entry of spores through a break in the skin following a burn or other skin trauma. Risk factors for mucormycosis include diabetes, steroid use, cancer, organ transplant, and COVID-19 infection (World Health Organization 2022).
Our group and the van Hoof laboratory reported recently that Mucorales species elaborate a variant of the fungal tRNA splicing machinery in which the RNA ligase is a stand-alone enzyme, homologous to the ligase domain of Trl1 (Fig. 1B), that is active in vivo in budding yeast in lieu of the Trl1 ligase domain (Ahammed and van Hoof 2024; Ghosh et al. 2024). Biochemical characterization of the Mucor circinelloides RNA ligase affirmed its requirement for a 2′-PO4 terminus in the end-joining reaction (Ghosh et al. 2024). At first glance, neither group was able to identify the missing Mucorales end-healing enzymes. Thus, doubt was raised (Ahammed and van Hoof 2024) as to the suitability of any putative inhibitors of the kinase and CPD domains of the canonical fungal Trl1 enzyme for activity against Mucorales. In the present study, we identify and characterize the elusive Mucorales RNA 5′-kinase as a stand-alone enzyme of the P-loop phosphotransferase superfamily. The M. circinelloides kinase, and the homologous kinases of Mucorales species Rhizopus azygosporus and Lichtheimia corymbifera, resemble the kinase domains of the trifunctional fungal Trl1 enzymes with respect to their specificity for GTP as the phosphate donor, thereby fortifying the case for the tRNA-splicing RNA kinase as a potential antifungal target. We present a crystal structure of the Rhizopus kinase that reveals the basis for its guanosine nucleotide specificity.
RESULTS
Mucorales species encode a stand-alone polynucleotide kinase
Mucorales comprise an order within the phylum Mucoromycota, subphylum Mucoromycotina, class Mucoromycetes. Our initial PSI-Blast search of Mucorales taxa with S. cerevisiae Trl1 (827 aa) retrieved proteins from M. circinelloides (372 aa), R. azygosporus (370 aa), and L. corymbifera (390 aa) homologous to the N-terminal RNA ligase domain but did not return convincing homologs of the Trl1 kinase or CPD modules. By performing a PSI-Blast search of the kingdom Fungi with M. circinelloides RNA ligase (MciRNL 372 aa), we found that Mucoromycota taxa within subphylum Glomeromycotina, class Glomeromycetes encode a canonical trifunctional Trl1 protein. Thus, the organization of the tRNA splicing apparatus diverged during the branching of the Mucoromycota subphyla. We then deployed the kinase-CPD segment of the Trl1 homolog from Glomeromycetes species Gigaspora margarita (GenBank KAF0554865.1) to search Mucoromycotina and thereby retrieved a 208-aa M. circinelloides protein (GenBank EPB87039.1) with moderate homology (36% amino acid identity/similarity) to the kinase domain of the Gigaspora bait protein, including conservation of the NTP-binding P-loop motif (GSGKST). We did not retrieve a credible CPD homolog.
A search of Mucorales with the candidate M. circinelloides kinase readily identified homologs from R. azygosporus (174 aa) and L. corymbifera (163 aa). A primary structure alignment of the three Mucorales proteins shows that they share 106 positions of amino acid side chain identity/similarity (denoted by dots in Fig. 2A). The Mucorales candidate kinases include the P-loop element (shaded cyan in Fig. 2A) that engages the phosphates of the NTP donor and the divalent cation cofactor and a downstream aspartate (shaded yellow in Fig. 2A) that engages the 5′-OH of the polynucleotide acceptor in other well-characterized polynucleotide kinases (Eastberg et al. 2004; Das et al. 2014; Dikfidan et al. 2014).
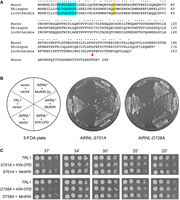
Mucorales encode a stand-alone kinase active in tRNA splicing in vivo. (A) Alignment of the primary structures of candidate polynucleotide kinases from M. circinelloides (GenBank EPB87039.1), R. azygosporus (GenBank RCH95593.1), and L. corymbifera (GenBank CDH52573.1). Positions of side chain identity/similarity are indicated by dots. The signature P-loop motif that engages the NTP phosphate donor is highlighted in cyan. A conserved aspartate implicated in the activation of the polynucleotide 5′-OH nucleophile is shaded in yellow. A conserved serine (Ser146), implicated in the present study as a determinant of the GTP donor specificity of the Mucorales kinases, is denoted by a red arrowhead. (B) Complementation of S. cerevisiae trl1Δ was assayed by plasmid shuffle (Sawaya et al. 2003; Schwer et al. 2004). Yeast trl1Δ p360-TRL1 (URA3 CEN) cells were cotransformed with: (i) an empty CEN HIS3 vector and an empty CEN TRP1 vector (negative control); (ii) an empty CEN HIS3 vector and a CEN TRP1 plasmid encoding S. cerevisiae TRL1 (positive control); (iii) an empty CEN HIS3 vector and a CEN TRP1 plasmid expressing either of two kinase-defective AtRNL* mutants of the Trl1-like plant tRNA ligase (AtRNL-S701A or AtRNL-D726A); (iv) the CEN TRP1 AtRNL* plasmid and a CEN HIS3 plasmid expressing the KIN-CPD module of S. cerevisiae TRL1 (aa 389–827); and (v) the CEN TRP1 AtRNL* plasmid and a CEN HIS3 or 2µ HIS3 plasmid expressing MciKIN. Trp+ His+ transformants were selected at 30°C and then streaked on agar medium containing 0.75 mg/mL FOA. The plates were photographed after 4 days at 30°C. (C) Aliquots (3 µL) of serial fivefold dilutions of FOA-resistant trl1Δ strains expressing the indicated genes were spotted on YPD agar plates and incubated at 20, 25, 30, 34, and 37°C. Photographs of the plates are shown.
Mucor KIN is active in vivo in S. cerevisiae
We showed previously that fusion of the LIG, KIN, and CPD domains within a single polypeptide is not critical for S. cerevisiae tRNA splicing in vivo (Sawaya et al. 2003) and that expression of heterologous plant, viral, or bacterial RNA repair systems can complement the lethality of a trl1Δ knockout in S. cerevisiae (Schwer et al. 2004; Wang et al. 2006; Nandakumar et al. 2008; Tanaka et al. 2011). Complementation of budding yeast defective for one of the tRNA splicing reactions (achieved by plasmid shuffle using kinase-defective or CPD-defective missense mutants of AtRNL, a plant ortholog of yeast Trl1) provides a surrogate means to validate the RNA repair capacity of candidate end-healing enzymes from heterologous sources (Ramirez et al. 2008; Schwer et al. 2008; Jain and Shuman 2009). To query whether a Mucorales kinase candidate is a bona fide tRNA kinase, we cloned the ORF encoding Mucor circinelloides kinase (MciKIN) into yeast CEN HIS3 and 2µ HIS3 plasmids wherein its expression is driven by the S. cerevisiae TPI1 promoter. We tested complementation by plasmid shuffle in an S. cerevisiae trl1Δ p(CEN URA3 TRL1) strain (Sawaya et al. 2005). Complementation tests entailed cotransformation with: (i) an empty CEN HIS3 vector and an empty CEN TRP1 vector (negative control); (ii) an empty CEN HIS3 vector and a CEN TRP1 plasmid encoding S. cerevisiae TRL1 (positive control); (iii) an empty CEN HIS3 vector and a CEN TRP1 plasmid expressing either of two kinase-defective AtRNL* mutants of the Trl1-like plant tRNA ligase (AtRNL-S701A or AtRNL-D726A) that are specifically defective for RNA kinase activity in vivo and in vitro (Wang et al. 2006; Remus and Shuman 2013); (iv) the CEN TRP1 AtRNL* plasmid and a CEN HIS3 plasmid expressing the KIN-CPD module of S. cerevisiae TRL1 (aa 389–827); and (v) the CEN TRP1 AtRNL* plasmid and a CEN HIS3 or 2µ HIS3 plasmid expressing MciKIN. Whereas we recovered viable trl1Δ p(CEN TRP SceTRL1) strains that lacked the p(CEN URA3 TRL1) plasmid after selection for growth on medium containing 5-FOA (5-fluoroorotic acid), the trl1Δ cells cotransformed with AtRNL* mutants and an empty vector failed to yield viable colonies on 5-FOA medium (Fig. 2B). The two lethal kinase-defective AtRNL* mutations were complemented in trans by coexpression of the KIN-CPD domain of SceTRL1 (Fig. 2B). The instructive finding was that the trl1Δ MciKIN + AtRNL* cotransformants grew on 5-FOA medium (Fig. 2B). The trl1Δ AtRNL* MciKIN strains grew as well as the trl1Δ SceTRL1 control when spot-tested on rich agar medium at 25–37˚C (Fig. 2C). In contrast, the trl1Δ AtRNL* SceKIN-CPD strains were slow-growing at 37˚C (Fig. 2C). We conclude that MciKIN can execute the 5′-OH RNA kinase step of fungal tRNA splicing in vivo.
Polynucleotide kinase activity of recombinant MciKIN
We produced MciKIN in E. coli as a His10Smt3 fusion and purified it from a soluble extract by sequential Ni-affinity chromatography/imidazole elution, removal of the His10Smt3 tag by treatment with Ulp1 protease, recovery of the tag-free MciKIN protein in the flowthrough of a second Ni-affinity column, and a final Superdex-200 gel filtration step. SDS-PAGE analysis of the MciKIN preparation, comprising a single 24 kDa polypeptide, is shown in Figure 3A.
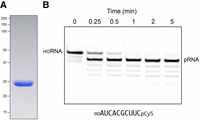
RNA kinase activity of recombinant MciKIN. (A) An aliquot (10 µg) of purified recombinant MciKIN was analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. (B) A reaction mixture (110 µL) containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM DTT, 1 mM MgCl2, 0.1 mM GTP, 100 nM 10-mer 5′-OH RNA substrate 3′-labeled with Cy5 (depicted at bottom; purchased from IDT), and 5 nM MciKIN was incubated at 37˚C. The reactions were initiated by adding MciKIN to a prewarmed reaction mixture. Aliquots (10 µL) were withdrawn prior to adding enzyme (time 0) or at the times specified after enzyme addition and quenched immediately with an equal volume of 90% formamide, 50 mM EDTA. The products were analyzed by electrophoresis (at 10 W constant power) through a 15 cm 20% polyacrylamide gel containing 7 M urea in 45 mM Tris-borate, 1 mM EDTA. The Cy5-labeled RNAs were visualized by scanning the gel with a Typhoon biomolecular imager (Cytiva). The positions of the 10-mer HORNA substrate and the 10-mer pRNA product are indicated.
To assay kinase activity, we deployed as substrate a synthetic 10-mer 5′-OH RNA oligonucleotide with a 3′-Cy5 fluorescent tag (Fig. 3B). The 5′-OH RNA (100 nM) was incubated at 37˚C with 5 nM MciKIN in the presence of 1 mM Mg2+ and 0.1 mM GTP for 0.25, 0.5, 1, 2, and 5 min. The products were analyzed by urea-PAGE and visualized by scanning with a Typhoon biomolecular imager (Fig. 3B). MciKIN converted the 10-mer 5′-OH RNA substrate into a more rapidly migrating 5′-PO4 product in a time-dependent fashion. The substrate preparation contained a minor ladder of serially shorter 3′-Cy5-labeled RNAs (comprising 14% of the total Cy5 signal) that were also phosphorylated during the kinase reaction (Fig. 3B). Analyzing the fluorescence intensity in ImageQuant TL indicated that 70% of the input 10-mer 5′-OH RNA was converted to 10-mer pRNA in 0.25 min, which translates into a turnover number of ∼54 min−1.
MciKIN is specific for GTP as the phosphate donor
No 5′ phosphorylation was detected in the absence of exogenous GTP (Fig. 4, lane 0). To test NTP substrate preference, 5′-OH RNA (100 nM) was reacted for 5 min at 37˚C with 5 nM MciKIN and 1 mM Mg2+ in the presence of 1, 10, 100, or 1000 µM ATP, GTP, CTP, or UTP (Fig. 4). Back-titrating the GTP concentration from 1000 to 10 µM had little effect on the yield of phosphorylated RNA. The fact that kinase activity was detectable (27% conversion of 5′-OH RNA 10-mer to pRNA 10-mer) at 1 µM GTP (i.e., 10-fold molar GTP excess over the input RNA substrate) attests to the efficiency with which GTP is used as the phosphate donor. In contrast, the extents of RNA phosphorylation with 1000 µM CTP (13% conversion) or UTP (17% conversion) were less than that seen with 1 µM GTP, signifying that CTP and UTP were at least three orders of magnitude less effective as phosphate donors. MciKIN generated no detectable pRNA in the presence of 100, 10, or 1 µM CTP and UTP. The extents of RNA phosphorylation with 100 µM ATP (14% conversion) and 1000 µM ATP (30% conversion) versus 1 µM GTP indicated that ATP was at least two orders of magnitude less effective than GTP as a phosphate donor for the Mucor kinase.
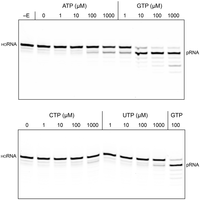
GTP is the preferred phosphate donor for MciKIN. Reaction mixtures (10 µL) containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM DTT, 1 mM MgCl2, 100 nM (1 pmol) 10-mer 5′-OH RNA substrate 3′-labeled with Cy5, 5 nM (50 fmol) MciKIN, and 0, 1, 10, 100, or 1000 µM of the indicated NTP were incubated at 37˚C for 5 min. MciKIN was omitted from the reaction mixture in lane –E. The reactions were quenched with formamide/EDTA, and the products were analyzed by urea-PAGE. The positions of the 10-mer HORNA substrate and the 10-mer pRNA product are indicated.
Divalent cation specificity
GTP-dependent RNA 5′-OH phosphorylation by MciKIN was strictly dependent on an added divalent cation (Fig. 5A). The metal cofactor requirement was satisfied by 1 mM Mg2+, Mn2+, and Ca2+, whereas 1 mM Co2+, Cu2+, Ni2+, and Zn2+ were ineffective (Fig. 5A). We proceeded to conduct a metal mixing experiment in which kinase reactions containing 1 mM Mg2+ were supplemented with 1 mM of another divalent cation. We found that Co2+, Cu2+, Ni2+, and Zn2+ inhibited MciKIN activity in the presence of magnesium (Fig. 5B). These findings lead us to speculate that: (i) the relatively “hard” metals magnesium and calcium, and also manganese, can form a MceKIN•GTP•Me2+ donor complex that is permissive for catalysis; and (ii) the inhibitory “soft” metals copper, nickel, zinc, and cobalt out-compete magnesium for occupancy of a divalent cation site on the enzyme-substrate complex, wherein engaged they are unable to support kinase reaction chemistry.
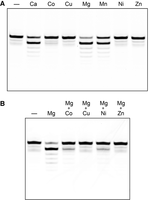
Divalent cation requirement. (A) Metal specificity. Reaction mixtures (10 µL) containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 0.1 mM GTP, 100 nM (1 pmol) 10-mer 5′-OH RNA substrate 3′-labeled with Cy5, 5 nM (50 fmol) MciKIN, and either no added divalent cation (lane –) or 1 mM CaCl2, CoCl2, CuCl2, MgCl2, MnCl2, NiCl2, or ZnCl2 as specified were incubated at 37˚C for 5 min. (B) Inactive metals are inhibitory. Reaction mixtures (10 µL) containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 0.1 mM GTP, 100 nM (1 pmol) 10-mer 5′-OH RNA substrate 3′-labeled with Cy5, 5 nM (50 fmol) MciKIN, and either no added divalent cation (lane –), 2 mM MgCl2, or 1 mM MgCl2 plus 1 mM CoCl2, CuCl2, NiCl2, or ZnCl2 as specified were incubated at 37˚C for 5 min. The reaction products were analyzed by urea-PAGE.
MciKIN can use RNA or DNA as the phosphate acceptor
In the experiment shown in Figure 6, we assayed GTP-dependent 5′-OH phosphorylation of 3′-Cy5-tagged 10-mer 5′-OH RNA and 5′-OH DNA substrates (with the same nucleotide sequence) as a function of input MciKIN during a 5 min reaction at 37˚C. Whereas RNA and DNA were phosphorylated to similar extents at saturating MciKIN, specific activity with DNA was ∼twofold lower than with RNA based on the titration profile.

RNA kinase versus DNA kinase activity. Reaction mixtures (10 µL) containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM DTT, 1 mM MgCl2, 0.1 mM GTP, 100 nM (1 pmol) 10-mer 5′-OH RNA or DNA substrate 3′-labeled with Cy5 (depicted at top), and MciKIN as specified were incubated at 37˚C for 5 min. The reactions were quenched with formamide/EDTA, and the products were analyzed by urea-PAGE.
Asp36 is essential for MciKIN activity
Asp36 is predicted to act as a general base catalyst that accepts a proton from the 5′-OH of the polynucleotide acceptor in the kinase reaction (Eastberg et al. 2004; Wang et al. 2002; Das et al. 2014; Dikfidan et al. 2014). To test this idea, we mutated Asp36 to alanine and asparagine (the latter being an isostere of aspartate that can engage in hydrogen bonding but not in proton transfer) and purified the recombinant D36A and D36N proteins (Fig. 7A). Whereas 50 fmol of wild-type MciKIN sufficed to phosphorylate all the input 5′-OH RNA substrate, the D36A mutant failed to form detectable pRNA product at up to 400 fmol of input protein (Fig. 7B). The D36N mutant displayed no activity at 50 fmol of input protein and only scant formation of pRNA by 400 fmol of enzyme (Fig. 7B). These results are consistent with a catalytic role for MciKIN Asp36, as proposed for other 5′-OH polynucleotide kinases.
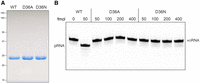
Asp36 is essential for MciKIN activity. (A) Aliquots (10 µg) of purified recombinant wild-type, D36A, and D36N MciKIN proteins were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. (B) Reaction mixtures (10 µL) containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM DTT, 1 mM MgCl2, 0.1 mM GTP, 100 nM (1 pmol) 10-mer 5′-OH RNA substrate 3′-labeled with Cy5, and wild-type, D36A, or D36N MciKIN proteins as specified were incubated at 37˚C for 5 min. The reactions were quenched with formamide/EDTA, and the products were analyzed by urea-PAGE. The positions of the 10-mer HORNA substrate and the 10-mer pRNA product are indicated.
GTP-dependent RNA kinase activity of Rhizopus and Lichtheimia KIN proteins
We produced R. azygosporus KIN (RazKIN; 174 aa), and L. corymbifera KIN (LcoKIN; 163 aa) in E. coli as His10Smt3 fusions and purified them from soluble extracts by sequential Ni-affinity chromatography/imidazole elution, removal of the His10Smt3 tag by treatment with Ulp1 protease, recovery of the tag-free RazKIN and LcoKIN proteins in the flowthrough of a second Ni-affinity column, and a final Superdex-200 gel filtration step. SDS-PAGE analysis showed that the RazKIN and LcoKIN preparations comprised single polypeptides of the expected size (Fig. 8A). LcoKIN and RazKIN (at 5 nM concentration) catalyzed efficient phosphorylation of 100 nM HORNA during a 5 min reaction at 37˚C with 1 mM Mg2+ in the presence of 10 µM GTP (93% and 82% conversion to pRNA, respectively), 100 µM GTP (94% and 91% conversion, respectively), or 1000 µM GTP (94% and 91% conversion, respectively (Fig. 8B,C). No activity was observed when GTP was omitted. Partial phosphorylation occurred when GTP was added at 1 µM concentration (a 10-fold molar excess over the HORNA substrate), to the extent of 72% conversion by LcoKIN and 58% conversion by RazKIN (Fig. 8B,C), thereby underscoring that these enzymes can scavenge limiting amounts of the GTP phosphate donor. In contrast, LcoKIN and RazKIN displayed feeble activity in the presence of the same concentrations of ATP, CTP, and UTP (Fig. 8B,C). The extents of RNA phosphorylation by LcoKIN and RazKIN observed in the presence of 1 mM ATP (63% and 69%, respectively), 1 mM CTP (74% and 52%, respectively), and 1 mM UTP (66% and 60%, respectively) were similar to that seen with 1 µM GTP. We surmise that GTP is between two and three orders of magnitude more effective than other ribonucleoside triphosphates as a substrate for LcoKIN and RazKIN.

RNA kinase activity of Rhizopus and Lichtheimia KIN proteins. (A) Aliquots (10 µg) of purified recombinant RazKIN and LcoKIN proteins were analyzed by SDS-PAGE. The Coomassie blue-stained gel is shown. The positions and sizes (kDa) of marker polypeptides are indicated on the left. (B,C) Reaction mixtures (10 µL) containing 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 2 mM DTT, 1 mM MgCl2, 100 nM (1 pmol) 10-mer 5′-OH RNA substrate 3′-labeled with Cy5, 5 nM (50 fmol) LcoKIN (B) or RazKIN (C), and 0, 1, 10, 100, or 1000 µM of the indicated NTP were incubated at 37˚C for 5 min. The reactions were quenched with formamide/EDTA, and the products were analyzed by urea-PAGE.
Crystal structure of RazKIN in complex with GDP•Mg2+
Crystals of RazKIN that had been preincubated with 5 mM GTP and 10 mM Mg2+ were grown by sitting drop vapor diffusion against a precipitant solution containing 0.2 M NH4Cl and 20% PEG-3350. Crystals diffracting to between 1.4 Å and 2.0 Å resolution were in space group P212121 and contained one RazKIN protomer in the asymmetric unit. The structure was solved by molecular replacement using 1.65 Å diffraction data from a single crystal and a RazKIN search model generated in AlphaFold (Abramson et al. 2024). The refined experimental structure model (Rwork/Rfree = 16.9/23.7, Table 1) comprised a continuous polypeptide (aa 1–172), with unambiguous density corresponding to GDP•Mg2+ in the active site (Fig. 9A). We surmise that the input GTP was hydrolyzed by the enzyme, either during crystal growth or in the crystal. A stereo view of the RazKIN•GDP•Mg2+ complex is shown in Figure 9B. The enzyme consists of a central 5-strand parallel β-sheet flanked by two α-helices on the left side and two α-helices and a 310 helix on the right side. The β1–α1 phosphate-binding loop (P-loop) contains the signature 13GSGKST18 motif that engages the triphosphate moiety of the NTP donor. In the RazKIN structure, P-loop Lys16-Nζ contacts the β phosphate and Thr18-OH coordinates the α phosphate (Fig. 10). In addition, the P-loop Gly13, Ser14, Gly15, Lys16, Ser17, and Thr18 amide nitrogens donate hydrogen bonds to the GDP α and β phosphates. The six ligands of the octahedral Mg2+ coordination complex are: a GDP β phosphate oxygen, Ser17-Oγ, and four waters (Fig. 10). The α4-loop-α5 segment forms a lid that covers the GDP phosphates (Fig. 9B). The lid α4 helix residue Arg99 makes a cation–π stack on the guanine base while lid loop residue Arg103 coordinates the α and β phosphates (Fig. 10). A “G-loop” element connecting the β5 strand and the α6 helix (named after an analogous loop in the Candida Trl1 RNA kinase domain) forms a pocket around the guanine base (Fig. 9B). The G-loop Pro149 main-chain carbonyl makes a hydrogen bond to the 2′-OH of the ribose sugar (Fig. 10). G-loop residue Ser146 (which is conserved in LcoKIN and MciKIN; denoted by the red triangle in Fig. 2A) makes a network of five hydrogen bonds from the main-chain carbonyl oxygen and amide nitrogen atoms and the side-chain hydroxyl group to the guanine N1, N2, and O6 atoms (Figs. 9B and 10) that would account for the observed guanine nucleotide specificity of the Mucorales kinases.
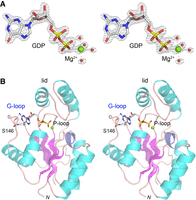
Structure of Rhizopus RNA kinase in complex with GDP•Mg2+. (A) Stereo view of an omit electron density map (gray mesh, contoured at 2σ) covering the GDP and Mg2+•(H2O)4 ligands in the kinase active site. GDP is rendered as a stick model with gray carbons and yellow phosphorus atoms. A magnesium ion engaging the GTP β phosphate is depicted as a green sphere. Waters occupying five of the six positions in the octahedral magnesium coordination complex are rendered as red spheres. (B) The tertiary structure of RazKIN is shown as a cartoon trace with β-strands colored magenta, α-helices colored cyan, and a 310 helix colored blue. The helix-loop-helix “lid” overlying the GDP and the P-loop that engages the GDP phosphates are indicated in black font. The “G-loop” that engages the guanine nucleobase is indicated in blue font. Hydrogen bonds to the guanine from Ser146 in the G-loop are denoted by dashed lines.
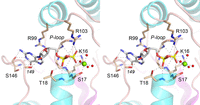
The kinase phosphodonor site and atomic contacts to GDP•Mg2+. A detailed stereo view of the kinase phosphodonor site is shown. Kinase side chain and main chain contacts to GDP and the Mg2+•(H2O)4 complex are depicted as dashed lines.
Crystallographic data and refinement statistics
A Dali search of the Protein Data Bank (Holm et al. 2023) with the RazKIN structure returned numerous P-loop phosphotransferase enzymes that share a common fold. The top hits (Z scores between 14.2 and 16.6) were: (i) the 5′ polynucleotide kinase domain of mammalian 5′-kinase/3′-phosphatase (PNKP), a DNA repair enzyme (PDB 1YJ5); (ii) Methanocaldococcus jannaschii O-phosphoseryl-tRNA kinase, an enzyme involved in selenocysteine synthesis (PDB 3ADD); (iii) Chaetomium thermophilum Kti12, an ATPase involved in tRNA anticodon modification (PDB 6QP0); (iv) the 5′ polynucleotide kinase module of Clostridium thermocellulum Pnkp, a trifunctional RNA repair complex composed of 5′ kinase, 2′,3′ phosphatase, and RNA ligase domains (PDB 4GP6); (v) the kinase domain of human 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PDB 2AXN); and (vi) the 5′ polynucleotide kinase domain of Candida albicans Trl1 (PDB 6U05).
DISCUSSION
The present study fortifies previous evidence that the Mucorales tRNA splicing machinery is an exception to the rule whereby fungal tRNA splicing is catalyzed by a trifunctional Trl1 enzyme composed of fused colinear healing and sealing domains. Rather, Mucorales deploy separately encoded stand-alone monofunctional RNA ligase and RNA kinase enzymes, structurally and functionally homologous to the LIG and KIN domains of Trl1, respectively. Here and previously (Ghosh et al. 2024), we showed by genetic complementation that the Mucor RNA kinase (MciKIN) and RNA ligase (MciRNL) are capable of performing the 5′ end-healing and end-sealing steps of budding yeast tRNA splicing in vivo. Biochemical characterization of the monofunctional Mucorales LIG and KIN enzymes revealed that they share the distinctive substrate specificities of the LIG and KIN domains of the canonical fungal Trl1 enzymes: (i) in the case of the ligase, a requirement for a 2′-PO4 for end joining; and (ii) for the kinase, a strong preference for GTP as the phosphate donor. Thus, we feel justified in applying abductive reasoning to surmise that Mucor MciRNL and MciKIN, and their orthologs in Rhizopus and Lichtheimia, are agents of tRNA splicing in those taxa. To wit, both enzymes pass the “duck test” whereby “if it looks like a duck, swims like a duck, and quacks like a duck, then it most likely is a duck.” Of course, definitive genetic proof would entail demonstration that RNL and KIN are essential for the viability of Mucorales fungi, followed by the creation of conditional Mucorales mutant strains with loss-of-ligase or loss-of-kinase function, followed by demonstration that such mutant fungi accumulate unligated tRNA exons under restrictive conditions. Such proof will not be readily forthcoming given that Mucorales are not genetically tractable à la budding yeast.
Our efforts to identify a functional Mucorales counterpart of the CPD domain of Trl1, a member of the 2H phosphoesterase family (Banerjee et al. 2019b), have not borne fruit. It is conceivable that: (i) a putative Mucorales 2H-type CPD has diverged from the Trl1 CPD to the point that it is not easily recognized; (ii) Mucorales rely on an entirely different flavor of enzyme to convert 2′,3′-cyclic phosphate tRNA exon ends into the 3′-OH,2′-PO4 ends required for end joining by the Mucorales RNA ligase; or (iii) Mucorales tRNA splicing endonuclease is capable of directly generating 3′-OH,2′-PO4 ends during intron removal, thereby obviating the need for a separate 3′ end-healing enzyme.
A key finding from our biochemical characterization is that the RNA kinases from three different Mucorales species display high specificity for GTP as the phosphate donor, akin to what was reported previously for the Trl1 kinase domains from Candida albicans, Aspergillus fumigatus, Coccidioides immitis, Kluyveromyces lactis, and S. cerevisiae (Sawaya et al. 2003; Remus and Shuman, 2014; Remus et al. 2016, 2017). This GTP preference distinguishes the fungal tRNA-splicing kinases from the kinase domain of the trifunctional plant tRNA ligase AtRNL, and from the kinase domain of the bacterial Hen1•Pnkp RNA repair complex, both of which are active with either ATP, GTP, CTP, or UTP as the phosphate donor (Das et al. 2013; Remus and Shuman 2013). Crystal structures of the Candida Trl1 kinase in complex with GTP•Mg2+ and GDP•Mg2+ highlighted a unique G-loop element that accounts for guanine nucleotide specificity and mutations of amino acids that contact the guanine nucleobase effaced kinase activity in vitro and Trl1 function in vivo (Remus et al. 2017; Banerjee et al. 2019b). It was suggested that the distinctive G-loop and guanine-binding pocket of Candida Trl1 kinase affords a plausible target for a small molecule that could selectively inhibit the fungal Trl1 kinase without affecting the many other P-loop superfamily phosphotransferases that either have no nucleobase preference or specifically use ATP as the phosphate donor. It would be optimal to identify or design a shared chemical scaffold capable of inhibiting the GTP-dependent RNA kinases from multiple fungal pathogens.
To evaluate the degree to which the principles of guanine nucleotide recognition might be conserved, we solved a crystal structure of RazKIN in complex with GDP and Mg2+. In most respects, the RazKIN•GDP•Mg2+ structure resembles the structure of Candida Trl1 kinase with GDP•Mg2+ (Dali Z score 14.2; 2.7 Å rmsd at 153 Cα positions). The most instructive feature of the RazKIN structure is the presence of a G-loop between β5 and α6 (analogous to, but not identical to, the G-loop in the Candida kinase) that makes a network of hydrogen bonds from the Ser146 side-chain hydroxyl, main-chain carbonyl, and main-chain amide to the guanine N2, N1, and O6 atoms (Fig. 9) that neatly account for the GTP donor specificity of the Mucorales kinases. As noted previously (Remus et al. 2017), the length and amino acid sequence of the G-loop segment varies between Trl1 kinase enzymes from different fungal genera. The Candida G-loop (KLSKDENSSKSS; with amino acids that contact guanine highlighted in boldface) differs from the G-loop conserved among the Mucorales kinases (DPSP[D/E][T/P][Y/V]CT; Ser146 in boldface).
Whereas the present study expands our understanding of the donor specificity of fungal tRNA splicing RNA kinases, the principles of RNA acceptor recognition are unknown. Closing this knowledge gap will require experimental structure determination of fungal RNA kinases from diverse genera, captured as their respective Michaelis complexes with GTP•Mg2+ and 5′-OH RNA.
MATERIALS AND METHODS
Expression plasmids for Mucorales RNA kinases
Synthetic DNA open reading frames (ORFs) encoding M. circinelloides RNA kinase MciKIN (GenBank EPB87039.1), L. corymbifera RNA kinase LcoKIN (GenBank CDH52573.1), and R. azygosporus RNA kinase RazKIN (GenBank RCH95593.1) were purchased from Integrated DNA Technologies. The synthetic genes, which were codon-optimized for expression in E. coli, were PCR-amplified with primers that introduced a BamHI site immediately flanking the start codon and an XhoI site downstream from the stop codon. The PCR products were digested with BamHI and XhoI. The KIN ORFs were inserted into pET28b-His10Smt3 to generate T7 RNA polymerase-based expression plasmids encoding the KIN polypeptides fused to an N-terminal His10Smt3 tag. Missense mutations D36A and D36N were introduced into the pET28b-His10Smt3-MciKIN plasmid by using the Agilent QuikChange II Site-Directed Mutagenesis kit. The MciKIN ORF was also inserted into yeast expression vectors under the transcriptional control of the yeast TPI1 promoter, thereby yielding plasmids p413-MciKIN (CEN HIS3 MciKIN) and p423-MciKIN (2µ HIS3 MciKIN). The plasmid inserts were sequenced to verify that no unwanted coding changes were introduced during PCR amplification and cloning.
MciKIN purification
The pET28b-His10Smt3-MciKIN plasmid was transformed into E. coli BL21(DE3) cells. A 1000 mL culture amplified from a single kanamycin-resistant transformant was grown at 37°C in Terrific Broth medium containing 60 μg/mL kanamycin until the A600 reached 0.8–1.0. The culture was chilled on ice for 1 h, adjusted to 2.2% (v/v) ethanol and 0.5 mM IPTG, and then incubated for 20 h at 17°C with constant shaking. Cells were harvested by centrifugation and stored at −80°C. All subsequent steps were performed at 4°C. Cells were thawed and resuspended in 50 mL of buffer A (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM DTT, 20 mM imidazole, 10% sucrose), containing 1 complete EDTA-free protease inhibitor cocktail tablet (Roche). Lysozyme was added to a concentration of 1 mg/ml. After incubation for 30 min, the lysate was sonicated to reduce viscosity, and the insoluble material was removed by centrifugation at 38,000g for 45 min. The supernatant was mixed for 1 h with 10 mL of Ni-nitrilotriacetic acid agarose resin (Qiagen) that had been equilibrated with buffer A. The resin was recovered by centrifugation and washed twice with 50 mL of buffer A, then with 20 mL of 50 mM Tris-HCl, pH 8.0, 3 M KCl, followed by 100 mL of buffer B (50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM DTT, 10% glycerol) containing 20 mM imidazole. The resin was centrifuged again, resuspended in 20 mL of buffer B with 20 mM imidazole, and poured into a column. The bound material was eluted with buffer B containing 300 mM imidazole, while collecting 5 mL fractions. The polypeptide compositions of the flowthrough and eluate fractions were monitored by SDS-PAGE. The 300 mM imidazole eluate fractions containing His10Smt3-MciRNL (15 mL volume) were supplemented with Smt3-specific protease Ulp1 (Ulp1/His10-Smt3-MciKIN ratio of 1:425 [w/w]) and then dialyzed overnight against 2000 mL of buffer C (50 mM Tris-HCl, pH 8.0, 250 mM NaCl, 20 mM imidazole, 1 mM DTT, 10% glycerol) containing 1 mM EDTA, during which time the His10Smt3 tag was cleaved. The dialysates were mixed for 1 h with 10 mL of Ni-nitrilotriacetic acid agarose resin that had been equilibrated with buffer C (lacking EDTA). Tag-free MciKIN protein was recovered in the flowthrough fractions, which were concentrated by centrifugal ultrafiltration (Amicon Ultra-15; 10 kDa cutoff) to 11 mL volume. Two aliquots of MciKIN (5 mL each) were gel-filtered through a 125 mL 16/60 HiLoad Superdex 200 column (GE Healthcare) equilibrated in buffer D (20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM DTT, 1 mM EDTA, 5% glycerol) at a flow rate of 0.5 mL/min while collecting 2 mL fractions. The peak fractions were pooled, concentrated by centrifugal ultrafiltration, and stored at −80°C. Protein concentrations were determined with Bio-Rad dye reagent using bovine serum albumin as the standard. The yield of wild-type MciKIN was ∼80 mg per liter of bacterial culture. The yields of the D36A and D36N mutants were 18 and 22 mg per liter of culture, respectively. RazKIN and LcoKIN were expressed and purified as described for MciKIN, with a final protein yield of 55 mg and 65 mg per liter of bacterial culture, respectively.
Crystal structure of RazKIN
A solution of RazKIN (7 mg/mL in buffer D), 5 mM GTP, and 10 mM MgCl2 was prepared on ice. An aliquot (110 nl) of the RazKIM solution was mixed with an equal volume of precipitant/well solution containing 0.2 M NH4Cl, 20% (w/v) PEG-3350. Crystals were grown by sitting drop vapor diffusion at room temperature in 96-well crystallization plates. Single crystals that grew after 2 days were cryoprotected with well solution containing 40% PEG-400 and flash-frozen in liquid nitrogen. Diffraction data were collected at the National Synchrotron Light Source II (NSLS-II) FMX 17-ID-2 beamline with 0.2° oscillation. Data were processed and scaled in XDS (Kabsch 2010), POINTLESS and AIMLESS in the CCP4 Program Suite (Agirre et al. 2023). An AlphaFold model of RazKIN without nucleotide was used as the search model for a molecular replacement solution in PHASER (McCoy et al. 2007). The resulting model had one RazKIN protomer in the asymmetric unit. The model was refined in REFMAC (Murshudov et al. 2011) and COOT (Emsley and Cowtan 2004). Omit difference maps of RazKIN, calculated in PHENIX (Adams et al. 2010), revealed unambiguous density for GDP and an adjacent octahedral Mg2+ coordination complex. The final model at 1.65 Å resolution was refined to Rwork/Rfree of 16.9/23.7, with no Ramachandran outliers (Table 1).
DATA DEPOSITION
Structure coordinates have been deposited in Protein Data Bank under accession code 9D8A.
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health (NIH) grants R35-GM126945 (S.S.) and R01-GM134021 (B.S.). The MSKCC structural biology core laboratory is supported by National Cancer Institute grant P30-CA008748. X-ray diffraction data were collected at the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The FMX beamline in the Center for BioMolecular Structure (CBMS) is primarily supported by the National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30-GM133893) and by the DOE Office of Biological and Environmental Research (KP1605010). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080247.124.
- Received August 27, 2024.
- Accepted September 24, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








