
Promoter-independent synthesis of chemically modified RNA by human DNA polymerase θ variants
- Taylor Tredinnick1,
- Tatiana Kent1,
- Leonid Minakhin1,
- Ziyuan Li2,
- Jozef Madzo3,
- Xiaojiang S. Chen2 and
- Richard T. Pomerantz1
- 1Department of Biochemistry and Molecular Biology, Sidney Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, USA
- 2Molecular and Computational Biology, USC Dornsife Department of Biological Sciences, University of Southern California, Los Angeles, California 90007, USA
- 3Coriell Institute for Medical Research, Camden, New Jersey 08103, USA
- Corresponding author: richard.pomerantz{at}jefferson.edu
Abstract
Synthetic RNA oligonucleotides composed of canonical and modified ribonucleotides are highly effective for RNA antisense therapeutics and RNA-based genome engineering applications utilizing CRISPR–Cas9. Yet, synthesis of synthetic RNA using phosphoramidite chemistry is highly inefficient and expensive relative to DNA oligonucleotides, especially for relatively long RNA oligonucleotides. Thus, new biotechnologies are needed to significantly reduce costs, while increasing synthesis rates and yields of synthetic RNA. Here, we engineer human DNA polymerase theta (Polθ) variants and demonstrate their ability to synthesize long (95–200 nt) RNA oligonucleotides with canonical ribonucleotides and ribonucleotide analogs commonly used for stabilizing RNA for therapeutic and genome engineering applications. In contrast to natural promoter-dependent RNA polymerases, Polθ variants synthesize RNA by initiating from DNA or RNA primers, which enables the production of RNA without short abortive byproducts. Remarkably, Polθ variants show the lower capacity to misincorporate ribonucleotides compared to T7 RNA polymerase. Automation of this enzymatic RNA synthesis technology can potentially increase yields while reducing costs of synthetic RNA oligonucleotide production.
Keywords
INTRODUCTION
The ability to synthesize long (>1000 nt) mRNA molecules with modified ribonucleotides (i.e., pseudouridine) for vaccine production has been optimized by developing modified bacteriophage RNA polymerases (RNAPs) (Chelliserrykattil and Ellington 2004; Rosa et al. 2021; Elkhalifa et al. 2022). In contrast, the synthesis of shorter (i.e., ∼16–150 nt) RNA oligonucleotides containing site-specific chemical modifications such as those used for antisense RNA therapeutics (i.e., gapmer and siRNA) and CRISPR genome engineering (sgRNA) remains inefficient, resulting in low yields, and is expensive, especially for kilogram production of antisense RNA therapeutics and therapeutic grade sgRNA (Roy and Caruthers 2013; Molina and Sanghvi 2019; Catani et al. 2020; Glazier et al. 2020; Crooke et al. 2021a,b). Such RNA oligonucleotides are synthesized using phosphoramidite chemistry which requires multiple steps per nucleotide addition, generates high levels of toxic waste, and is unable to generate relatively long (>50 nt) RNA at low cost and high yields, posing as a major obstacle for the development and manufacturing of synthetic RNA for genome engineering and other applications.
To overcome this limitation, we decided to engineer a promoter-independent DNA-dependent RNAP with the ability to accommodate various ribonucleotide analogs and chemically modified RNA primers. A similar strategy aimed at converting A-family Thermus aquaticus (Taq) DNA polymerase (DNAP) into a DNA-dependent RNAP via a steric-gate mutation previously failed owing to the enzyme's inability to fully extend A-form RNA/DNA which is significantly wider than B-form DNA/DNA (Ong et al. 2006). For example, although conversion of the characterized steric-gate residue Glu615 to glycine—known to reduce discrimination against ribonucleoside incorporation—enabled the enzyme to efficiently incorporate ribonucleotides, the enzyme failed to synthesize RNA >6–7 nt in length using a DNA/DNA primer–template substrate in the presence of Mg2+ (Ong et al. 2006). The addition of Mn2+ allowed for slightly further extension of a DNA primer in the presence of ribonucleotides (Ong et al. 2006). These results indicated that Taq DNAP is unable to fully accommodate A-form RNA–DNA which is wider than B-form DNA–DNA.
Our recent findings revealed that the related A-family DNA polymerase theta (Polθ) fully extends A-form DNA/RNA primer–templates due to a significant thumb subdomain conformational change that enables accommodation of the wider DNA/RNA hybrid (Chandramouly et al. 2021). Based on these findings, we hypothesized that a steric-gate Polθ mutant (E2335G; referred to as PolθRP1) would perform efficient promoter-independent RNA synthesis. The promiscuous nature of Polθ, for example, the use of noncanonical nucleotides, also suggested that PolθRP1 would incorporate various chemically modified ribonucleotides (Kent et al. 2016a; Chandramouly et al. 2021).
Here, we characterize the ability of Polθ variants containing steric-gate mutations to rapidly synthesize relatively long (>90 nt) RNA products by initiating RNA synthesis on either DNA/DNA or RNA/DNA primer–templates in vitro. We also demonstrate the ability of these engineered enzymes to synthesize long RNA products containing various ribonucleotide analogs commonly used for stabilizing RNA in cells for therapeutic and genome engineering applications.
RESULTS
Limited RNA synthesis activity by Taq DNAP E615G
Polθ is closely related to A-family DNAPs from bacteria, such as Taq DNAP and Escherichia coli DNAP I (Klenow fragment) with the exception of additional unstructured loops within the polymerase domain of Polθ (Zahn et al. 2015; Black et al. 2016; Malaby et al. 2017; Black et al. 2019). Superposition of Polθ (Zahn et al. 2015) with Taq DNAP (Li et al. 1999) is presented in Figure 1A. The superposition shows close alignment of their respective steric-gate residue which plays a major role in suppressing ribonucleotide incorporation for this A-family polymerase class. For instance, steric-gate mutants of Polθ, Taq DNAP, and related E. coli Klenow fragment have been shown to efficiently incorporate ribonucleotides (Astatke et al. 1998; Ong et al. 2006; Randrianjatovo-Gbalou et al. 2018). However, steric-gate Taq DNAP and Klenow fragment mutants showed very limited RNA synthesis, resulting in premature termination of RNA synthesis after incorporating ∼6–7 nt on a DNA template (Astatke et al. 1998; Ong et al. 2006). We hypothesized that the observed premature termination activity by steric-gate mutants of Taq DNAP and Klenow fragment in the presence of ribonucleoside triphosphates (NTPs) is due to the enzymes’ inability to fully accommodate the wider A-form DNA/RNA.

Structure–function analysis of Taq steric-gate variant. (A) Superposition of Taq DNAP and Polθ crystal structures. Superposition of Taq DNAP (blue; PDB ID: 1qss) and Polθ (green; PDB ID: 4x0q) bound to DNA/DNA templates with incoming ddGTP shows the close alignment of their respective steric-gate residues. (B) Denaturing gel showing premature termination of Taq DNAP E615G on the indicated DNA/DNA primer–template in the presence of NTPs (left). Denaturing gel showing exonuclease activity by Taq DNAP E615G on the indicated RNA/DNA primer–template in the presence of NTPs (right).
We tested the ability of a Taq DNAP steric-gate mutant (E615G) to synthesize RNA along DNA/DNA and RNA/DNA primer–templates. Consistent with prior studies, we found that a Taq DNAP steric-gate mutant (E615G) efficiently incorporated ribonucleotides on DNA/DNA; however, the enzyme exhibited strong termination after incorporating 6–7 nt, similar to prior studies (Fig. 1B, left; Ong et al. 2006). The Taq DNAP E615G mutant degraded the RNA primer on the RNA/DNA template under identical conditions with NTPs, likely due to its 5′–3′ exonuclease activity (Fig. 1B, right). For example, although Taq DNAP possesses a 3′–5′ proof-reading like domain, similar to the Klenow fragment, its activity has been inactivated by acquired mutations (Park et al. 1997). Here, we found that Taq DNAP exhibits robust nuclease activity on an RNA/DNA primer, likely due to its 5′–3′ exonuclease activity. Taken together, these data further confirm the difficulties of converting Taq DNAP into a proficient promoter-independent DNA-dependent RNAP that is capable of synthesizing relatively long RNA.
Robust RNA synthesis by Polθ steric-gate variants
Our prior studies elucidated the ability of Polθ to efficiently utilize DNA/RNA A-form primer–templates as substrates which revealed its ability to perform reverse transcriptase activity, similar to retroviral reverse transcriptases (Chandramouly et al. 2021). Structural analysis of Polθ bound to a DNA/RNA primer–template compared to a DNA/DNA primer–template is reviewed in Figure 2A. The superposition of Polθ bound to DNA/RNA over the prior Polθ DNA/DNA ternary complex revealed that the thumb subdomain of Polθ undergoes an unprecedented structural rearrangement in order to accommodate the wider A-form DNA/RNA hybrid in its active center (Chandramouly et al. 2021). Approximately 57% of the thumb subdomain residues were converted from alpha helices to loops (Chandramouly et al. 2021). The superposition suggests that lack of this conformational change would result in a clash between the thumb subdomain and the wider RNA/DNA hybrid. The ability of Polθ to fully accommodate DNA/RNA A-form nucleic acid in an active configuration suggests that Polθ steric-gate variants may utilize RNA/DNA primer–template substrates and synthesize long RNA products in a DNA template-dependent manner. Although, the possibility exists that Polθ adopts a different configuration on an RNA/DNA compared to a DNA/RNA primer–template. Furthermore, Polθ has been shown to be highly permissive in incorporating noncanonical nucleotides (Kent et al. 2016a,b). Thus, we hypothesized that the polymerase can also accommodate various ribonucleotide analogs that are used for increasing the half-life of RNA in cells. The superposition of the two structures also revealed a significant 42° rotation of the fingers domain outward in the Polθ:DNA/RNA complex, indicating that the enzyme was captured in an open or partially open configuration (Fig. 2A).

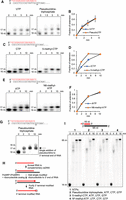
Polθ steric-gate variants exhibit robust RNA synthesis activity. (A) Superposition of Polθ:DNA/RNA and Polθ:DNA/DNA crystal structures. Superposition of Polθ:DNA/RNA (red; PDB ID: 6XBU) and Polθ:DNA/DNA (pink; PDB ID: 4x0q) with incoming ddGTP reveals significant conformational changes in the thumb and fingers subdomain of Polθ when bound to A-form DNA/RNA. (B) Denaturing gel showing efficient extension of RNA/DNA by WT Polθ in the presence of dNTPs. (C) Denaturing gel showing premature termination by WT Polθ on RNA/DNA in the presence of NTPs. (D–F) Denaturing gels showing efficient DNA-dependent RNA synthesis by PolθRP1 and PolθRP1 variants on the indicated primer–templates in the presence of NTPs, MgCl2, and indicated NaCl titration. (G) Denaturing gel showing RNase H degradation of the RNA portion of the RNA/DNA hybrid synthesized by PolθRP2. (H) Denaturing gel showing T7 RNAP promoter-dependent synthesis of RNA with an expected length of 95 nt.
We first investigated the ability of WT Polθ (polymerase domain: residues to 1792–2590) to incorporate deoxyribonucleoside monophosphates (dNMPs) on an RNA/DNA primer–template where the primer was composed of RNA. In this scenario, the enzyme must initiate DNA synthesis on A-form RNA/DNA. As predicted from prior studies of Polθ on an A-from DNA/RNA primer–template, we observed efficient activity of the wild-type enzyme on the RNA/DNA primer–template, resulting in DNA-dependent DNA synthesis initiating from an RNA primer (Fig. 2B). Hence, these data demonstrate the ability of Polθ to extend an RNA primer annealed to a DNA template, and confirm the ability of the enzyme to function on A-form nucleic acid. Next, we investigated the ability of WT Polθ to incorporate ribonucleotides on the same RNA/DNA template. As expected, the WT enzyme showed inefficient DNA-dependent RNA synthesis activity which confirms its ability to strongly discriminate against ribonucleotides, like most DNAPs (Fig. 2C).
To significantly reduce Polθ’s discrimination against incorporating ribonucleotides, we mutated its steric-gate residue E2335. Two different mutant versions of Polθ were generated. The first included a single mutation in the steric-gate (PolθRP1; E2335G). The second variation (PolθRP2; E2335G, I2326F) included the steric-gate mutation in addition to a second mutation (I2326F) predicted to further increase the enzyme's promiscuity based on prior studies with Taq DNAP mutants (Ong et al. 2006). A previous study showed efficient ribonucleotide incorporation by Polθ steric-gate variants on single-strand DNA substrates (Randrianjatovo-Gbalou et al. 2018). However, Polθ steric-gate mutant activity on primer–templates has not been investigated.
In contrast to the equivalent steric-gate mutation in Taq DNAP, PolθRP1 (E2335G) efficiently extended DNA/DNA and RNA/DNA in the presence of NTPs, resulting in RNA 95 nt in length (Fig. 2D). The double mutant PolθRP2 also showed efficient RNA synthesis on RNA/DNA primer–templates, resulting in RNA products 95 and 135 nt in length (Fig. 2E). PolθRP1 synthesis of a 200 nt RNA is also demonstrated (Fig. 2F). Here, the addition of NaCl promoted full-length RNA product formation. RNase H treatment following the synthesis reaction with NTPs unequivocally demonstrated that PolθRP2 synthesizes RNA as expected (Fig. 2G). As a comparison, T7 RNAP synthesis of the identical 95 nt RNA sequence by initiating from its canonical promoter was highly impure due to its inherent abortive RNA synthesis activity during the incomplete transition from transcription initiation to the elongation phase (Fig. 2H). This demonstrates a major drawback for synthesizing pure RNA oligonucleotides with T7 RNAP. These data demonstrate Polθ steric-gate mutants as effective promoter-independent DNA-dependent RNAPs, and confirm that Polθ is highly active on A-form RNA/DNA.
Comparison of Polθ and T7 RNAP on RNA/DNA primer–templates
Several studies have shown the ability of T7 RNAP to form an elongation complex (EC) on short RNA/DNA primer–templates and these complexes have been used to study the biochemistry, fidelity and structure biology of the T7 RNAP EC (Tahirov et al. 2002; Pomerantz et al. 2006). Considering that T7 RNAP is used to synthesize mRNA vaccines and RNA for basic research, we further compared this prototypical promoter-dependent RNAP to PolθRP1 on an RNA/DNA primer–template using identical conditions. T7 RNAP was inactive compared to PolθRP1 on an RNA/DNA primer–template comprised of a 16 nt RNA and 200 nt DNA template (Fig. 3A). Reducing the RNA primer length to 7 nt on a shorter DNA template allowed minor (10%–13%) RNA extension activity by T7 RNAP compared to PolθRP1 (Fig. 3B, left). Next, we preincubated both enzymes on the RNA/DNA primer–template for 10 min prior to the addition of NTPs since T7 RNAP is known to form an EC under these conditions (Tahirov et al. 2002; Pomerantz et al. 2006). T7 RNAP was still deficient in extending the RNA/DNA template even after 20 min, whereas PolθRP1 showed nearly full extension after 5 min (Fig. 3B, right). Next, we compared the relative efficiencies of ribonucleotide misincorporation by PolθRP1 and T7 RNAP. Prior studies have shown the ability to measure the fidelity of the T7 RNAP EC on short RNA/DNA primer–templates (Pomerantz et al. 2006). Thus, we used similar conditions and compared correct (UMP) versus incorrect (GMP) ribonucleotide incorporation by T7 RNAP EC following the necessary 10 min preincubation period. Consistent with prior studies, T7 RNAP misincorporated two GMPs via a template strand misalignment mechanism (Fig. 3C, right; Pomerantz et al. 2006). Also consistent with prior studies, after T7 RNAP correctly incorporated UMP, it proceeded to misincorporate UMP (Fig. 3C, left; Pomerantz et al. 2006). Using identical conditions, we unexpectedly found that PolθRP1 showed little to no capacity to misincorporate GMP (Fig. 3D, right). PolθRP1 showed no ability to misincorporate UMP after the initial correct incorporation step (Fig. 3D, left). These findings suggest PolθRP1 exhibits higher fidelity DNA template-dependent RNA synthesis compared to T7 RNAP and are consistent with our prior studies demonstrating that PolθRP1 exhibits higher fidelity on A-form versus B-form primer–templates (Chandramouly et al. 2021). High-throughput sequencing and analysis of PCR amplified cDNA synthesized from PolθRP1 RNA products showed that 71% of sequencing reads aligned perfectly to the reference sequence, whereas 17% of the reads had only one base variation (Supplemental Fig. 1).

Comparison of Polθ steric-gate variant to T7 RNAP. (A,B) Denaturing gels showing time courses of PolθRP1 and T7 RNAP on the indicated RNA/DNA primer–templates in the presence of canonical NTPs. T7 RNAP and PolθRP1 were preincubated with the RNA/DNA template for 10 min in panel B where indicated. % RNA extension indicated (B). (C,D) Denaturing gels showing RNA extension by PolθRP1 and T7 RNAP following a 10 min preincubation period on the indicated RNA/DNA with the indicated NTPs.
Polθ variant synthesis of chemically modified RNA
Chemically modified ribonucleotide analogs enable the stabilization of RNA in cells and reduce immunogenicity. For example, pseudouridine significantly increases the half-life of synthetic mRNA by reducing its immunogenicity and enzymatic cleavage by RNase L, and later developments led to the use of N1-methylpseudouridine in mRNA vaccines (Kariko et al. 2008, 2012; Anderson et al. 2011; Zhao and He 2015; Nance and Meier 2021). Hence, this single chemical modification in synthetic RNA may be among the most important developments in modern RNA biotechnology. Additional naturally occurring ribonucleotides in cellular RNA include 5-methyl-cytidine and N6-methyl-adenosine, among many others (Zhang et al. 2019; Boo and Kim 2020; McCown et al. 2020).
We demonstrate that PolθRP2 efficiently incorporates pseudouridine, 5-methyl-cytidine, and N6-methyl-adenosine monophosphates with similar efficiency as canonical ribonucleotides (Fig. 4A–F). Importantly, time-dependent extension of RNA/DNA with nucleotide analogs enabled the incorporation of single nucleotide analogs, resulting in specific 3′-terminal RNA chemical modifications. For example, the right panels in Figure 4A,C, and E show the ability to incorporate single ribonucleotide analogs (pseudouridine, 5-methyl-cytidine, N6-methyl-adenosine) in a time-controlled manner. Further experimentation with pseudouridine-triphosphate revealed 5 min as optimal for incorporating a single pseudouridine monophosphate at the 3′-terminal end of RNA (Fig. 4G). The ability of PolθRP2 to incorporate single modified ribonucleotides in a time-dependent manner enables rapid modification of the 3′-terminal end of RNA molecules via enzymatic activity. For example, following annealing of the 3′ end of RNA to a complementary ssDNA oligonucleotide allows for single ribonucleotide incorporation by Polθ steric-gate mutants (Fig. 4H). The modified RNA species may then be purified and used for downstream biotechnology applications. We also envision the ability to add multiple consecutive chemically modified ribonucleotide analogs in a template and time-dependent manner at the 3′-terminal end of RNA which will be useful for reducing nuclease activity or immunogenicity of RNA used for therapeutic and genome engineering applications. Because PolθRP2 exhibits efficient incorporation of base-modified ribonucleotide analogs, we tested whether the mutant enzyme can synthesize relatively long RNA with the substitution of a canonical ribonucleotide with a base-modified analog. The results show that PolθRP2 exhibits efficient synthesis of RNA in the presence of pseudouridine-triphosphate, 5-methyl-CTP and N6-methyl-ATP substituted for UTP, CTP, and ATP, respectively (Fig. 4I). Controls show that the Polθ steric-gate variant fails to synthesize these RNA products when only three NTPs are added to the reaction (Supplemental Fig. 2), consistent with a poor capacity to misincorporate ribonucleotides as shown in Figure 3D. Additional controls show that PolθRP1 does not show a preference for misincorporation during primer extension with NTP analogs (Supplemental Fig. 3). Taken together, the results presented in Figure 4 demonstrate the ability of the Polθ steric-gate variant to efficiently incorporate ribonucleotide analogs with modified base moieties and synthesize RNA with base modifications.
Polθ steric-gate variant efficiently incorporates ribonucleotide analogs with base modifications. (A,C,E,G) Denaturing gels showing PolθRP2 time-dependent incorporation of the indicated canonical and base-modified ribonucleotides on the indicated RNA/DNA primer–templates. (B,D,F) Scatter plots comparing the relative rates of PolθRP2 incorporation of the indicated canonical and base-modified ribonucleotides. n = 3, ±SD. (H) Schematic representation of a method for modifying the 3′-terminal end of RNA by Polθ steric-gate variants. (I) Denaturing gels showing time courses of DNA-dependent RNA synthesis by PolθRP2 in the presence of the indicated NTPs.
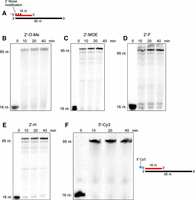
Various 2′ ribonucleoside modifications are widely used at the terminal ends of RNA-based antisense therapeutics for their ability to enhance complementary strand binding via higher TM, reduce immunogenicity, and suppress nuclease digestion (Khvorova and Watts 2017). Polθ steric-gate mutants appear to utilize 2′-O-methyl-NTPs with relatively low efficiency. For example, we found that PolθRP1 exhibited slow RNA extension in the presence of three 2′-O-methyl-NTPs and showed strong pausing or premature termination events (Fig. 5B, left). The addition of MnCl2 instead of MgCl2 improved the rate of extension with 2′-O-methyl-NTPs (Fig. 5B, right). This was expected since polymerases are known to exhibit more promiscuous activity in the presence of MnCl2. We additionally examined PolθRP1 activity with 2′-O-methyl-NTPs in a different sequence context in Figure 5C. Significant pausing and/or premature termination events are also observed on this template, demonstrating that Polθ steric-gate mutants exhibit the limited capacity to synthesize RNA with consecutive 2′-O-methyl ribose modifications.

Polθ steric-gate variant incorporation of 2′-O-modified ribonucleotides. (A) Schematic of RNA/DNA primer–templates. Red text, RNA; black text, DNA. (B,C) Denaturing gels showing time courses of PolθRP1 extension of the indicated RNA/DNA templates in the presence of the indicated 2′-O-Me-NTPs and divalent cation.
Some biotechnology applications may require RNA to be fully composed of modified ribonucleotides for superior stability and/or cellular uptake. For example, phosphorothioate modification of RNA is known to enhance the stability of synthetic RNA against nucleases and improve RNA uptake (Khvorova and Watts 2017). The phosphorothioate linkage represents a sulfur replacement of a nonbridging phosphodiester oxygen, and is one of the most widely used nucleic acid modifications (Eckstein 2014). Along with 2′-O-methyl and 2′-O-methoxyethyl modifications, phosphorothioate modifications are among the most frequently used chemical modifications for RNA-based therapeutics in clinical trials (Shen and Corey 2018). Furthermore, it was recently shown that phosphorothioate mRNA modification accelerates the translation initiation rate, resulting in higher efficiency of protein synthesis. Thus, phosphorothioate-modified mRNA is likely to increase the efficacy of mRNA vaccines (Kawaguchi et al. 2020).
Notably, PolθRP1 mediated full-length RNA synthesis in the presence of all four α-phosphorothioate-NTPs, although the rate of RNA synthesis was significantly slower compared to canonical NTPs (Fig. 6). For example, although the enzyme was able to fully extend the RNA primer in the presence of all four α-phosphothio-NTPs, the full-length product was not observed for at least 60 min at 37°C. The reduced RNA synthesis rate may be due to slower phosphodiester bond formation in the presence of α-phosphothio-NTPs.
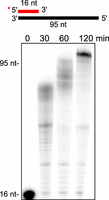
Polθ steric-gate variant synthesis of phosphorothioate-modified RNA. Denaturing gel showing a time course of PolθRP1 DNA-dependent RNA synthesis in the presence of all four 1′-phosphorothioate-NTP analogs with 1 mM DTT, 5 mM MgCl2, and 1 mM MnCl2.
As a strategy to enable rapid synthesis of relatively long RNA with 5′-terminal 2′ ribose chemical modifications, we examined the extension of 5′ modified RNA primers by PolθRP1 in the presence of canonical NTPs. The results show that PolθRP1 efficiently synthesizes RNA products by extending RNA primers containing multiple consecutive 2′ ribonucleotide modifications (2′-O-Me, 2′-MOE, 2′-F) at the 5′ terminus (Fig. 7B–D). We also demonstrate the ability of the mutant enzyme to synthesize RNA with a DNA–RNA chimeric primer containing six consecutive deoxyribonucleotides at the 5′ terminus (Fig. 7E). PolθRP1 also extends an RNA primer containing a 5′-terminal Cy3 fluorophore as expected. In this case, the RNA was visualized via Cy3 fluorescence imaging (Fig. 7F). These data demonstrate the utility of synthesizing relatively long RNA with various 5′-terminal modifications which may be useful for biotechnology and biomedical applications, and basic RNA research.
Polθ steric-gate variant extension of RNA primers with 5′ modifications. (A) Schematic of RNA/DNA template. Black stars indicate the location of three consecutive 2′-ribose modifications at the 5′ terminus. (B–F) Denaturing gels showing time courses of PolθRP1 extension of the indicated 5′ modified RNA/DNA template in the presence NTPs and MgCl2.
DISCUSSION
This report demonstrates a rapid promoter-independent enzymatic synthesis of long RNA oligonucleotides with canonical NTPs and various ribonucleotide analogs using engineered Polθ steric-gate mutants by extending preannealed DNA/DNA and RNA/DNA primer–templates. We also demonstrate the ability to enzymatically synthesize synthetic RNA containing site-specific chemical modifications by using two strategies. The first enables 5′-terminal modifications by utilizing a synthetic RNA primer with 5′-terminal modified ribonucleotides (Fig. 7). The second strategy enables the incorporation of 3′-terminal ribonucleotide modifications by annealing synthetic RNA to a sequence-specific DNA template and incorporating a specific number of ribonucleotides in a time-dependent manner (Fig. 4H). These enzymatic mechanisms and strategies pave the way for next-generation RNA oligonucleotide synthesis methods. For example, automation and high-throughput implementation of the methods described herein, and related enzymatic methods, have the potential to lower the production costs of synthetic RNA with 5′ and 3′ chemical modifications, increase yields, and reduce turnaround time. Specifically, the use of liquid handler-based robotics and nucleic acid surface attachment methods can conceivably enable repeated use of immobilized DNA templates which can potentially increase the yield of enzymatic synthesis of RNA oligonucleotides. Hence, an automated solid-phase enzymatic RNA synthesis platform based on this technology can conceivably be scaled up for kilogram scale production of synthetic RNA for antisense and genome engineering applications. Additionally, the high-throughput development of this RNA synthesis technology can possibly enable rapid synthesis of RNA oligonucleotide libraries for discovery-based research. Finally, engineering additional classes of promoter-independent DNA-dependent RNAPs with distinct ribonucleotide substrate specificities and enzyme characteristics may broaden the capabilities of promoter-independent enzymatic RNA synthesis for the production of chemically modified synthetic RNA oligonucleotides for antisense therapeutics, RNA aptamers, and genome engineering applications. For instance, novel bioengineered promoter-independent DNA-dependent RNAPs that exhibit higher efficiency of incorporating ribonucleotides with 2′-OH modifications may be beneficial for the production of highly stable synthetic RNA for therapeutics and genome engineering.
MATERIALS AND METHODS
Proteins
All site-directed mutations were generated using Agilent Quickchange II following the manufacturer's protocol.
WT Polθ, PolθRP1, and PolθRP2 were expressed and purified as described (Hogg et al. 2011), with the following changes. The eluted fractions from 5 mL HisTrap column (Cytiva) were diluted to 180 mM NaCl, loaded onto a 5 mL Heparin HiTrap column (Cytiva) and eluted with a gradient to buffer C (50 mM Hepes 8.0; 10% Glycerol; 0.005% NP-40) containing 1 M NaCl. Fractions with target protein were pooled, mixed with 10 units of suitable SUMO protease (LifeSensors) and dialyzed overnight against buffer C containing 300 mM NaCl and 20 mM β-mercaptoethanol. The digested fractions were then further purified over a HisTrap column by separating the cleaved His-tag and undigested protein fraction.
T7 RNAP was purified as described (McDevitt et al. 2018).
Purification of Taq DNAP E615G: pET28a vector (Addgene) expressing an amino-terminally 6HIS-tagged version of the Taq DNAP E615G was transformed into BL21(DE3) cells (Invitrogen). The E615G mutation was generated via site-directed mutagenesis using a QuikChange II XL Site-Directed Mutagenesis Kit (Agilent). Freshly grown colonies were inoculated into a starter culture of 40 mL LB supplemented with 50 µg/mL kanamycin and were shaken overnight at 37°C. Next, the overnight cells were added to 4 L of LB with 50 µg/mL kanamycin and grown at 37°C until OD600 ∼ 0.5. Then the shaker temperature was turned to 18°C, and the cells were growing for the next 1 h followed by the addition of IPTG to a final concentration of 0.2 mM. The cells were further shaken overnight, next pelleted in a centrifuge at 4°C (30 min at 3000g) and frozen at −80°C. Frozen pellets (20 g) were thawed on ice and resuspended in 200 mL of lysis buffer containing 50 mM HEPES pH8.0, 0.5 M NaCl, 10% glycerol, 10 mM imidazole pH 8.0, 5 mM βME, 1.5% Igepal CA630 supplemented with 2 mM PMSF and four tablets of SIGMAFAST EDTA-free protease inhibitor cocktail (Sigma). The cells were sonicated on ice, heated in a water bath for 40 min at 65°C and centrifuged for 60 min at 25,000g. The cleared lysate was loaded onto a 5 mL HisTrap FF crude column (Cytiva) and washed with buffer A (50 mM HEPES pH8.0, 0.5 M NaCl, 10% glycerol, 35 mM imidazole pH 8.0, 5 mM βME, 0.005% Igepal). The bound protein was eluted with buffer B containing 200 mM imidazole. The fractions containing Taq DNAP were pooled and dialyzed against 1 L of buffer C (50 mM HEPES pH8.0, 0.1 M NaCl, 10% glycerol, 5 mM βME, 0.005% Igepal) overnight at 4°C. The protein was then loaded onto a 5 mL HiTrap Heparin HP column (Cytiva) and eluted with a NaCl gradient (from 0.1 M to 1 M) in buffer C. Fractions containing Taq DNAP were pooled, concentrated on a spin concentrator Amicon Ultra with 30,000 MWCO (Sigma), centrifuged 10 min at 20,000g and loaded onto a size exclusion column Superdex200 Increase 10/300 (GE Healthcare). The desired protein fractions were combined, aliquoted and frozen at −80°C.
Nucleic acids
Primer strands were 5′-phosphorylated with T4 polynucleotide kinase (New England Biolabs) and (γ-32P) ATP (PerkinElmer) in 1× T4 polynucleotide kinase buffer (New England Biolabs) at 37°C for 60 min. DNA/DNA and RNA/DNA primer–templates were annealed by mixing a ratio of 1:1.5 of primer to template then heating to 95°C–100°C followed by slow cooling to room temperature. The primer in Figure 7F was 5′ conjugated with a Cy3 fluorophore. All oligonucleotides were purchased from Integrated DNA Technologies (IDT) and sequences (5′–3′) are listed below.
RP635:GCGGAGGGCGATAACG
RP635R:rGrCrGrGrArGrGrGrCrGrArUrArArCrG
RP273:AGACTCCGTATCGTAAAGTGACCGACGGTGTTGT
AACTGACGAAATTCACTACCTGTCTGCTATCGAAGAA
GGCAACTACGTTATCGCCCTCCGC.
RP273B:/Biotin/AGACTCCGTATCGTAAAGTGACCGACGG
TGTTGTAACTGACGAAATTCACTACCTGTCTGCTATCG
AAGAAGGCAACTACGTTATCGCCCTCCGC.
RP643:TCCTGGCCAATGAGATGGCAGCTGCCAATGGCT
GGGCACACAGACTCCGTATCGTAAAGTGACCGACGG
TGTTGTAACTGACGAAATTCACTACCTGTCTGCTATCG
AAGAAGGCAACTACGTTATCGCCCTCCGC.
RP644:AGGCAACCGCGTTATCGCCCTCCGC.
RP645:AGGCAACGTCGTTATCGCCCTCCGC.
RP665:AGGCAACGCCGTTATCGCCCTCCGC.
RP668:GATCGATTAATACGACTCACTATAGGGCGGAGG
GCGATAACGTAGTTGCCTTCTTCGATAGCAGACAGGT
AGTGAATTTCGTCAGTTACAACACCGTCGGTCACTTT
ACGATACGGAGTCT.
RP668C:AGACTCCGTATCGTAAAGTGACCGACGGTGTT
GTAACTGACGAAATTCACTACCTGTCTGCTATCGAAG
AAGGCAACTACGTTATCGCCCTCCGCCCTATAGTGA
GTCGTATTAATCGATC.
RP670: TCGAGCACGTTATCGCCCTCCGC
TKD200:CCACTTTTCAAGTTGATAACGGACTAGCCTTAT
TTAACTTGCTATGCTGTTTTGAATGGTTCCCAAAACAG
CATAGCTCTAAAACACAGTTCCTGACTACGAAAGAGA
CTCCGTATCGTAAAGTGACCGACGGTGTTGTAACTG
ACGAAATTCACTACCTGTCTGCTATCGAAGAAGGCA
ACTACGTTATCGCCCTCCGC.
R7: rGrCrGrGrCrGrA
TS01: GGGTCCTGTCTGAAATCGACATCGCCGC
Primer extension assays
Figure 1B: Primer extension assays with 100 nM of Taq DNAP E615G were incubated with 15 nM of radiolabeled DNA/DNA (RP635R/RP665) or RNA/DNA (RP635R/RP665) in buffer A (20 mM Tris-HCl pH 7.5, 0.01% NP-40, 0.1 mg/mL BSA, 10% glycerol, 10 mM MgCl2) and 150 µM NTPs at 37°C for 30 min.
Figure 2B–E: Primer extension assays were performed with 50 nM WT Polθ (Figure 2B,C), 100 nM PolθRP1 (Figure 2D), and 100 nM PolθRP2 (Figure 2E) and were incubated with 15 nM 5′-radiolabeled RNA/DNA (RP635R/RP273) or DNA/DNA (RP635/RP273) for the indicated times in buffer A with 150 µM dNTPs (Figure 2B) or NTPs (Figure 2C–E) at 37°C.
Figure 2F: Primer extension assays were performed with 200 nM PolθRP1 and were incubated with 15 nM 5′-radiolabeled RNA/DNA (RP635R/TKD200) for 60 min in buffer A with the indicated concentrations of NaCl and 300 µM dNTPs at 37°C.
Figure 2G: Primer extension assays were performed with 100 nM PolθRP2 and were incubated with 15 nM of 5′-radiolabeled DNA/DNA (RP635/RP273) for the indicated times in buffer A with 150 µM NTPs at 37°C. Two units of RNase (New England Biolabs) were added as indicated for an additional 30 min at 37°C.
Figure 3A: Primer extension assays with 100 nM of PolθRP1 and T7 RNAP were incubated with 50 nM of radiolabeled RNA/DNA (RP635R/TKD200) primer/template for the indicated time in minutes within a buffer containing (0.01% NP-40, 0.1 mg/mL BSA, 10 mM MgCl2, 10% glycerol) with 300 µM NTPs at 37°C for the indicated time periods. PolθRP1 assays also contained 20 mM Tris at pH 7.5 and 0.5 mM DTT, while T7 RNAP assays also contained 40 mM Tris at pH 7.9 and 1 mM DTT.
Figure 3B: Primer extension assays with 100 nM of PolθRP1 and T7 RNAP were incubated with 50 nM of radiolabeled RNA/DNA (R7/TS01) primer/template for the indicated time in minutes within a buffer containing (0.01% NP-40, 0.1 mg/mL BSA, 10 mM MgCl2, 10% glycerol) at 37°C for the indicated time periods with or without a 10 min preincubation at 25°C before NTPs were added for a final concentration of 300 µM. PolθRP1 assays also contained 20 mM Tris at pH 7.5 and 0.5 mM DTT, while T7 assays also contained 40 mM Tris at pH 7.9 and 1 mM DTT.
Figure 3C,D: Primer extension assays with 200 nM of PolθRP1 and T7 RNAP were incubated with 100 nM of radiolabeled RNA/DNA (R7/TS01) primer/template for the indicated time in minutes within a buffer containing (0.01% NP-40, 0.1 mg/mL BSA, 10 mM MgCl2, 10% glycerol) with 300 µM the indicated nucleotides at 37°C for the indicated time periods. PolθRP1 assays also contained 20 mM. Tris at pH 7.5 and 0.5 mM DTT, while T7 RNAP assays also contained 40 mM Tris at pH 7.9 and 1 mM DTT.
Figure 4A,B: Primer extension assays were performed with 40 nM PolθRP2 and were incubated with 60 nM 5′-radiolabeled RNA/DNA (RP635R/RP273) for the indicated times in buffer A supplemented with 10 mM MgCl2 along with 150 µM of UTP or pseudouridine-triphosphate at 25°C.
Figure 4C–F: Primer extension assays were performed with 20 nM of PolθRP2 and were incubated with 60 nM of 5′-radiolabeled RNA/DNA (RP635R/RP644 [Figure 4C,D]; RP635R/RP645 [Figure 4E,F]) for the indicated times in buffer A with 150 µM CTP or 5-methyl-deoxycytidine triphosphate (Figure 4C,D) and 150 µM ATP or N6-methyl-adenosine triphosphate (Figure 4E,F) at 25°C. Scatter plots represent the average % extension at the indicated times. Data represent mean (n = 3) ±SD. % extension was calculated by dividing the intensity of the sum of the extended products by the sum of the intensity of the extended and unextended products then multiplying by 100. Relative gel band intensities were determined using ImageJ.
Figure 4G: Primer extension assays were performed with 40 nM PolθRP2 and were incubated with 60 nM 5′-radiolabeled RNA/DNA (RP635R/RP273) for the indicated times in buffer A with 150 µM pseudouridine-triphosphate at 25°C.
Figure 4I: Primer extension assays were performed with 100 nM PolθRP2 and were incubated with 15 nM 5′-radiolabeled RNA/DNA (RP635R/RP273) for the indicated times in buffer A with 150 µM of the indicated nucleotides at 37°C.
Figure 5B: Primer extension assays were performed with 100 nM PolθRP1 and were incubated with 20 nM of 5′-radiolabeled RNA/DNA (RP635R/RP273) for the indicated times in buffer A supplemented with 5 mM MgCl2 or 5 mM MnCl2 with 150 µM of the indicated 2′-O-methyl-nucleoside triphosphates at 37°C.
Figure 5C: Primer extension assays were performed with 100 nM PolθRP1 and were incubated with 20 nM 5′-radiolabeled RNA/DNA (RP635R/RP644) for the indicated times in buffer A with 150 µM of the indicated 2′-O-methyl-nucleoside triphosphates at 37°C.
Figure 6: Primer extension assays were performed with 100 nM of PolθRP1 and were incubated with 20 nM of 5′-radiolabeled RNA/DNA (RP635R/RP273) for the indicated times in buffer A (supplemented with 5 mM MgCl2,1 mM MnCl2, and 1 mM DTT) along with 150 µM of α-phosphothioate-NTPs at 37°C.
Figure 7B–E: Primer extension assays were performed with 400 nM of PolθRP1 and were incubated with 20 nM of 5′-radiolabeled RNA/DNA (RP635R/RP273 with the indicated 5′-terminal chemical modifications to the RNA primer) in buffer A for the indicated times with 250 µM of NTPs at 37°C.
Figure 7F: Primer extension assays were performed with 400 nM of PolθRP1 and were incubated with 20 nM of 5′-Cy3 conjugated RNA/DNA (RP635R-Cy3/RP273) for the indicated times in buffer A with 250 µM NTPs at 37°C.
All primer extension assays were terminated with 25 mM EDTA and 45% formamide. Radiolabeled DNA and RNA products and Cy3 conjugated RNA products were resolved in urea denaturing 15% polyacrylamide gels and visualized by phosphorimager.
In vitro transcription
Figure 2G: In vitro transcription assays were performed with 100 nM T7 RNAP and were incubated with 20 nM double-strand DNA (RP668/RP668C) for the indicated times in buffer B (40 mM Tris-HCl pH 7.9, 0.01% NP-40, 0.1 mg/mL BSA, 6 mM MgCl2, 1 mM DTT, and 10% glycerol) with 300 µM NTPs, along with 32P-α-ATP (PerkinElmer) at 37°C. Assays were terminated with 25 mM EDTA and 45% formamide, then radiolabeled RNA was resolved in urea denaturing 15% polyacrylamide gels and visualized by phosphorimager.
RNA sequencing and analysis
RNA synthesis was performed by preincubating 75 nM PolθRP1 with 20 nM 635R/273B RNA/DNA primer–template in buffer 25 mM Tris-HCl pH 7.8, 0.01% NP-40, 10% glycerol, 5 mM MgCl2, 0.1 mg/mL BSA for 5 min at 37°C. RNA synthesis was initiated by adding 200 µM NTPs for an additional 30 min. The reaction was then mixed with Dynabeads M-280 streptavidin prewashed with buffer (described above) in the presence of 1 M NaCl. The RNA/DNA product was incubated with the beads for ∼10 min, then the beads were washed three times using a magnetic holder with buffer plus 1 M NaCl using 500 µL volumes to remove unbound protein and nucleic acid. Following washing, beads were suspended in 50 mM Tris-HCl pH 8.8, boiled for 10 min, then placed on ice for 5 min. The supernatant was collected and nucleic acid purified using Zymo Research RNA Clean and Concentrator kit at room temperature. Purified RNA was used for cDNA synthesis using Multiscribe Reverse Transciptase (Applied Biosystems) using manufacturer's protocol. Nucleic acid products were purified using Zymo Oligo Clean and Concentrator kit. The purified cDNA was then amplified via PCR (Phusion High-Fidelity PCR Master Mix with HF Buffer [Thermo Scientific]) using PCR primers RP635, RP672 resulting in 95 bp product as observed on an agarose gel. Next, the 95 bp DNA product was amplified using nested PCR primer RP635A and RP672A to increase the length to 145 bp for downstream high-throughput sequencing. The 145 bp product was purified using Zymo Oligo Clean and Concentrator kit. Finally, the 145 bp PCR DNA products were submitted for next-generation high-throughput sequencing (Genewiz from Azenta Life Sciences). Reads were aligned to the 145 bp reference sequence with bowtie2 aligner; samtools were used to extract the tags from bam files representing the distance from the template, substitution and gap opening (NM, XM, XO). All plots were generated from extracted counts using ggplot in R.
Nucleotide analogs
2′-O-methyl-NTPs, N6-methyl-ATP, 5-methyl-CTP, and pseudouridine-triphosphate were purchased from Trilink Biotechnologies. 2′-F-NTPs were purchased from Jena Biosciences.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
R.T.P. is a cofounder and chief scientific officer of Recombination Therapeutics, LLC. X.S.C. is a cofounder of Recombination Therapeutics, LLC. The affiliation does not affect the authors’ impartiality, adherence to journal standards and policies, or availability of data. A provisional patent application on this process and findings was filed by R.T.P.
ACKNOWLEDGMENTS
This research was supported by National Institutes of Health (NIH) grants (1R01GM137124, 1R01HG011669) to R.T.P.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079396.122.
- Received August 5, 2022.
- Accepted April 7, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Taylor Tredinnick is the first author of this paper, “Promoter-independent synthesis of chemically modified RNA by polymerase θ variants.” Taylor is a Research Assistant in the Pomerantz laboratory at Thomas Jefferson University. The laboratory focuses on DNA repair, cancer drug development, and more recently, methods in RNA biotechnology.
What are the major results described in your paper and how do they impact this branch of the field?
In our paper, we characterized the ability of DNA polymerase θ mutants to synthesize RNA oligonucleotides in a promoter-independent fashion. We also found that these enzymes were able to incorporate several modified nucleotides (NTPs) into the RNA oligonucleotide chain. As these modified NTPs have recently been used with success in RNA-based therapeutics and vaccines, the ability to incorporate them into synthetic RNA using our bioengineered enzyme should prove useful for researchers and manufacturers in this area. We believe that our paper outlines a potential method for future enzymatic production of chemically modified RNA.
What led you to study RNA or this aspect of RNA science?
RNA-based research, therapeutics and vaccines have made a substantial impact on the world in recent years. As the field continues to grow, cheaper, more efficient, and greener methods for producing chemically modified synthetic RNA will be needed. Discovering new methods that meet these criteria is our goal in developing new enzymatic RNA synthesis technologies.
If you were able to give one piece of advice to your younger self, what would that be?
Make an effort to meet more people in your field of interest and use your connections to find a good mentor, then absorb all you can from them.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
The Principal Investigator of my laboratory, Richard Pomerantz, has taught me much about biomedical research over the past few years. However, what I learned from him the most is, if you want to do high-quality science and make an impact on the world, you need to take the time to find the mission that drives you. Once you find it, you'll work as hard as you need to in order to achieve your goals.
What are your subsequent near- or long-term career plans?
I will be transferring to another university to gain experience in methods of translational research. Ultimately, my mission is to become a physician-scientist and medical educator.








