
Abolished frameshifting for predicted structure-stabilizing SARS-CoV-2 mutants: implications to alternative conformations and their statistical structural analyses
- 1Department of Biotechnology, National Institute of Pharmaceutical Education and Research (NIPER-R)-Raebareli, Lucknow 226002, India
- 2Department of Chemistry, New York University, New York, New York 10003, USA
- 3Courant Institute of Mathematical Sciences, New York University, New York, New York 10012, USA
- 4NYU-ECNU Center for Computational Chemistry, NYU Shanghai, Shanghai 200062, P.R. China
- 5NYU Simons Center for Computational Physical Chemistry, New York University, New York, New York 10003, USA
- 6Department of Biology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA
- Corresponding author: rf.abhishek.dey{at}niperrbl.ac.in
-
Handling editor: Peter Stadler
Abstract
The SARS-CoV-2 frameshifting element (FSE) has been intensely studied and explored as a therapeutic target for coronavirus diseases, including COVID-19. Besides the intriguing virology, this small RNA is known to adopt many length-dependent conformations, as verified by multiple experimental and computational approaches. However, the role these alternative conformations play in the frameshifting mechanism and how to quantify this structural abundance has been an ongoing challenge. Here, we show by DMS and dual-luciferase functional assays that previously predicted FSE mutants (using the RAG graph theory approach) suppress structural transitions and abolish frameshifting. Furthermore, correlated mutation analysis of DMS data by three programs (DREEM, DRACO, and DANCE-MaP) reveals important differences in their estimation of specific RNA conformations, suggesting caution in the interpretation of such complex conformational landscapes. Overall, the abolished frameshifting in three different mutants confirms that all alternative conformations play a role in the pathways of ribosomal transition.
Keywords
INTRODUCTION
Programmed ribosomal frameshifting (PRF) is used by multiple RNA viruses to translate proteins from overlapping open reading frames (ORF) of their genome (Brierley 1995; Dinman 2006). This mechanism involves repositioning of the translating ribosome by 1 or 2 nt (nucleotides), thus effectively changing the frame of translation (Staple and Butcher 2003; Atkins et al. 2016). PRF is basically of many types: The most common are −1 PRF (backtracking of ribosomes by 1 nt) (Atkins et al. 2016) and +1 PRF (skipping of 1 nt by translating ribosomes) (Harger et al. 2002). The additional rarer kind of PRF involves +1 and −2 frameshifting of translating ribosomes (Napthine et al. 2017). All frameshifting mechanisms are regulated by one or more different kinds of mechanisms (Napthine et al. 2017). −1 PRF happens when the ribosome is forced to move back by 1 nt toward the 5′ direction and further continues protein synthesis by reading ORF in the −1 frame, or in other words, ribosomes read a different codon sequence (Dinman 2012). Three RNA-specific elements regulate −1 PRF (Napthine et al. 2017). These are a slippery sequence, a spacer region, and an RNA secondary structure. The slippery sequence typically consists of a ZZZNNNH motif with Z as any three identical nucleotides; N could be U or A, and H could be A, C, and U (Kelly et al. 2020). Codons involved in the formation of a slippery sequence have very rarely associated tRNA, thus inducing the slippage of translating ribosomes and enabling −1 PRF (Harger et al. 2002). This slippery sequence is connected to an RNA secondary structure (another regulatory element of −1 PRF) through a spacer. This secondary structure enhances the effect of a slippery sequence by stalling the ribosome during translation and forcing it to relocate by −1 nt in the 5′ direction (Brierley 1995). This kind of regulation enables the virus to encode multiple proteins (Bhatt et al. 2021). Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) uses this −1 PRF to regulate the translation of proteins from its alternative overlapping ORFs, ORF1a and ORF1b (Kelly et al. 2020; Bhatt et al. 2021; Schlick et al. 2021a,b; Zhang et al. 2021).
The genetic makeup of SARS-CoV-2 is composed of single-stranded positive-sense RNA. Out of its 10 ORFs, partially overlapped ORF1a and ORF1b of SARS-CoV-2 code for nonstructural proteins, including RNA-dependent RNA polymerase (RdRP), an essential enzyme critical for maintaining the viral life cycle inside the host cell (Subissi et al. 2014; Malone et al. 2022). Translation of RdRP from ORF1b is facilitated by the −1 PRF, which is triggered by a specific mRNA structure present at the interface of ORF1a and ORF1b (Subissi et al. 2014). Additionally, a hepta-nucleotide sequence known as “slippery site” is joined to this RNA structural element at its 5′ end by a short spacer region, thus constituting the RNA frameshift signaling element (FSE) which regulates the −1 PRF in SARS-CoV-2 (Kelly et al. 2020). As mentioned above, pausing during ribosomal translation is followed by subsequent unfolding in the downstream RNA structure to continue protein translation from a new ORF.
Previous biophysical studies have determined that the three-stem H-type pseudoknot is the most likely RNA structure present in the FSE of SARS-CoV-2 RNA genome (Bhatt et al. 2021; Roman et al. 2021; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022). A 6.9 Å cryo-EM structure of 88 nt long SARS-CoV-2 FSE forms a λ-shaped conformation, with the 5′ end being flexible enough to switch between “threaded” and “unthreaded” conformation, thus regulating the FSE activity (Zhang et al. 2021). Two recent crystal structures of SARS-CoV-2 FSE also identified it as a H-type pseudoknot; however, these RNA FSE include deletions of certain residues to stabilize their crystal structures (PDB ID: 7MLX and 7LYJ) (Roman et al. 2021; Jones and Ferré-D'Amaré 2022). Our earlier study, which includes the combination of RNA as graph-based modeling (RAG), two-dimensional structure prediction algorithms, and chemical probing and mutational profiling studies on multiple FSE lengths of SARS-CoV-2, highlighted the presence of at least three conformations that SARS-CoV-2 FSE can adopt (Schlick et al. 2021a).
Significantly, we identified three alternative RNA FSE structures in our prior study of 77 nt RNA constructs (Schlick et al. 2021a). In addition to two different H-type pseudoknots (3_6 and 3_3 dual graphs), an unknotted three-way junction (3_5 graph) was also identified in the FSE landscape (Schlick et al. 2021a).
The extensive body of existing literature on the SARS-CoV-2 FSE makes it an ideal model system for understanding the role of multiple conformations in RNA processes. Here, we study multiple FSE constructs, including structure stabilizing mutants predicted earlier in Schlick et al. (2021a,b), by DMS probing coupled with mutational profiling data and corresponding frameshifting efficiency assays. The abolished frameshifting on these mutants and consensus analysis using several algorithms that deconvolute the DMS chemical reactivity into multiple conformations (including DANCE-MaP [Olson et al. 2022], DREEM [Tomezsko et al. 2020], and DRACO [Morandi et al. 2021]) underscores the importance of conformational transitions during frameshifting, and the potential for new therapeutic strategies that can be developed based on selected RNA fold distributions.
RESULTS
Analysis of DMS-informed structures in the context of cryo-EM structures
We begin our investigation of the SARS-CoV-2 FSE secondary structure landscape by evaluating solved three-dimensional structures. In Figure 1A, we show the structural model 87 nt SARS-CoV-2 FSE construct, including the slippery sequence based on the electron density obtained from Cryo-EM (PDB ID 6XRZ) (Zhang et al. 2021). As can be seen in Figure 1B, several structural models were able to fit the cryo-EM density (PDB ID 6XRZ) (Zhang et al. 2021). There is high level structural agreement in the FSE region of these 87 nt FSE Cryo-EM models (PDB ID 6XRZ), with slightly more flexibility in the slippery site (Fig. 1A,B). Previous molecular dynamics simulations of this construct agree with this level of conformational dynamics, especially in stem helix 2 (Rangan et al. 2021; Schlick et al. 2021b). In all these models, however, the consensus secondary structure is the three stem 3_6 pseudoknot (H-type pseudoknot) (Fig. 1C).
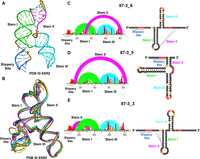
Comparison of experimentally derived secondary and tertiary conformations for the 86 nt SARS-CoV-2 FSE. (A) 3D structure displaying 3_6 H-type pseudoknot conformation in λ form obtained through cryo-EM (PDB ID 6XRZ). (B) Multiple cryo-EM derived structural models (PDB ID 6XRZ) display conformational flexibility in 87 nt FSE. The 5′ end consisting of a slippery site is highly flexible; however, all alternative 3D conformations adopt the same 3_6 H-type pseudoknot secondary structure conformation. (C) Arc plot and radial layout computed through DMS-MaP and SHAPEknots for 86 nt FSE 3_6 H-type pseudoknot secondary structure (D) 3_5 three-way junction and (E) 3_3 H-type pseudoknot. Slippery site, Stem I, Stem II, and Stem III are annotated in the arc plot and radial layout. The three alternative conformations depicted in C, D, and E were also observed in a previous SHAPE study (Schlick 2021a).
The 3_6 notation refers to the RNA-As-Graphs (RAGs) notation of secondary structures, where dual graphs are indexed according to stem number at decreasing compactness, as introduced in Fera et al. (2004), Jain et al. (2020), and Schlick et al. (2021a,b). RAGs are graphical representations of RNA secondary structure models (Jain et al. 2020). These are coarse-grained graphs where the helices in RNA are represented by vertices while loops are shown as edges (Jain et al. 2020). The major advantages of these graphs are that they make RNA structure comparison more robust and efficient. Our DMS-MaP (dimethyl sulfate-mutational profiling) experiments on this 87 nt construct in Figure 1C indicate low (black), medium (yellow), and high (red) nucleotides based on their reactivity against DMS (Mustoe et al. 2019). Low (black) nucleotides indicate low DMS reactivity, meaning nucleotides are base-paired. Medium (yellow) nucleotides represent moderate DMS reactivity, meaning nucleotides are either base-paired or unpaired, while high (red) nucleotides represent high DMS reactivities, meaning nucleotides are unpaired and easily accessible to DMS (Dey et al. 2021). We previously collected SHAPE-MaP (Selective 2′ Hydroxyl Acylation followed by Primer Extension-Mutational Profiling) data on this same construct (Schlick et al. 2021a). Both DMS and SHAPE reactivities can be used as a pseudo-free energy term in thermodynamic folding algorithms to significantly improve structure prediction (Deigan et al. 2009; Greenwood and Heitsch 2020).
Our two DMS-MaP biological replicates in Supplemental Figure S1 are quantitatively reproducible (R2> 0.85). When used for structure prediction with SHAPEknots (Hajdin et al. 2013), we obtained three conformations, shown in Figure 1C–E. They correspond to 3_6, 3_5, and 3_3 RAG topologies, and their relative folding energies are within 3 kcal/mol (see Table 1). This result is remarkably consistent with our previous SHAPE-MaP data on 77 nt, 87 nt, and 144 nt constructs, suggesting that the SARS-CoV-2 FSE adopts multiple conformations (Schlick et al. 2021a). These data are also consistent with other groups’ analyses of the FSE secondary structure (Rangan et al. 2021; Schlick et al. 2021b; Lan et al. 2022; Yan et al. 2022; Pekarek et al. 2023). We have emphasized in our prior work that not only can the RNA fold into different conformations for the same length (Schlick et al. 2021a,b; Yan et al. 2023), the fold distribution is also highly sensitive to length, when residues are either added or deleted (Yan et al. 2023). Fundamentally, these results suggest that the SARS-CoV-2 FSE likely adopts more than one conformation, consistent with the results obtained from other group's analyses of the FSE secondary structure (Rangan et al. 2021; Schlick et al. 2021a,b; Lan et al. 2022; Yan et al. 2022; Pekarek et al. 2023).
Minimum free energies computed for shorter SARS-CoV-2 FSE constructs after DMS-MaP and ShapeKnots
DMS data of FSE crystallization constructs
We further collected DMS data on crystal constructs (PDB ID 7MLX and 7LYJ) (Roman et al. 2021; Jones and Ferré-D'Amaré 2022). As can be seen in Fig. 2A, the 66 nt construct of 7LYJ adopts the 3_6 pseudoknot. Our DMS data, when used as a pseudo-free energy restraint term, also leads to the same single low energy structure (Fig. 2B; Table 1) in 3_6 topology. The 65 nt construct (PDB ID 7MLX) (Fig. 2C) is also predicted to adopt the 3_6 conformation (Fig. 2D). However, DMS data also point to an alternative 3_1, higher in energy conformation (Table 1). These data and analyses confirm the dominance of the 3_6 pseudoknot for short FSE lengths, which does not contain any slippery sites. Furthermore, from these data and structural analysis, it is clear that the crystallization constructs were optimized to favor the 3_6 conformation, which likely made them more amenable to obtaining high-resolution crystal structures in this conformation.
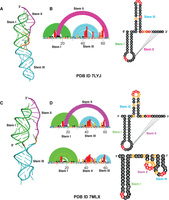
DMS-MaP for crystal structure constructs of SARS-CoV-2 FSE. (A) 3D crystal structure (PDB ID 7LYJ) of 66 nt FSE construct in 3_6 H-type pseudoknot conformation. (B) Arc plot and radial layout of 7LYJ computed through DMS-MaP and SHAPEknots in 3_6 topology. (C) 3D crystal structure (PDB ID 7MLX) of 65 nt FSE construct in 3_6 H-type pseudoknot conformation. (D) Arc plot and radial layout of 7MLX computed through DMS-MaP and SHAPEknots in 3_6 topology. A minor 3_1 conformation for 7MLX is also present. Stem I, Stem II, and Stem III are annotated in the arc plot and radial layout.
Predicted 77 nt FSE conformation-stabilizing mutants uniformly decrease frameshifting efficiency
As mentioned above, our previous SHAPE chemical probe 5NIA (5-nitroisatoic anhydride) analysis on 77 nt FSE (without the slippery sequence) also favors the 3_6 pseudoknot (Roman et al. 2021; Schlick et al. 2021a; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022). When alternatively probed by DMS, 3_6 emerges as the lowest energy conformation followed by 3_5 and 3_3 (Supplemental Fig. S2; Table 1).
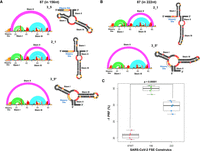
Using RAG-IF (inverse folding programs for graphs) (Fera et al. 2004; Jain et al. 2020; Schlick et al. 2021a,b), we have computationally designed three 77 nt mutant systems with up to six mutations to selectively stabilize each of these three conformations. Our DMS data on these three constructs (Fig. 3A–C) here confirm that each of these mutants selectively stabilize one of the three available conformations (Table 1). Indeed, SHAPEknots predict a single conformation for each of these mutants (Table 1). Moreover, subjecting these 77 nt mutant constructs to functional assays using dual-luciferase reporter systems (see Materials and Methods) reveals essentially abolished frameshifting (Fig. 3D). This is surprising, because despite the dominance of the 3_6 pseudoknot suggested by crystallography, Cryo-EM, SHAPE-MaP/DMS-MaP reactivity data, and computational predictions (Bhatt et al. 2021; Roman et al. 2021; Schlick et al. 2021a,b; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022), all mutants appear to lose frameshifting entirely. In fact, the abolished frameshifting from the 3_6 stabilizing mutant is the most puzzling result initially; however, this result can be explained by the required availability of other folds to the frameshifting process (see Discussion).
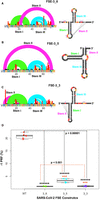
DMS-MaP and frameshifting efficiency of FSE conformation stabilizing mutants. Arc plot and radial layout computed by DMS-MaP and SHAPEknots for (A) FSE-3_6 mutants stabilizing 3_6 H-type pseudoknots, (B) FSE-3_5 mutants stabilizing 3_5 three-way junctions, and (C) FSE-3_3 mutants stabilizing 3_3 H-type pseudoknots. Stem I, Stem II, and Stem III are annotated in the arc plot and radial layout. Arrows in arc plot and asterisks in radial layout represent point mutations. (D) Frameshifting efficiency of 3_6, 3_5, and 3_3 stabilizing mutants highlighting substantial decrease in frameshifting efficiency when compared to WT 87 nt FSE construct. Frameshifting efficiency was calculated using dual-luciferase assays.
As a control, we also DMS probed, modeled, and measured the frameshifting efficiency of two additional longer WT constructs (156 nt and 222 nt long) (Fig. 4A, B, and C, respectively). Our previous work has shown that longer constructs of SARS-CoV-2 FSE tend to adopt other conformations due to a competing Stem 1 and attenuator hairpin (Yan et al. 2022, 2023) (S Lee, S Yan, A Dey, et al., pers. comm.). As can be seen in Figure 4C, both the 156 nt and 222 nt constructs have increased frameshifting compared to the short FSE. The 156 nt construct shows ∼15% more frameshifting efficiency, while the 222 nt construct had ∼10% more frameshifting efficiency than the 87 nt WT construct (Fig. 4C). The 156 nt construct, which according to DMS guided thermodynamic modeling adopts the three alternative conformations (Fig. 4A; Tables 2 and 4), has the highest frameshifting efficiency of all the constructs we tested. Even more puzzling, neither the 156 nt nor 222 nt constructs are predicted to adopt the 3_6 cryo-EM/crystal conformation (Table 2). The explanation here likely stems from the ease of refolding into related RNA folds during ribosomal translation (S Lee, S Yan, A Dey, et al., pers. comm., Fig. 8) (more in Discussion). Overall, the above result suggests that the actual mechanism of frameshifting does not depend on a single conformation, but in fact requires all three conformations switching continuously into one another.
Conformational flexibility and frameshifting efficiency of FSE in larger constructs. Arc plot and radial layout computed by DMS-MaP and SHAPEknots for (A) 156 nt construct in 3_5, 2_1, and 3_5* topology and for (B) 222 nt construct in 2_1 and 3_5* topology. Slippery site, Stem I, Stem II, and Stem III are annotated in the arc plot and radial layout. (D) Frameshifting efficiency of 156 nt and 222 nt construct stabilizing mutants highlighting substantial increase in frameshifting efficiency when compared to WT 87 nt FSE construct. Frameshifting efficiency was calculated using dual-luciferase assays.
Minimum free energies computed for longer SARS-CoV-2 FSE constructs after DMS-MaP and ShapeKnots
Correlated mutational profiling for multiple conformations
So far, our analysis of FSE structures has relied on a traditional coupling of thermodynamic folding parameters with a pseudo-free energy term correction using DMS data. This approach is optimized for improved accuracy of minimum free energy predictions and was trained on highly structured RNAs, such as ribosomal RNAs (rRNAs). Yet, applications to model alternative secondary structures poses challenges, likely explaining the large differences in predicted free energies of folding across different constructs (Tables 1 and 2). Three recently developed alternatives to thermodynamic structure modeling have been proposed to deconvolute alternative structures using correlated mutation in DMS-MaP data: DANCE-MaP (Olson et al. 2022), DREEM (Tomezsko et al. 2020), and DRACO (Morandi et al. 2021).
Analyses of our DMS-MaP data using these packages with SHAPEknots predictions yield conformer distributions shown in Tables 3 and 4. For the 87 nt construct, DANCE-MaP, and DREEM computed 3_6 as predominant conformations (34%, and 89%, respectively, Table 3). However, DRACO only predicted 16% of 3_6 conformations as minor conformations and 57% of 3_3 conformations as major fractions. In accordance with the above results, DANCE-MAP also predominantly identified 3_6 conformations of 77 nt constructs (99%, Table 3). For 77 nt FSE, all three programs identified 3_6 as the major conformation, but also suggest small (<6%) amounts of 3_5, 3_3, and 2_1 (2_1 is a simple two-stem structure).
Boltzmann fractions computed for shorter SARS-CoV-2 FSE constructs using Dance-MaP, DREEM, and DRACO
Boltzmann fractions computed for longer SARS-CoV-2 FSE constructs using Dance-MaP, DREEM, and DRACO
For our structure stabilizing mutants, the combined analyses in Table 3 validate the changed distribution in the conformational landscape, except for the 3_6 mutant where only DREEM favors a simple two-stem structure (2_1), and the 3_3 mutant, where only DRACO favors a 3_8 pseudoknot, which usually forms at long lengths due to competing alternatives to stem 1 (S Lee, S Yan, A Dey, et al., pers. comm.). Testing these three programs on the crystal structure data confirms these imperfect agreements.
Finally, our DMS-MaP data for longer constructs using these packages suggest a variety of conformations, including 3_6, 3_5, 2_1, 3_3, and 3_2 (Table 4). These also include some additional conformational variants like 3_5*, 2_1*, and 3_2*, which are similar to 3_5, 2_1, and 3_2 topology with only differences in base-pairing arrangements (Table 4). This increase of folds at larger lengths is consistent with our recent finding of structural heterogeneity at increasing FSE lengths (S Lee, S Yan, A Dey, et al., pers. comm.). Comprehensively, correlated mutational analysis using different deconvolution algorithms illustrates the conformational variability in the SARS-CoV-2 frameshifting element critical for regulating its translational efficiency.
DISCUSSION
As discussed in this work, the SARS-CoV-2 FSE not only plays a critical role in coronavirus replication and virulence (Iserman et al. 2020; Sanders et al. 2020; Bhatt et al. 2021; Roman et al. 2021; Schlick et al. 2021a,b; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022; Yan et al. 2022, 2023), but is also of intense interest because of the intriguing repertoire of length-dependent conformations. Prior studies have reported many possible conformations (Iserman et al. 2020; Manfredonia et al. 2020; Sanders et al. 2020; Bhatt et al. 2021; Roman et al. 2021; Schlick et al. 2021a; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022) for the SARS-CoV-2 FSE, although cryo-EM and crystallographic studies have identified the H-type pseudoknot (3_6) at short lengths (Roman et al. 2021; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022). However, none of these studies have fully characterized the functional consequences of changing relative fractions of different suboptimal ensembles. Our work here has probed the FSE structural abundance by subjecting several wild-type sequences (77 nt, 87 nt, 156 nt, and 222 nt) and three predicted structure-stabilizing mutants (Iserman et al. 2020; Manfredonia et al. 2020; Sanders et al. 2020; Bhatt et al. 2021; Roman et al. 2021; Schlick et al. 2021a,b; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022) to DMS probing and mutational analysis profiling (DMS-MaP).
Different FSE constructs chosen for this current study are based on previous construct lengths earlier used for simulation and 5NIA probing studies (Schlick et al. 2021a,b). We now compare our results obtained from current analyses in context with these other studies (Iserman et al. 2020; Manfredonia et al. 2020; Morandi et al. 2021; Roman et al. 2021; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022). By using these experimental data and analysis packages designed to handle multiple conformations (DANCE-MaP [Olson et al. 2022], DREEM [Tomezsko et al. 2020], and DRACO [Morandi et al. 2021]), we characterized the structural distributions in these variable systems. We also performed functional assays for the mutants and showed abolished frameshifting. The wild-type system characterization emphasized the prevalence of the 3_6 pseudoknot but also the emergence of alternative forms: 3_3 H-type pseudoknot and 3_5 three-way junction (Figs. 1 and 2; Supplemental Fig. S2; Tables 1 and 3).
These findings are in agreement with our earlier SHAPE studies (Schlick et al. 2021a), and a recent single-molecule structural study where the architecture of the core FSE (86 nt) element was divided in two cluster conformations with 3_6 and 3_5 as the dominant topology (Pekarek et al. 2023). These findings also agree with our recent characterization of length-dependent FSE systems that show the emergence of an alternative stem 1, along with simple stem–loop folds (3_1, 3_2, 2_1) at long lengths (S Lee, S Yan, A Dey, et al., pers. comm.). Clearly, SARS-CoV-2 FSE is highly flexible in nature and can exist in multiple conformations.
The extension of these analyses to the three RAG-designed mutants (Iserman et al. 2020; Manfredonia et al. 2020; Sanders et al. 2020; Bhatt et al. 2021; Roman et al. 2021; Schlick et al. 2021a,b; Zhang et al. 2021; Jones and Ferré-D'Amaré 2022) suggests markedly different distributions, namely, emphasizing the dominance of the 3_6, 3_3, and 3_5 conformations in turn, although the three deconvolution analysis packages show variations. It is important to note that the structure stabilizing mutations for the 3_6, 3_5, and 3_3 topologies were one of multiple thermodynamically equivalent possible combinations (Supplemental Table S1). Thus, from a mutational perspective, it is far more challenging to identify mutations that preserve the essential equilibrium between these structures than to stabilize each one individually. However, intriguingly, the essential loss of frameshifting efficiency for these structure stabilizing mutants (Fig. 3D) suggests that suppression of structural transitions, or the ability of the RNA to sample alternative conformations, is essential for frameshifting. Indeed, our conformational landscape viewpoint has revealed essential residues to stabilize central conformations, while others add variability into the FSE conformational repertoire (Schlick et al. 2021a,b; Yan et al. 2022,2023). But while it is plausible that 3_5 and 3_3 stabilizing mutants may not frameshift, why would the 3_6 stabilizing mutant not frameshift significantly?
The frameshifting ability must be an intricate function of the folding and refolding of variable FSE RNA segments interacting with multiple rRNAs during translation. As we proposed in S Lee, S Yan, A Dey, et al. (pers. comm.), the 3_6 conformation is dominant at short lengths when the slippery site is constrained and the 5′-end sequence from FSE is short. Otherwise, the alternative stem 1 and attenuator hairpin AH consort to block the pseudoknot 3_6 and favor 2_1 or 2_2 stem–loops and/or a different pseudoknot 3_8 (S Lee, S Yan, A Dey, et al., pers. comm.). Once the 3_6 pseudoknot is recovered upon ribosomal unwinding, it competes with the 3_3 pseudoknot through structural interconversions (S Yan and T Schlick, in prep.). All these structural interchanges must occur fluidly and quickly, involving energy barriers that are lowered by ribosomal intervention and winding/unwinding forces. Thus, a new intervention avenue for suppressing infection might involve tampering with this conformational cascade via mutations and/or structure stabilizing small drug compounds. Pursuing such designs by experiments and computations using FSE systems that yield specific conformational distribution will be fascinating.
Three recent approaches, DREEM, DRACO, and DANCE-MaP, leverage correlated DMS modifications in single reads to identify alternative sub-populations of RNA structures. We collected DMS data on wild-type, crystal, cryo-EM, and alternative conformation stabilizing mutants and evaluated the predicted percentage of alternative conformations using these three different algorithms. Earlier, we performed conformational landscape analysis on different length constructs on SARS-CoV-2 FSE through 5NIA probing and mutational profiling (Schlick et al. 2021a). However, SHAPE reactivities obtained from 5NIA probing are insufficient to perform deconvolution studies and obtain the relative fractions of each alternative conformation. Thus, to deconvolute any alternative conformations, different deconvoluting algorithms were executed on the DMS-MaP data obtained for various FSE constructs in the current study.
In addition, we measured frameshifting efficiency of these constructs. Our data and analysis reveal that stabilizing any of the three major secondary structures predicted to exist in the wild-type FSE effectively abolishes function. Furthermore, through correlated mutation analysis methods, we show that the FSE adopts multiple conformations, but that there are important differences in the relative estimates of abundance. Our work, therefore, illustrates that estimating the relative abundance of alternative conformations in RNAs remains a computational challenge, while also revealing that these methods are powerful tools for studying the role of these alternative conformations in RNA function.
Finally, the notion that RNAs are versatile molecules that adopt many possible conformations has been well appreciated for riboswitches and noncoding long RNAs (Serganov and Patel 2012; Bose et al. 2024). Yet, characterizing their conformational variability is challenging. We used the free energies calculated by ShapeKnots for each alternative ensemble obtained after running DMS-MaP seq and other deconvoluting algorithms (DREEM, DRACO, and DanceMapper) for each construct to model them. Tables 1 and 2 report the free energies calculated for each SARS-CoV-2 FSE ensemble. However, the free energies calculated after running deconvoluting algorithms were further converted into Boltzmann fractions to determine the relative occupancy of each ensemble in the milieu (Tables 3 and 4). DMS-data deconvoluting packages as used here certainly help in examining the conformational flexibility that SARS-CoV-2 FSE exhibits, but results among the programs are not consistent, pointing to many areas of future improvement. Clearly, considering an extended landscape of length-dependent structures with various programs along with other experimental and computational data is important. Viruses such as coronaviruses or HIV (Mouzakis et al. 2013) provide particularly interesting examples of the variability of structures found in very short segments. Indeed, that both use the −1 frameshifting mechanism suggests that frameshifting viral systems define ideal platforms for interrogating the intricate landscape of multiple RNA folds. Our study has thus emphasized the importance of these multiple forms and the challenges associated with their analyses. New therapeutic strategies to affect frameshifting also naturally emerge.
MATERIALS AND METHODS
SARS-CoV-2 sequence
29,891 nt long SARS-CoV-2 reference sequence was retrieved from GISAID (Elbe and Buckland-Merrett 2017) (accession ID: EPL_ISL_402124). The 87 nt FSE occupies the residues 13462-13548.
In vitro RNA chemical probing read by mutational profiling
Various SARS-CoV-2 FSE constructs were synthesized as G-blocks from Integrated DNA Technologies (IDT). These constructs differ in their lengths and were chosen based on our earlier simulation studies and 5NIA-based mutational profiling (Schlick et al. 2021a,b). These were chosen to investigate the role of additional nucleotide sequences and lengths on the overall conformation landscape of the SARS-CoV-2 FSE. All of these constructs were flanked at both 5′ and 3′ ends by RNA cassettes (Wilkinson et al. 2006). The T7 promoter region was added to the 5′ end of each construct for in vitro transcription of RNA, using T7 RNA polymerase from a T7 HiScribe RNA Synthesis Kit (New England Biolabs). Synthesized RNA was subjected to DNase treatment (TURBODNase) and was further purified using Purelink RNA Mini Kit (Invitrogen) and quantified using NanoDrop.
For chemical probing, 6 μg of purified in vitro transcribed RNA was denatured at 65°C for 5 min and snap-cooled in ice. Following denaturation, folding buffer (100 mM KCl, 10 mM MgCl2, 100 mM Bicine, pH 8.3) was added to the denatured RNA and the whole reaction was incubated at 37°C for 10 min. The total reaction volume was 100 μL. The folded RNA was further treated with 10 μL of 1:10 ethanol-diluted dimethyl sulfate (DMS). For control, an equivalent volume of ethanol was added to the folded RNA. Probing was initiated by incubating the reaction mixture at 37°C for 5 min and was quenched afterward by adding 100 μL of 20% β-mercaptoethanol (β-ME). DMS is known to only modify unpaired adenine and cytosine leaving guanine and uracil (Mustoe et al. 2019); however, in the presence of Bicine buffer (pH 8.0), DMS probes all the 4 nt simultaneously, as shown earlier (Mustoe et al. 2019). Modified and unmodified RNAs were purified using a PureLink RNA Mini Kit and quantified using NanoDrop.
Library construction, sequencing, and data processing
Following chemical probing, both modified and unmodified RNA were reverse transcribed using specific primer complementary
to the 3′ RNA cassette (Dey 2023) and Superscript II reverse transcriptase under error prone conditions, as previously described (Smola et al. 2015). The generated cDNA was purified using a G50 column (GE Healthcare) and subjected to second-strand synthesis (NEBNext Second
Strand Synthesis Module). For constructing next-generation sequencing libraries, the double-stranded (ds) cDNA was PCR amplified
with primers directed against 5′ and 3′ RNA cassettes and NEB Q5 HotStart polymerase (NEB). To introduce unique barcodes,
secondary PCR was performed using TrueSeq primers (NEB) (Smola et al. 2015). Amplified products were purified using Ampure XP (Beckman Coulter) beads, and quantification of the libraries was done
using a Qubit dsDNA HS Assay Kit (Thermo Fisher). Purified libraries were quality checked using Agilent Bioanalyzer. These
libraries were finally sequenced as 2 × 151 paired end reads on the Illumina MiSeq platform. All the samples recorded sequencing
depth of > 0.6 million reads (Supplemental Table S2). ShapeMapper2 algorithm (Busan and Weeks 2018) was used to calculate mutation frequency in both chemically modified (DMS treated) and control/unmodified (ethanol treated)
RNA samples. Chemical modifications on each RNA nucleotide were calculated using the following equation:
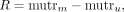 where R is the chemical reactivity, mutrm is the mutation rate calculated for chemically modified RNA, and mutru is the mutation rate calculated for unmodified control RNA samples (Smola et al. 2015; Smola and Weeks 2018). ShapeMapper2 was also used to calculate the parse mutations from the sequencing data. Chemical reactivity obtained from
ShapeMapper 2.0 was used to inform a minimum free energy structure using ShapeKnots (Hajdin et al. 2013). The resultant model was visualized using VARNA (Darty et al. 2009). RNA arc and secondary structure models were generated using RNAvigate (Irving and Weeks 2024).
where R is the chemical reactivity, mutrm is the mutation rate calculated for chemically modified RNA, and mutru is the mutation rate calculated for unmodified control RNA samples (Smola et al. 2015; Smola and Weeks 2018). ShapeMapper2 was also used to calculate the parse mutations from the sequencing data. Chemical reactivity obtained from
ShapeMapper 2.0 was used to inform a minimum free energy structure using ShapeKnots (Hajdin et al. 2013). The resultant model was visualized using VARNA (Darty et al. 2009). RNA arc and secondary structure models were generated using RNAvigate (Irving and Weeks 2024).
DANCE-MaP analysis on SARS-CoV-2 FSE constructs
The DANCE-MaP (deconvolution and annotation of ribonucleic conformational ensembles) algorithm uses maximum likelihood (ML) strategy to extract large amounts of the total information available from single chemical probing experiments (Olson et al. 2022). DANCE-MaP measures chemical reactivities on all 4 nt simultaneously, including base-pairing and tertiary interactions, which enables it to measure and detect possible RNA ensembles at single nucleotide resolution. Parsed files (for both modified and unmodified data sets) were used in the DANCE-MaP pipeline to calculate and validate alternative conformations for SARS-CoV-2 FSE constructs. The resultant reactivity profile was then used as a constraint in ShapeKnots (Hajdin et al. 2013) to generate probable SARS-CoV-2 FSE ensembles for different constructs.
DREEM analysis on SARS-CoV-2 FSE constructs
The “Detection of RNA folding ensembles using expectation–maximization” (DREEM) algorithm (Tomezsko et al. 2020) was run directly on the sequencing reads of DMS modified SARS-CoV-2 FSE constructs to determine the presence of alternative structures in the constructs. Using an expectation–maximization technique, DREEM clusters the sequencing reads into discrete groups based on patterns of DMS-induced mutations. Log-likelihood is maximized to obtain the DMS modification rate per base for each cluster. In this study, a maximum of K = 3 clusters were used to group the bit-vectors. The resulting DMS reactivities for each cluster were then used as constraints for ShapeKnots (Hajdin et al. 2013) predictions. Hence, distinct structural clusters with their relative ratios result in different folds, which represent the heterogeneity of RNA secondary structure. To predict the 87 nt FSE structure for long constructs of 156 nt and 222 nt, DMS reactivity constraints of the 87 nt region retrieved from the normalized reactivity profile by DREEM for the complete constructs were used.
DRACO analysis on SARS-CoV-2 FSE constructs
We also applied the DRACO algorithm (Morandi et al. 2021), which performs deconvolution of alternative RNA conformations from mutational profiling experiments with a combination of spectral clustering and fuzzy clustering, to validate the structure predictions of SARS-CoV-2 FSE. Spectral clustering is performed for the sliding windows along the transcript, allowing the optimal number of coexisting conformations (clusters) to be automatically identified from the eigen gaps. Following the determination of the number of clusters, fuzzy clustering is carried out to allow bases to be weighted according to their affinity to each cluster. DRACO then reconstructs overall mutational profiles by merging overlapping windows with the same number of clusters. DRACO reports consecutive sets of windows with varying amounts of clusters separately. The paired-end reads were merged by PEAR (Zhang et al. 2014) and mapped to the reference sequence using the rf-map tool (Incarnato et al. 2018) (parameters: -b2 -cqo -ctn -mp “–very-sensitive local”). The resulting BAM files were then analyzed with the rf-count tool to produce MM files (-r -m -mm -na -ni). MM files were analyzed with DRACO (Morandi et al. 2021) (parameters: –allNonInformativeToOne –nonInformativeToSurround –minClusterFraction 0.1), and deconvoluted mutation profiles were extracted from the resulting JSON files. Normalized reactivity profiles were obtained by first calculating the raw reactivity scores via the scheme by DMS-MaP (Zubradt et al. 2017) as the per-base ratio of the mutation count and the read coverage at each position, and then by 90% Winsorizing as a normalization method, using the rf-norm tool (Incarnato et al. 2018) (parameters: -sm 4 -nm 2 -rb AC -mm 1). Data-driven RNA structure prediction was performed using ShapeKnots (Hajdin et al. 2013) and the normalized reactivity profiles.
Dual-luciferase assay of −1 PRF
pJD2514-87, a test SARS-CoV-2 reporter plasmid of 87 nt construct and pJD2257, a 0-frame readthrough control plasmid for dual-luciferase reporter assay, were obtained as a kind gift from Professor Jonathan D. Dinman, University of Maryland. A Q5 Site-Directed Mutagenesis Kit (NEB) was used to generate various SARS-CoV-2 FSE conformation stabilizing constructs and other length-dependent constructs, and their sequences were confirmed through Sanger sequencing (Eurofins Genomics). HEK293 cells were used as host cells to transfect the SARS-CoV-2 FSE dual-luciferase reporter plasmid using Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific). The −1 PRF efficiency was assayed in cultured HEK293, as described previously (Kelly et al. 2020), using the Dual-Luciferase Reporter Assay System Kit (Promega). At 24 h posttransfection, HEK293 cells were washed with 1× PBS buffer and then lysed with 1× passive lysis buffer (Promega). The cells were cleared of any cell debris by centrifugation at 14,000 rpm for 10 min at 4°C. Each cell lysate, both test [pJD2514] and 0-frame readthrough control [pJD2257], were assayed in triplicate in a 96-well plate, and luciferase activity was quantified using a CLARIOstar (BMG Labtech) Plate Reader. SARS-CoV-2 frameshifting efficiency was calculated by averaging the triplicates of firefly and Renilla luciferase per samples and calculating the ratio of firefly to Renilla luciferase of each sample. Percent frameshifting was subsequently calculated for test samples by comparing their luminescence ratio (as calculated above) with the 0-frame readthrough control set at 100%. This ratio of ratios provided the percent −1 PRF efficiency of each SARS-CoV-2 FSE construct. Statistical analyses were conducted with Welch's two sample t-test using R software.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by U.S. National Institutes of Health grants NHLBI R01 HL111527 and NIGMS R35 GM140844 to A.L., National Science Foundation Award DMS 2151777 from the Division of Mathematical Sciences to T.S. and A.L., and National Institutes of Health R35GM122562 from the National Institute of General Medical Sciences to T.S. Dr. Dey acknowledges the Department of Biotechnology, Government of India for the Ramalingaswami Re-entry fellowship (BT/RLF/Re-entry/02/2021). The authors also acknowledge Professor Jonathan D. Dinman for kindly providing reporter plasmids for dual-luciferase assays to assess the −1 PRF efficiency of SARS-CoV-2 FSE constructs.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080035.124.
-
Freely available online through the RNA Open Access option.
- Received March 27, 2024.
- Accepted July 21, 2024.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Abhishek Dey is the first author of this paper, “Abolished frameshifting for predicted structure-stabilizing SARS-CoV-2 mutants: implications to alternative conformations and their statistical structural analyses.” Dr. Dey is a Ramalingaswami Re-entry Fellow at the National Institute of Pharmaceutical Education and Research (NIPER)-Raebareli, a prestigious institution of national significance. The Ramalingaswami Re-entry Fellowship was awarded to Dr. Dey by the Department of Biotechnology, Government of India. Understanding RNA architecture and epigenetic changes that can govern RNA's function in regulating gene regulation and other cellular processes is Dr. Dey's primary area of study interest. Additionally, his goal is to create RNA nanoparticles as the next wave of biomedical treatments. He intends to target different RNA structures and alterations for medicinal and diagnostic applications with these nanoparticles.
What are the major results described in your paper and how do they impact this branch of the field?
This work identifies the previously unknown conformational landscape of the SARS-CoV-2 frameshifting element. Multiple interchanging conformations adopted by SARS-CoV-2 FSE are critical for controlling the programmed ribosomal frameshift. This regulation is necessary as the virus regulates the expression of late and early phase proteins. This study also focuses on identifying these variable conformations through chemical probing and mutational profiling and using various structure deconvoluting algorithms, and the challenges associated with their analyses. Overall, the findings from this study will be used to design novel therapeutic interventions targeting these structural antiviral elements of the SARS-CoV-2 frameshifting element.
What led you to study RNA or this aspect of RNA science?
Because of RNA's unusual structural variability and functional diversity, as well as its pivotal role in gene expression and regulation, I became interested in the field of RNA science. I was always captivated by the fact that RNA not only acts as a messenger between proteins and DNA, but also carries out a number of regulatory tasks. My interest was piqued further by the discovery of noncoding RNAs and their role in intricate biological processes, including the mechanisms behind disease. Furthermore, there are now more opportunities to investigate the potential of RNA in therapies and diagnostics thanks to the quick developments in RNA technologies, such as chemical probing and next-generation sequencing. My research was motivated by the intriguing possibility of using RNA for medicinal purposes, such as creating RNA-based nanoparticles for targeted therapy. I am passionate about contributing to this fascinating and rapidly developing subject of RNA science, and my background in RNA biology has given me the means to explore these complex facets of the science.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
Indeed, unexpected results can be obtained from RNA chemical probing and mutational profiling tests, and these methods also present unique problems that shape future directions in study. I ran into the following situations:
-
Unexpected RNA structures: RNA structures that are more complicated than expected or that were not predicted can be found using chemical probing studies. To better understand the links between RNA structure and function, these findings may prompt researchers to investigate novel RNA folding algorithms or biological assays.
-
Biological insights: Unexpected outcomes from RNA experiments could reveal novel RNA regulatory elements, RNA–protein interactions, or RNA changes that impact gene expression, among other new biological insights.
-
Integration with computational approaches: Computational models and bioinformatics technologies used with experimental data integration can uncover previously unknown patterns or relationships. This multidisciplinary approach can influence computational tactics and future research objectives.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
I have always been driven by an intense interest about the natural world, even in my childhood days. This lifelong curiosity with natural occurrences shaped my knowledge and directed my scientific career. It also laid the groundwork for my lifelong journey into the scientific community.
My early interest in science was nurtured by a series of inspirational mentors and experiences. As I continued my academic journey from high school to university, advanced biology, biochemistry and chemistry classes, coupled with laboratory work, allowed me to hone my skills and deepen my knowledge.
While pursuing my doctoral degree, I had the privilege of working alongside experienced researchers who mentored me and provided valuable insights into the scientific process. Co-authoring papers and presenting my research at conferences were important turning points that strengthened my resolve to pursue a career in science.
Today, as a scientist, I continue to be driven by the same curiosity that ignited my passion in childhood. My research focuses on understanding RNA structures, modifications and their role in gene regulation, particularly in the context of diseases. The journey from observing natural phenomena as a curious child to investigating complex biological processes as a researcher has been both challenging and rewarding.
If you were able to give one piece of advice to your younger self, what would that be?
One piece of advice I would give to my younger self is to “embrace curiosity and never stop learning.” The spark that ignites invention and discovery is curiosity. It's what motivates academics and scientists to push the limits of knowledge and discover new areas. You can make a significant contribution to research and continuously deepen your understanding of the world by fostering your curiosity and remaining receptive to new ideas.
What are your subsequent near- or long-term career plans?
My immediate goals are to build a strong research program that explores the structural and functional properties of RNA, especially how it affects gene expression and causes disease. Using my experience and the most recent developments in RNA technology, my objective is to create novel RNA-based nanoparticles for therapeutic and diagnostic uses.
In the long run, I want to broaden the scope of my work by working with interdisciplinary teams and combining structural biology, computational biology, and clinical research to get my results into useful applications. My goal is to head a research group that not only contributes to our growing knowledge of RNA biology but also leads the way in creating RNA-based treatments for a range of illnesses. In the end, I hope to make a positive impact on the field by serving as a mentor to the next generation of scientists, encouraging creativity, and advancing the development of RNA-based biological therapies.








