
Phase-separated ribosome-nascent chain complexes in genotoxic stress response
- Orsolya Németh-Szatmári1,
- Bence Nagy-Mikó1,
- Ádám Györkei2,3,
- Dániel Varga4,
- Bálint Barna H. Kovács4,
- Nóra Igaz1,
- Bence Bognár1,
- Zsolt Rázga5,
- Gábor Nagy1,
- Nóra Zsindely1,
- László Bodai1,
- Balázs Papp2,
- Miklós Erdélyi4,
- Mónika Kiricsi1,
- András Blastyák6,
- Martine A. Collart7,
- Imre M. Boros1 and
- Zoltán Villányi1
- 1Department of Biochemistry and Molecular Biology, University of Szeged, 6726 Szeged, Hungary
- 2Institute of Biochemistry, Biological Research Centre, 6726 Szeged, Hungary
- 3Section for Physiology and Cell Biology, Department of Biosciences, University of Oslo, 0316 Oslo, Norway
- 4Department of Optics and Quantum Electronics, University of Szeged, 6720 Szeged, Hungary
- 5Department of Pathology, Faculty of Medicine, University of Szeged, 6720 Szeged, Hungary
- 6Institute of Genetics, Biological Research Centre, 6726 Szeged, Hungary
- 7Department of Microbiology and Molecular Medicine, Institute of Genetics and Genomics Geneva, Faculty of Medicine, University of Geneva, 1211 Geneva 4, Switzerland
- Corresponding author: villanyi.zoltan{at}bio.u-szeged.hu
Abstract
Assemblysomes are EDTA- and RNase-resistant ribonucleoprotein (RNP) complexes of paused ribosomes with protruding nascent polypeptide chains. They have been described in yeast and human cells for the proteasome subunit Rpt1, and the disordered amino-terminal part of the nascent chain was found to be indispensable for the accumulation of the Rpt1-RNP into assemblysomes. Motivated by this, to find other assemblysome-associated RNPs we used bioinformatics to rank subunits of Saccharomyces cerevisiae protein complexes according to their amino-terminal disorder propensity. The results revealed that gene products involved in DNA repair are enriched among the top candidates. The Sgs1 DNA helicase was chosen for experimental validation. We found that indeed nascent chains of Sgs1 form EDTA-resistant RNP condensates, assemblysomes by definition. Moreover, upon exposure to UV, SGS1 mRNA shifted from assemblysomes to polysomes, suggesting that external stimuli are regulators of assemblysome dynamics. We extended our studies to human cell lines. The BLM helicase, ortholog of yeast Sgs1, was identified upon sequencing assemblysome-associated RNAs from the MCF7 human breast cancer cell line, and mRNAs encoding DNA repair proteins were overall enriched. Using the radiation-resistant A549 cell line, we observed by transmission electron microscopy that 1,6-hexanediol, an agent known to disrupt phase-separated condensates, depletes ring ribosome structures compatible with assemblysomes from the cytoplasm of cells and makes the cells more sensitive to X-ray treatment. Taken together, these findings suggest that assemblysomes may be a component of the DNA damage response from yeast to human.
Keywords
INTRODUCTION
Assembly of protein complexes in the dense eukaryotic cytoplasm can be challenging. Cotranslational assembly of proteins can facilitate the assembly process and is essential in specific cases. In the last few years some examples for this process were reported, such as the synthesis of two adjacent proteins within the proteasome base, namely Rpt1 and Rpt2. While these proteins interact in their native context, they do not interact in the yeast two-hybrid assay and are not even soluble when produced separately (Barrault et al. 2012; Fu et al. 2001). Instead, Rpt1 and Rpt2 are produced with ribosome pausing within EDTA- and RNase-resistant granules that contain the largest subunit of the Ccr4–Not complex, Not1 (Panasenko et al. 2019). These granules, referred to as Not1-containing assemblysomes (NCAs), are distinct from other known granules, such as processing bodies (P-bodies) and stress granules (SG), as evidenced with fluorescent microscopy (Panasenko et al. 2019). P-bodies contain components of the mRNA decay machinery and are RNase- and cycloheximide- (CHX) sensitive (Hubstenberger et al. 2017; Youn et al. 2019). SG contains translation initiation components and their size is between 0.1 and 1 µm. Both P-bodies and SGs are dense entities and can be sedimented with moderate-speed centrifugation (8–10,000g) (Jain et al. 2016; Hubstenberger et al. 2017). NCAs are distinct from both of these granules as they contain ribosomes paused in translation with protruding nascent polypeptide chains, can be sedimented only by high-speed ultracentrifugation and are resistant to EDTA and CHX treatment (Jain et al. 2016; Hubstenberger et al. 2017; Panasenko et al. 2019; Youn et al. 2019). Table 1 summarizes the fundamental attributes of SGs, P-bodies, and NCAs.
Basic attributes of stress granules, P-bodies, and assemblysomes
Ribosome profiling indicated ribosome pause sites on the RPT1 and RPT2 mRNAs and it was determined that their translation in NCAs resumes only when the nascent chains of the partners interact (Panasenko et al. 2019). The amino-terminal domains of both Rpt1 and Rpt2 are suggested to protrude out of the ribosome exit tunnel in the context of stalled translation and these are the interacting helices of the two proteins. Both amino-terminal protruding domains contain disordered regions, a feature important for the accumulation of the paused ribosome-nascent chain complexes (RNCs) in assemblysomes (Panasenko et al. 2019). While paused ribosomes can provoke ribosome collisions and thereby risk being eliminated by the ribosome quality control (RQC) mechanism, stalled Rpt1 and Rpt2 are very stable in NCAs indicating that NCAs protect the paused ribosomes from RQC (Inada 2013). This remarkable feature of NCAs most likely contributes to promoting the cotranslational interaction of the partner proteins.
The structure of NCAs, and how widespread they are, is not known. What we know so far about NCAs is limited to their discovery in the context of the cotranslational assembly of Rpt1 and Rpt2 (Panasenko et al. 2019). A recent study using ribosome profiling revealed the pivotal role of Not1 in regulating ribosome pausing on numerous mRNAs, indicating that the NCA regulation might be widespread (Gillen et al. 2021; Allen et al. 2023). However, maybe not all mRNAs translated in assemblysomes show detectable ribosome pausing by ribosome profiling, because assemblysomes are RNase-resistant (Panasenko et al. 2019). It is not yet even clear if Not1 is present in all assemblysomes. Taking into consideration these limitations of earlier studies, we tried an independent approach, wherein we exploited known features of assemblysomes to conduct a global in silico analysis to find protein complexes that may fall under assemblysome regulation. Then, taking as an example yeast Sgs1, a positive hit obtained in the in silico analysis which is a protein implicated in DNA damage response, we show that Sgs1 ribosome-associated nascent chains are indeed a component of assemblysomes. Furthermore, we observe that perturbation of phase separation aggravates DNA damage sensitivity. By using a number of biochemical and microscopic approaches, we demonstrate the high similarity of stalled Sgs1–RNA–ribosome complexes to Rpt1-containing assemblysomes. Finally, by experiments conducted on human cell lines of tumorous origin, we present pieces of evidence to show that the role of assemblysomes in the DNA damage response is likely a conserved phenomenon.
RESULTS
Assemblysome formation requires ubiquitylation
In order to understand the basic principles of assemblysome formation, we took advantage of the published construct which models Rpt1 stalling and assemblysome formation. This construct consists of a copper-inducible CUP1 promoter, followed by sequences encoding a Flag-tag, the amino-terminal 135 amino acids of Rpt1 and then 12 lysine (K12) codons that result in ribosome stalling (Fig. 1A; Matsuda et al. 2014). The Rpt1 nascent chain expressed from this construct migrates on SDS-PAGE with a unique and discrete size, but at a higher apparent molecular weight than the expected 25 kDa (Fig. 1A). This shift in size is the result of ubiquitylation, and nascent Rpt1 is quantitatively modified in this context as revealed by nickel affinity purification performed on extracts of cells expressing histidine-tagged ubiquitin (Fig. 1A). To clarify the role of ubiquitylation, we mutated the first six lysine residues of Rpt1, and observed that the nascent Rpt1 with these mutations migrated with a size approximately 8 kDa smaller, indicating that it was no longer ubiquitinated (Fig. 1B). We tested the sedimentation profile of ubiquitinated and nonubiquitylated Rpt1 on sucrose gradients. As previously determined, the nascent Rpt1 was detected in the ribosome-containing fractions of the sucrose gradient, including the very heavy polysome-containing fractions (Fig. 1C; Panasenko et al. 2019). However, the nonubiquitinated nascent Rpt1 was detected in ribosome-free fractions of the sucrose gradient, indicating that the ubiquitination of the nascent chain was essential to ensure its ribosome association and presence in heavy sedimenting particles (Fig. 1C). Presence of the nonubiquitinated nascent Rpt1 in free fraction indicates abortive translation, possibly a result of RQC, or translation through the stalling sequence to the stop codon (Fig. 1C). We hypothesized that ubiquitylation may counteract such events to facilitate assemblysome formation in the context of other proteins too, and used the possibility of mono-ubiquitylation, as a tenet to screen for candidates by using bioinformatics. Upon Flag-immunoprecipitation to enrich nascent amino-terminally tagged Rpt1, it was revealed that it is mostly present in ubiquitinated form as revealed by ubiquitine antibody, but low levels of faster migrating Rpt1 were detectable with Flag antibody. This faster migrating form was not detectable with antibodies to ubiquitin (Supplemental Fig. S1).
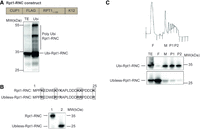
Amino-terminal nascent chain ubiqutination is important for NCA formation. (A) Ubiquitinated proteins were affinity purified by nickel beads from cells coexpressing a stalled nascent Rpt1 chain (RNC) (RNC construct above blot) and His6-tagged ubiquitin from the CUP1 promoter. The input extract (TE) or Ubi-affinity proteins (Ubi) were detected by western blotting with antibodies to the amino-terminal Flag-tag. The discrete Rpt1-RNC is ubiquitinated (Ubi-Rpt1-RNC). Polyubiquitinated forms of the Rpt1-RNC are additionally visible (Poly Ubi-Rpt1-RNC). (B) Expression of the Rpt1-RNC without (1) and with (2) the first six lysines mutated to arginine (mutations shown above the blot) (Ubiless-Rpt1-RNC) monitored by western blotting with anti-Flag antibodies. (C) Polysome profiling of cells expressing the Rpt1-RNC or the Ubiless-Rpt1-RNC (as in B). Fractions were visualized with anti-Flag antibodies. (TE) Total extract, (F) free fractions, (M) 80S monosomes, (P1) light polysomes, (P2) heavy polysomes (the profile for the Ubi-Rpt1-RNC is shown above the blots with an indication of the analyzed fractions).
Global scale in silico screen for yeast amino-terminal disordered proteins predicts DNA damage response complexes as possible assemblysome-regulated candidates
Having given that the amino terminus was found to be important for NCA formation in the case of Rpt1 and Rpt2, we performed a global scale prediction of potentially assemblysome-associated nascent proteins by ranking all known protein complex subunits of the yeast Saccharomyces cerevisiae according to their amino-terminal disorder propensity (Supplemental Table S2; Panasenko et al. 2019). The possibility for mono-ubiquitylation as a second tenet was applied to exclude those proteins from the analysis that lack lysine residues within 25 amino acids in their amino terminus, which likely rules them out as a target for this modification.
Stalling of RNCs of both RPT1 and RPT2 mRNAs happen to occur at a rare codon pair. Moreover, it was revealed recently that Not5—an enigmatic subunit of the Ccr4–Not complex—modulates the ribosome's fate through binding ribosomes with a vacant A site at a nonoptimal codon, and this binding facilitates dynamic condensate formation (Allen et al. 2021). Hence, as a third filter, we looked for the presence of rare codon pairs among the top transcripts that were positive hits by applying the two previous filters. Among the candidates we identified three subunits of distinct complexes playing a role in the DNA damage response: Rad10, Rad14, and Sgs1 (Supplemental Fig. S2). Rad10 and Rad14 together with Rad1 are components of the Nucleotide Excision Repair Factor 1 (Nef1) complex. Rad14 was shown to target the Rad1–Rad10 nuclease to UV-induced DNA damage sites in vivo (Guzder et al. 2006). Sgs1 is part of the DNA helicase-topoisomerase complex that together with Top3 and Rmi1 is an important player in DNA double-strand break repair (Gravel et al. 2008; Mimitou and Symington 2008; Zhu et al. 2008).
Certain mRNAs accumulate in EDTA-resistant ribosome pellets in a manner affected by UV treatment
In order to substantiate the in silico results, we chose to follow SGS1, RAD10, and RAD14 mRNAs and to test their presence in EDTA-resistant granules. As both P-bodies and SGs can be sedimented with moderate-speed (8–10,000g) centrifugation, ultracentrifugation of yeast extracts was performed after such prior purification of the lysate to separate these entities from assemblysomes (Jain et al. 2016; Hubstenberger et al. 2017). Ultracentrifugation of the supernatant obtained by the moderate-speed centrifugation was performed using a 60% sucrose cushion and resulted in two fractions: one which passes through the cushion and forms a pellet and the other that remains in or at the top of the cushion. This procedure was performed in the presence (or in the absence) of EDTA, which is known to disconnect the small and large subunits of the ribosome and helps to get rid of polysomes, since they would otherwise cosediment with assemblysomes (Fig. 2A; Panasenko et al. 2019). The utility of this experimental setup was verified by checking the major RNA species in various fractions on agarose gel. For example, SG is known to contain the 40S ribosome subunit and the abundance of 18S rRNA is evident in the moderate-speed (8000g) SG enriched fraction (this sample was treated with UV to provoke SG formation) (Fig. 2B; Kwon et al. 2007; Moutaoufik et al. 2014). The stronger 18S rRNA band in UV-treated compared to nontreated condition reflects that indeed SGs are enriched in the 8000g first pellet, as SGs contain the small subunit of the ribosomes (Fig. 2B, compare 18S rRNA bands marked with an arrow; Hubstenberger et al. 2017). High-speed pellets depleted from SGs obtained in the absence of EDTA contain substantial and apparently stoichiometric amounts of 18S and 28S rRNAs along with tRNAs, while in the presence of EDTA most of these RNAs remain in the supernatant. Still, however, significant quantities of them remain in the EDTA-resistant pellet indicating the presence of ribosomes in a structure distinct from their translationally competent form (Fig. 2B).
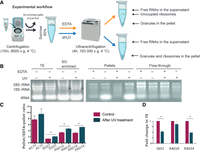
SGS1, RAD10, and RAD14 mRNAs are stored in EDTA-resistant, phase-separated granules. (A) Experimental workflow to pellet granules or granules and polysomes with ultracentrifugation of yeast total protein extracts on 60% sucrose cushions with or without EDTA, respectively. Ultracentrifugation of soluble total extracts on 60% sucrose cushions leads to an enrichment of polysomes in the pellet. If the experiment is performed in the presence of EDTA, the pellet contains no polysomes and is enriched in EDTA-resistant granules. SGs and P-bodies are pelleted before ultracentrifugation and EDTA treatment. (B) Total RNA extracted from yeast cells treated or not with UV were separated on 60% sucrose cushion in the presence or absence of EDTA and separated on 1% nondenaturing agarose gel. (TE) Total extracts. Separated RNA species are highlighted (tRNAs, 18S and 28S rRNAs). Arrows show 18S rRNA species to compare in order to reveal SGs that are provoked by UV and contain small ribosome subunits. (C) RT-qPCR analysis of mRNAs from pellets originating from identical extracts treated with or without EDTA. Results are expressed as pellet/EDTA-pellet ratio of mRNA quantities. Change in ACT1, SGS1, RAD10, and RAD14 and pellet/EDTA-pellet ratio of mRNA quantities after the UV treatments are shown next to the same ratio measured in the untreated control. Values in the chart represent the average of two independent measurements of biological replicates with error bars representing standard deviation. Statistical significance is determined by a two-tailed Student's t-test ([*] P < 0.05, [**] P < 0.01). (D) RT-qPCR analysis of mRNAs from the total extract of control and UV-treated yeast cells. Fold change in mRNA quantities in UV-treated samples are compared to the control in each case, and ACT1 mRNA was used for normalization. Statistical significance is determined by a two-tailed Student's t-test ([*] P < 0.05).
Having established that different ribonucleoprotein entities can be separated and distinguished by our experimental procedure we decided to quantitate specific mRNAs in pellets obtained with and without EDTA by RT-qPCR. By calculating ratios of the mRNA content of the samples, we can get information regarding the quantity of two different translationally engaged forms of a given mRNA; one is associated with ongoing translation, and the other is albeit initiated but stays stalled and awaits for resumption in assemblysomes. For example, a pellet/EDTA-pellet ratio around 1 indicates that the mRNA is associated with assemblysomes. This is on one hand because assemblysomes are the only mRNA-containing entities in the precleared extract which can pass the 60% sucrose cushion during ultracentrifugation in the presence of EDTA. On the other hand, ribosome pellets prepared without EDTA also contain assemblysomes, therefore in theory the ratio is exactly 1 if all mRNAs are in assemblysomes. On the contrary, high pellet/EDTA-pellet ratios are characteristic for mRNAs that are bound to translating ribosomes as EDTA can disconnect these. mRNA species not translated are not considered by the subsequent calculations. According to our in silico prediction, ACT1 mRNA is an unlikely component for assemblysome formation as actin is known to form polymers after translation, and thus it was chosen as a control. Indeed, the pellet/EDTA-pellet ratio is high for ACT1 (Fig. 2C). This indicates that the ribosome-bound ACT1 mRNA is mostly associated with polysomes rather than present in assemblysomes (Fig. 2C). In contrast, the SGS1 mRNA ratio of ±EDTA samples is around 1 indicating enrichment of SGS1 mRNA in the EDTA fraction, compared to that of the control ACT1 mRNA. The same ratio for RAD10 and RAD14 mRNAs was higher than the one observed with SGS1 but far from the control, this indicates RAD10 and RAD14 mRNA enrichment in the EDTA-resistant fraction compared to ACT1, while assemblysome presence is not as remarkable as for SGS1 mRNA (Fig. 2C).
Evidently, the need for a full-length DNA repair protein is mostly needed in case of DNA damage. Therefore, we UV-irradiated exponentially growing wild-type yeast cultures, not once but twice, to mimic acute UV stress and calculated the mRNA ratios of ±EDTA samples as above. Remarkably, ratios for SGS1, RAD10, and RAD14 mRNAs increased upon UV treatment in all cases indicating a shift of EDTA-resistant mRNAs to the EDTA-sensitive fraction. Meanwhile, total mRNAs did not increase indicating the lack of significant de novo mRNA synthesis (Fig. 2D). The most likely explanation for the above observation is that translation in the assemblysome-enriched EDTA-resistant pellets resumes, and moves toward polysome formation upon DNA damage to fulfill the demands of DNA damage response processes (Fig. 2C).
Granules contain multiple identical RNCs and their density is ORF-dependent
In order to provide visual evidence that the disordered amino-terminal of Sgs1 is capable to render the RNC to form assemblysomes, we created a similar construct as the Rpt1-RNC that was used to demonstrate the existence of EDTA-resistant NCAs (Fig. 3A; Panasenko et al. 2019). We chose to clone the first 135 amino acid encoding sequence of Sgs1 before the stalling sequence because the rare codon pairs were located exactly after the 135th codon in the case of Sgs1 (Fig. 3A). This is by chance identical to the length of the Rpt1-RNC which was also 135 amino acid long before the stalling sequence (Fig. 1A). In the case of Rpt1-RNC, the length of the construct was determined previously by analyzing ribosome stall sites with ribosome profiling (Panasenko et al. 2019). To compare granule forming capabilities of these similar inducible constructs and to obtain information about the physical properties of RNC granules in the cytoplasm, we performed dSTORM imaging on yeast cells transformed with the Rpt1- and Sgs1-RNC expressing plasmids. The approximate size of the ribosome is 30 nm in diameter and the resolution of dSTORM microscopy makes it possible to estimate (van de Linde et al. 2011; Nieuwenhuizen et al. 2015) the number of RNCs even if ribosomes are closely packed within granules. dSTORM microscopy revealed that expression of both constructs results in a clustered staining in the cytoplasm, suggesting that the nascent chains are not present as individual soluble RNCs that would stain the whole cytoplasm dispersedly but are rather present in granules with multiple RNCs inside (Fig. 3B). The clustered staining of Sgs1-RNC and Rpt1-RNC was very similar and confirmed that the amino-terminal of Sgs1 is also capable to form condensates in the cytoplasm similarly as was reported for Rpt1-RNC (Panasenko et al. 2019). We noticed minor differences in granule number and density between the two constructs (Fig. 3A). The lateral size (area) of the granules was larger with a similar median in Sgs1-RNC expressing cells than in Rpt1-RNC (Fig. 3C,D). Medians are identical due to the high number of individual RNCs that are not in granules (Fig. 3D–F). We found no typical geometric shape for a granule, but their density was different between the two construct-expressing cells, according to the average distance of the nearest granule (Fig. 3E). The average epitope number representing the density of epitopes inside granules was also slightly different between the two studied constructs. This value ranged from the average of five found in Rpt1-RNC granules to the average of 8 found in Sgs1-RNC granules (Fig. 3F), in parallel these values correlated with the slight differences in the lateral size of the granules in the range of several thousand square nanometers (compare Fig. 3D and F for both constructs). This latter comparison reveals that granules contain several nanometer wide entities for every epitope representing a nascent chain compatible with the size of ribosomes (∼30 nm).
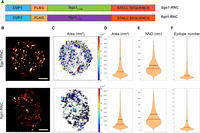
Rpt1- and Sgs1-RNCs form granules in the cytoplasm. (A) Schematic map of the two constructs expressing either Rpt1- or Sgs1-RNC that are identical apart from their first 135 amino acid encoding sequences generated for this study. (B) Two representative reconstructed dSTORM images reveal the granular staining of amino-terminally Flag-tagged Rpt1-RNCs and Sgs1-RNCs. Staining was performed using M2 Flag antibody followed by a secondary Alexa647 antibody. Scale bar: 1 µm. (C) Map of the cluster analyzed localizations via the DBSCAN algorithm. The clusters are color-coded based on the size of their area. Violin plots represent the different distribution of clustered Flag signal area in nm2 (D), of nearest neighbor distance (NND) in nm (E), and the distribution of the epitope number/cluster (F), comparing 36 different Sgs1-RNC expressing cells and 68 different Rpt1-RNC expressing cells, from two (Rpt1-RNC) and three (Sgs1-RNC) independent STORM measurements done on both constructs. Vertical lines represent the median (red) and the mean (black) values.
EDTA-pellets retrieved from MCF7 human breast adenocarcinoma cell line are enriched in mRNAs involved in DNA damage response
The in silico prediction done in yeast led to the discovery that Sgs1 is similarly present in EDTA-pellets and the nascent protein–ribosome complex forms similar cytoplasmic foci as Rpt1-RNC. We were curious to find out whether these cytoplasmic entities are conserved in higher eukaryotes or not. Although it is not possible to find rare codon pairs in the case of human gene products as codon optimality differs from cell line to cell line, we noticed that upon simply ranking the human complex subunits according to their amino-terminal disorder propensity, proteins involved in DNA repair—including BLM, the human ortholog of yeast Sgs1, and PSMC1, the human ortholog of Rpt2—are at the 3247 and 3513 positions, respectively, out of 23,391 proteins present in the list (Supplemental Table S3). Moreover, identical GO categories—including many involved in DNA repair—were enriched both at the top of human and yeast complex subunit amino-terminal disorder propensity rankings (Supplemental Table S4).
Intrigued by the above, we decided to sequence mRNA libraries of pellets and EDTA-pellets obtained from MCF7 tumor cells to reveal transcripts that are possibly regulated by assemblysomes. We divided the read counts obtained from sequencing the ribosome pellet library with read counts obtained from the EDTA-pellet library to express the mRNA ratio of ±EDTA pellets genome-wide (Supplemental Tables S5, S6). The lower this ratio is for each individual transcript the more likely that the mRNA is in assemblysomes as in the case of the yeast SGS1 (Fig. 2). We performed the sequencing in biological duplicates that showed good reproducibility for both the pellet and EDTA-pellet libraries (Supplemental Fig. S3).
We took the top and bottom 3000 mRNAs of the list generated by ranking them according to their ±EDTA ratio of reads in pellets and subjected both of these groups to gene ontology (GO) analysis. Interestingly, the top 3000 mRNAs of the list (ratio above 0.94), which we refer to as EDTA-sensitive pellet mRNAs, were enriched in mRNAs connected to translation, translation regulation, as well as mRNAs encoding proteins with mitochondrion-related functions (Table 2 “pellet GOs,” Supplemental Tables S6, S7 “pellet selected”). These same GO terms appeared in a former study when analyzing mRNAs associated with Not1 (Gupta et al. 2016). Moreover, GO terms connected to stress response were markedly underrepresented in this group (Table 2 “pellet GOs,” Supplemental Table S7 “pellet selected”). This is expected as the cells were harvested under ideal conditions in their exponential growth phase. Actually, EDTA-sensitive pelleted mRNAs are associated with actively translating polysomes, and under steady-state conditions there is no need to translate stress-responsive proteins. The bottom 3000 mRNAs of the ranking (±EDTA ratio below 0.5) considered to represent mRNAs associated with EDTA-resistant ribosomes, presumably assemblysomes, were enriched in mRNAs having cytoskeletal or microtubule-related functions, are involved in the stress response, assembly processes, related to autophagy, and importantly, play a role in DNA repair including BLM, the human ortholog of yeast Sgs1 (Table 2 “EDTA-pellet GOs,” Supplemental Tables S6, S7 “EDTA-pellet selected”). We refer to these mRNAs as EDTA-pellet mRNAs from here on.
GO term analysis on ribosome pellet and EDTA-pellet associated mRNAs
mRNAs encoding amino-terminal disordered proteins are enriched in EDTA-pellets
We checked with a permutation test if the proportionality of mRNAs encoding proteins with disordered amino terminus versus ordered amino terminus was above average for the EDTA-pellet mRNAs (Fig. 4). By taking 10,000 times 3000 random mRNAs, we show that the ratio of mRNAs encoding proteins with amino-terminally disordered domains versus mRNAs encoding proteins with nondisordered protein-encoding ones develops a normal distribution. The vertical line in Figure 4 is far off to the right of the normal distribution and represents the ratio observed in the sequenced 3000 EDTA-pellet associated MCF7 cell-derived mRNA group. Thus, these results show that the EDTA-pellet is enriched in amino-terminally disordered protein-encoding mRNAs.
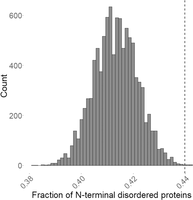
Permutation test reveals that mRNAs in EDTA-pellets are enriched in amino-terminally disordered protein-encoding ones. Taking 10,000 times randomly 3000 proteins from the UniProt database leads to a normal distribution of counts where each value at the x-axis represents the ratio of amino-terminally disordered and nondisordered protein counts. Protein disorder propensity at their 25 amino acid long amino-terminal was determined using the AlphaFold database (Jumper et al. 2021). Vertical line represents the ratio of disordered and nondisordered protein counts encoded by the 3000 mRNAs that were selected previously as mRNAs sedimenting in EDTA-pellet based on their pellet/EDTA-pellet read count ratio.
Moreover, we noticed that the number of identical GO categories between the top 3000 most amino-terminally disordered human complex subunits (Supplemental Table S3) and the EDTA-pellet associated mRNAs sequenced in the MCF7 cell line were markedly high (Supplemental Table S7). One hundred and fifty six GO categories were identical out of 448 enriched in EDTA-pellet associated mRNAs and out of 645 enriched among the top 3000 most amino-terminally disordered human complex subunits. Many of the identical GOs are related to stress response and to DNA repair (Supplemental Table S7, “EDTA-pellet common w. predicted”).
The distribution in the number of disordered amino acids among the first 50 was markedly different between the top 3000 and bottom 3000 hits of the ranking according to ±EDTA ratio of reads in pellets showing that EDTA-pellet mRNAs are encoding more disordered proteins as compared to translated mRNAs (Supplemental Fig. S4).
These results confirm the in silico prediction that amino-terminal disorder propensity indicates that a gene product is more likely to be present in EDTA-pellets.
1,6-hexanediol dissolves EDTA-resistant ribosome assemblies
Phase transitions of RNCs might have the potential to protect both the mRNA and the nascent chain from degradation as suggested earlier (Panasenko et al. 2019). Disordered protein structures were reported previously to be important for phase transitions (Alberti 2017). Because amino-terminal disordered protein structure appears to be important for the formation of condensates that presumably protects both the protein and the mRNA from degradation, we considered the possibility that some ribosomes might phase separate if a disordered nascent chain is protruding and exposed from them.
To follow up on this interesting hypothesis we decided to test if 1,6-hexanediol (HEX) dissolves condensates containing ACT1, SGS1, RAD10, and RAD14 mRNAs in yeast. HEX was reported to efficiently inhibit the phase transition of proteins, because it interferes with weak hydrophobic protein–protein or protein–RNA interactions that are required for dynamic, liquid-like assemblies to form (Kroschwald et al. 2017). The ultracentrifugation experiments were thus executed next in the presence of HEX to determine whether the mRNAs in EDTA-pellets are in granules of phase-separated origin. Upon HEX treatment, levels of both SGS1, RAD10, and RAD14 mRNAs in the EDTA-pellets were highly reduced in UV-irradiated yeast cells suggesting that phase separation is indeed involved in creating the granules (Supplemental Fig. S5). HEX-sensitive sedimentation of the control ACT1 mRNA in EDTA-pellets was much less obvious giving credit to the approach (Supplemental Fig. S5).
Phase-separated granules are present in the cytoplasm of A549 human alveolar adenocarcinoma cells
Our findings with yeast cells may represent a general phenomenon, and the formation of NCA-like phase-separated granules could be important in the context of radiation resistance in higher-ordered eukaryotes as suggested by genomic analysis (Table 2). In order to substantiate this idea, we looked for a radiation-resistant cell type. The A549 lung adenocarcinoma line was chosen for investigating tightly packed ribosomal assemblies, similar to those of phase-separated Rpt1- and Sgs1-RNC containing granules of yeast cells. Ribosomes have distinct morphology in electro micrographs (Igaz et al. 2020), so we chose to follow their possible higher-order arrangement with transmission electron microscopy (TEM). On electron micrographs of fixed A549 cells, closely packed ribosomes mostly in ring-like orientation were detected (Fig. 5A). These structures were around 100 nm in diameter in accord with the size of 8–9 ribosomes closely associated that was revealed with STORM by following the Flag-tagged nascent Sgs1-RNC sticking out of the ribosome (Fig. 3E, upper panel). To validate that these ribosomes are constituents of phase-separated granules, we performed the staining with A549 cells that were treated with 2% v/v HEX prior to fixation. In fact, occurrence of ring-oriented ribosomes was markedly reduced in the cytoplasm of HEX-treated A549 cells compared to nontreated ones (Fig. 5B,C).
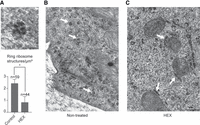
Closely packed ribosomes sensitive to 1,6-hexanediol treatment are detectable in the cytoplasm of A549 cells. (A) Typical example of a ring-like ribosome structure revealed by TEM after fixing with glutaraldehyde (top) and diagram of quantification of combined results of three independent experiments where ring ribosomes were counted on at least 1500 µm3 cell space for each condition analyzing at least three cells/biological replicates (with/without HEX) (below). Statistical significance is determined by two-tailed Student's t-test ([*] P < 0.05). Scale bar, 50 nm. (B) Cells were fixed with glutaraldehyde and examined with TEM. Scale bar, 500 nm. White arrows indicate typical ring-like ribosome structures, the most common appearance of neighboring ribosomes in nontreated cells. (C) Cells were treated with HEX for 1 h and fixed with glutaraldehyde for TEM as in panel B. Same magnification as in panel B. White arrows indicate ribosomes arranged in a linear fashion, the most common appearance of neighboring ribosomes in HEX-treated cells.
1,6-hexanediol sensitizes A549 cells to ionizing radiation
So far, we found that like in yeast cells expressing stalled ribosome-associated nascent chains, human cell lines contain small EDTA-resistant and phase-separated ribosomal assemblies. Next, we looked for the role of these in the DNA damage response. Equal numbers of A549 cells were treated or not with HEX and then irradiated with either a single 1 Gy dosage, or with consecutive treatments of 1 Gy with a 30 min incubation period between irradiations. Then we compared the number of survival colonies (Fig. 6). While a single irradiation clearly decreased cell viability/colony forming capacity, HEX-treated cells were no more sensitive to ionizing radiation. In the case of consecutively irradiated cells without HEX treatment, their viability was comparable to that of cells after single irradiation, despite the double dose of radiation they received. However, the viability of HEX-treated cells after double irradiation was significantly lower compared to either the HEX-treated and only once irradiated cells, and also to the non-HEX treated, consecutively irradiated cells (Fig. 6). These results point toward a mechanism that is required for DNA damage response and is regulated by phase separation in these cells.
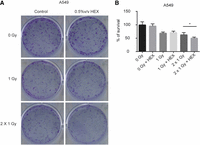
Phase separation is involved in DNA damage response. (A) 700–700 A549 cells were plated and treated or not with HEX in 0.5% v/v and exposed or not to 1 Gray (1 Gy) or two times 1 Gy irradiation dose with a 30 min incubation time between the two treatments (2 Gy). (B) Chart showing the combined results of the experiment described in A with three independent biological replicates. Survivals are expressed in % of survivals of nonirradiated plates. Statistical significance in the case of comparing 2 Gy to 2 Gy + HEX is determined by a two-tailed Student's t-test ([*] P < 0.05).
DISCUSSION
Assemblysomes are distinct phase-separated granules containing ribosomes paused in elongation
In eukaryotes, the two major steps of gene expression are physically separated by compartmentalization. Transcription of chromosomal genes is a nuclear process, whereas the translation of the resulting mRNA is solely cytoplasmic. There are numerous steps between these two major processes providing the possibility for quality control mechanisms and tuning gene expression post-transcriptionally dependent upon demand by challenges of exogenous origin, such as stress. Compartmentalization by phase separation has recently emerged as a mechanism to regulate gene expression post-transcriptionally, and translation in particular, and the formation of phase-separated granules such as P-bodies and SGs are important for mRNA quality control and stress responses, respectively (Decker and Parker 2012; Luo et al. 2018; Guzikowski et al. 2019).
NCAs have been described recently as another type of granules that have a role in the cotranslational assembly of certain multiprotein complexes, such as the proteasome (Panasenko et al. 2019). According to our earlier and current results, the following attributes are important for assemblysome formation: ribosome pausing during translation, an amino-terminally disordered structure and nascent chain ubiquitylation. Based on these attributes, we used a bioinformatics approach to predict the set of proteins in S. cerevisiae that might be the subject of assemblysome formation. We showed for one such candidate protein, Sgs1, that artificially stalled nascent Sgs1 is indeed present in phase-separated granules and importantly, the endogenous mRNA is detectable in HEX-sensitive phase-separated granules. This study validated that simply ranking proteins according to their amino-terminal disorder propensity reveals subunits of complexes reported to assemble cotranslationally, as they were enriched in the top 350 hits and represent ∼15% of the list of 2325 protein complex subunits studied in yeast (Supplemental Table S2). For example, Rpt1, Rpt2, Spt20, Taf10, and Set1 are all at the top of the list of our predicted hits and were all reported previously as subunits that are cotranslationally assembling with their partner (Halbach et al. 2009; Kassem et al. 2017; Kamenova et al. 2019; Panasenko et al. 2019).
We conclude that assemblysomes indeed represent a type of phase-separated granules distinct from other known granules based upon the following observations. First, unlike P-bodies and SGs, assemblysomes are relatively light assemblies of nascent chain-ribosome complexes and cannot be pelleted by moderate, but only high-speed centrifugation (Jain et al. 2016; Hubstenberger et al. 2017; Panasenko et al. 2019). Consistently, Sgs1-containing granules observed by STORM microscopy are smaller than either P-bodies or SGs (Nissan and Parker 2008; Van Treeck and Parker 2019). Second, assemblysomes, but not P-bodies and SGs, are CHX- and EDTA-resistant assemblies, and P-bodies, but not assemblysomes, are RNase-sensitive (Teixeira et al. 2005; Khong et al. 2017; Panasenko et al. 2019). Third, assemblysomes contain large ribosomal subunits absent from SGs (Jain et al. 2016). Finally, assemblysomes are not formed, but actually dismantled upon UV treatment (Table 1; Fig. 2). This observation is particularly exciting as will be discussed further below. We cannot rule out that there are transitions from assemblysomes to, for example, SGs, similar to the communication of P-bodies and SGs (Buchan et al. 2008). Altogether, assemblysomes are clearly a distinct type of granule with distinct behavior upon stress, and therefore having a distinct function.
Assemblysomes contain mRNAs of proteins involved in DNA repair
Our bioinformatic prediction of proteins likely to be translated in assemblysomes identified components of the DNA damage response in yeast. The fact that phase separation is critical for the DNA damage response may provide the clue in understanding the possible role of assemblysomes. Not only Sgs1, but other proteins that are involved in environmental stress responses, including players in genotoxic stress response, were significantly enriched among the top 15% of amino-terminally disordered proteins according to bioinformatical prediction using yeast and human databases. Moreover, it was confirmed by sequencing that mRNAs involved in stress response, and DNA repair in particular, was enriched among the bottom 3000 mRNAs of a ±EDTA read ratio in pellets ranking (ratio below 0.5). This ranking appears to be biologically relevant even when looking at the other extremity. The top 3000 mRNAs of the ranking (ratio above 0.94) for instance are enriched in mitochondrial and ribosomal protein (RP) mRNAs that were previously shown to be engaged with Not1 in yeast (Gupta et al. 2016). This suggests a Not1 function that regulates the movement of mRNAs between EDTA-sensitive and -resistant ribosomes. In this latter study, it was revealed that Not1 engagement with RP mRNAs correlates with their enhanced translation (Gupta et al. 2016). This is in accord with the fact that mRNAs with a high ±EDTA read ratio in pellets are most likely engaged with EDTA-sensitive translating ribosomes rather than in EDTA-pellets, which are presumably in condensates. Interestingly, stress-responsive mRNAs are underrepresented in the soluble ribosome-bound pool in the case of MCF7 cells, but on the contrary they are markedly enriched in EDTA-pellets according to GO analysis (Table 2; Supplemental Table S7). This result supports our qPCR approach performed under UV stress in yeast, and the idea that assemblysomes store mRNAs for a quick gene expression response in eukaryotes (Fig. 2). This is important for competitive reasons, given that in prokaryotes transcription and translation processes are going on in parallel, and therefore, the rate-limiting step of a gene expression response is determined only by the speed of translation. It is rather unlikely that evolution permitted for eukaryotic cells to perform the two major gene expression processes to respond to stress solely in a consecutive manner.
Stress responses require the production and/or activation of effectors, and this demand for new proteins may not be squared due to the transcriptional arrest and defect in translational initiation caused by the stress itself (Holcik and Sonenberg 2005; Lavigne et al. 2017). However, by storing premade stress-responsive mRNAs in the form of RNCs in assemblysomes, cells can efficiently cope with the situation, as such RNCs can continue their translation to produce proteins without the need of de novo transcription and/or translational initiation. As combined transcription and translation are relatively time-consuming processes, regulating protein synthesis at a late stage of their production has the potential of a fast and timely response to correct, for example, DNA damage even if genes required for the process are themselves affected by the damage, temporarily.
It is important to note that the stress response cannot be managed by the ubiquitous and continuous expression of certain effectors. For example, concerning the context of the DNA damage response, overexpression of Sgs1, a DNA helicase involved in double-strand DNA break repair, is toxic in yeast (Gangloff et al. 1994; Sinclair et al. 1997), or overexpression of the Rad1–Rad10 subunits of Nef1 leads to gross chromosome rearrangements (Hwang et al. 2005). The clear advantage of their delayed completed translation could be that gene products required for DNA repair, otherwise detrimental concerning unperturbed conditions, are expressed in an active form only when the DNA damage appears, avoiding the detrimental effects of unnecessary or excess expression.
Assemblysomes are membraneless compartments in eukaryotes with roles in DNA damage response
Finally, we present evidence that assemblysomes characterized in yeast are not only present in human A549 cells as well, as previously shown (Panasenko et al. 2019), but may also be relevant for the DNA damage response in human cells, thus have a conserved functional role. Indeed, we found that the otherwise radioresistant A549 cells become sensitive to DNA damage upon HEX treatment to interfere with phase separation. We used HEX in a concentration (0.5 v/v%) which has no effect on kinases and phosphatases (Düster et al. 2021). We noticed no adverse effects of HEX on growth of the A549 cell line, and even after a 1 Gy radiation dose we found no change in the number of survivals between HEX-treated or control A549 cell cultures. However, the change is significant after a second 1 Gy treatment that followed the first after a 30 min incubation period when the cells had time to form assemblysomes. Moreover, upon sequencing MCF7 ribosome pellets and EDTA-pellets, we identified genes belonging to DNA repair GO terms among the bottom 3000 of a ±EDTA read count ratio in pellets ranking of ribosome-associated mRNAs (Table 2). These discoveries suggest that EDTA-resistant condensates are responsible for a quick gene expression response to stress—and DNA damage response in particular—in eukaryotes. The process is competitive to gene expression response to stress in prokaryotes where transcription is happening parallel with the rate-limiting translation process. In theory, destroying condensates including assemblysomes responsible for translational repression of stress-responsive gene products might serve as a solution for sensitizing chemo- or radioresistant tumors in the future.
MATERIALS AND METHODS
In silico identification and analysis of yeast protein complexes
The list of protein complexes was downloaded from the UniProt database along with the protein and nucleotide sequences. Protein disorder data were retrieved from the MobiDB (Piovesan and Tosatto 2018) database and three different predictors were used: PONDR vsl2b (Peng et al. 2006), iUpred (Dosztányi et al. 2005a) long disorder, and Espritz-Xray (Walsh et al. 2012), from which the disorder content up to the first 50 amino acids was retrieved. Amino-terminal lysine content and codon pairs that significantly slow down or stall translation (Ghoneim et al. 2019) were identified from the protein and the nucleotide sequences, respectively, using an in-house Perl script. We used three different assays in the in silico prediction of disordered amino-terminal domains to generate the final ranking, but we highlight the results of altogether six different prediction methods (Supplemental Table S2; Linding et al. 2003a,b; Dosztányi et al. 2005a,b; Peng et al. 2006).
Yeast strains and culture conditions
All yeast strains used for this work are listed in Supplemental Table S1. Wild-type yeast cells were grown at 30°C in yeast extract peptone dextrose (YPD), while yeast cells transformed with plasmids containing genes under the regulation of copper-inducible CUP1 promoter were grown in synthetic dropout (SD) medium to exponential phase and induced for 10 min with 0.1 mM CuSO4 before harvesting.
Cell culture
MCF7 breast and A549 lung adenocarcinoma as well as DU-145 prostate cancer cell lines were purchased from ATCC and maintained in Roswell Park Memorial Institute 1640 (RPMI) medium (Biosera) complemented with 10% fetal bovine serum (FBS) (EuroClone), 2 mM glutamine (Sigma-Aldrich), 0.01% streptomycin, and 0.006% penicillin (Biowest). Cells were cultured under standard conditions in a 37°C incubator at 5% CO2 and 95% humidity. When indicated, cells were treated with 1,6-hexanediol at a final concentration of 2% v/v for 30 min.
Cloning
All plasmid and primers used for this work are listed in Supplemental Table 1. NEBuilder HiFi DNA Assembly (New England Biolabs) was used for clonings. Plasmid for stalled nascent Sgs1 (Sgs1-RNC) was from pOP164, described previously (Panasenko et al. 2019) and obtained by PCR with oligonucleotides specific for different ORFs and cloning by NEB HiFi. The first six lysines of the Rpt1 ORF in pOP164 (pMAC1152) were generated by PCR using an oligonucleotide bearing the mutations as a forward primer. All clones were verified by sequencing, and plasmids were transformed into the BY4741 yeast strain for subsequent work.
Polysome fractionation
Ribosomes were fractionated on a 12 mL 7%–47% sucrose gradient as in Panasenko and Collart (2012). Briefly, 100 mL of yeast in the exponential growth phase were treated or not with 100 µg mL−1 of CHX, harvested, washed with cold water, and resuspended in buffer A (20 mM HEPES, pH 8.0, 50 mM KCl, 10 mM MgCl2, 1% Triton X-100, 1 mM DTT, 1 mM PMSF, and protease inhibitor cocktail) with or without 100 µg mL−1 of CHX. Cells were broken with 0.5 mL of glass beads in 0.5 mL of buffer A for 15 min at 4°C. The lysates were clarified by centrifugation at 14,000g for 10 min. An amount of 0.2 mL of lysates containing 2–3 mg of total protein was applied on a 12 mL 7%–47% sucrose gradient in 20 mM HEPES, 50 mM KCl, 10 mM MgCl2 with or without 100 µg mL−1 of CHX and centrifuged for 150 min at 220,000g at 4°C. Fractions were collected using a UA/6 detector (ISCO), precipitated with TCA and separated by SDS-PAGE. When indicated, polysomes were dissociated by treatment with 25 mM EDTA added instead of CHX, in the lysis buffer and in the gradient.
In vivo ubiquitination assay
The assay was performed as previously described (Panasenko et al. 2006). Briefly, cells expressing His6-ubiquitin under the control of a copper-dependent promoter were grown in medium containing 0.1 mM CuSO4, and 50 A600 units were collected at the exponential growth phase. Cell pellets were resuspended in G-buffer (100 mM sodium Pi, pH 8.0, 10 mM Tris-HCl, 6 M guanidium chloride, 5 mM imidazole, 0.1% Triton X-100) to 50 mg mL−1. An amount of 1 mL of cell suspension was disrupted with 0.6 mL of glass beads during 6 min at 4°C and spun for 20 min at 13,000g. To remove guanidium chloride, 20 µL of the supernatants were diluted in 1.2 mL of water and concentrated with StrataClean resin (Stratagene) and eluted with 50 µL of Laemmli SB. A total of 3–5 µL of TE was analyzed by western blot with the relevant antibodies. The rest of the supernatant was incubated with 30 µL of nickel-nitrilotriacetic acid-agarose (Qiagen) for 2 h at room temperature with mild rotation. The agarose beads were washed three times with 0.5 mL of U-buffer (100 mM sodium Pi, pH 6.8, 10 mM Tris-HCl, 8 M urea, 0.1% Triton X-100). His6-ubiquitinated proteins were eluted with 50 µL of 2× Laemmli SB, and 12–15 µL of samples were analyzed by western blot with the relevant antibodies.
Flag immunoprecipitation
Fifty OD600 units of logarithmically growing yeast cells transformed with amino-terminally Flag-tagged Sgs1-RNC expressing plasmid were harvested and resuspended in lysis buffer (20 mM HEPES [pH 7.5], 20 mM KCl, 10 mM MgCl2, 1% Triton X-100, 1 mM DTT, protease inhibitor cocktail, 25 mM EDTA). Cell lysis was performed as described above. Whole protein lysate was used for immunoprecipitation with anti-Flag M2 antibody (F1804 Sigma-Aldrich) (1:500). Dynabeads M-280 Sheep anti-Rabbit IgG (11204D, Invitrogen) beads were used according to the manufacturer's instructions with some modifications. First, the beads were incubated for 1 h with 0.1% BSA in PBS with gentle tilting. Before use, beads were washed three times in PBS. A modified washing buffer was used (20 mM HEPES [pH 7.5], 20 mM KCl, 10 mM MgCl2, 2% Triton X-100, 1 mM DTT, protease inhibitor cocktail, 1 mM EDTA). Beads were resolubilized in 2× SDS loading buffer and eluted by boiling (10 min, 95°C).
Investigation of repeated UV treatment of yeast cells in the presence or absence of 1,6-hexanediol
Exponentially growing liquid yeast cell cultures (0.4–1 OD600) were UV-treated in Petri dishes. Each UV irradiation lasted for 2 min (18 mJ cm−2 dose). 1,6-hexanediol (SIGMA) at a final concentration of 5% v/v in 140 µL ethanol or ethanol vehicle alone was added after the first UV treatment. An incubation time (10 min) between two UV treatments was allowed to facilitate the formation of granules. Working in the dark during sample collection for UV-treated and for their control samples is a necessity to avoid the effect of the efficient visible light-inducible DNA repair system available in single-cell organisms (Kelner 1949).
Lysate preparation and ribosome fractionation for mRNA, rRNA, tRNA, and for protein examination
Twenty OD600 units of logarithmically growing yeast cells transformed or not with plasmids containing genes under the regulation of copper-inducible CUP1 promoter were harvested and broken with 0.5 mL of glass beads in 250 µL lysis buffer. Lysis buffer was used for cell lysis. To pellet ribosomes and granules we used lysis buffer (20 mM HEPES [pH 7.5], 20 mM KCl, 10 mM MgCl2, 1% Triton X-100, 1 mM DTT, protease inhibitor cocktail); to pellet EDTA-resistant granules we supplemented lysis buffer with EDTA to reach a 25 mM final concentration. RNase inhibitor was added to the buffers in cases when mRNA was to be followed after cell lysis. Each sample was vortexed with glass beads for 15 min at 4°C. Samples were centrifuged first with a short spin to pull the lysate off the beads followed by 10 min at 8000g at 4°C to get rid of cell debris, nuclei, aggregated proteins, and SGs. Supernatants of 100 µL were either treated or not with 25 mM EDTA layered on top of 500 µL 60% sucrose cushion (20 mM HEPES [pH 7.5], 20 mM KCl, 10 mM MgCl2, and 60% sucrose; either with 25 mM EDTA or without). Samples were ultracentrifuged for 4 h at 50,000 rpm at 4°C in a Sorvall MX 120/150 Plus Micro-Ultracentrifuge (Thermo Fisher Scientific) in S55A2 rotor. Pellets from ultracentrifugation were resolubilized in lysis buffer for RNA examination.
The mRNAs extracted from different fractions were analyzed by quantitative real-time PCR. For RNA extraction to examine the different rRNA and tRNA species of total extract, SG enriched, flow-through and pellet samples on nondenaturing 1% agarose gel, RNA was extracted with TRI reagent (ZYMO Research) according to the manufacturer's instructions.
In the case of human cell lines, 60% confluent cultures were harvested. Cell lysis was performed by 15 min incubation on ice in lysis buffer (100 mM KCl, 50 mM Tris-HCl [pH 7.4], 1.5 mM MgCl2 1 mM DTT, 1.5% NP-40, protease inhibitor cocktail, with or without 25 mM EDTA as indicated). Ultracentrifugation and downstream analysis were similar as above using the following buffer for the sucrose cushion: 100 mM KCl, 50 mM Tris-HCl (pH 7.4), 1.5 mM MgCl2, and 60% sucrose either with or without 25 mM EDTA. For RNA extraction, TRI reagent (ZYMO Research) was used according to the manufacturer's instructions. RNA from the entire pellets were extracted and solubilized.
RNA extraction and quantitative real-time PCR
After the ultracentrifugation step of yeast total protein extracts on 60% sucrose cushion with or without EDTA, total RNA was isolated from extracts obtained from different fractions using the NucleoSpin TriPrep Kit (Macherey-Nagel) following the requirements of the manufacturer. RNA concentration, was measured by NanoDrop spectrophotometer. Equal volumes from each fraction containing 0.1 ng–5 µg of total RNA were reverse transcribed with oligo(dT) primers using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Diluted cDNAs were mixed with GoTaq qPCR Master Mix (Promega) and analyzed with Piko-Real 96 Real-Time PCR System (Thermo Fisher Scientific). Gene-specific primers were used to detect SGS1, RAD10, RAD14, and ACT1.
Western blot
Total protein samples or immunoprecipitated samples were separated with 10% Tris-Tricine-SDS-PAGE (Schägger 2006) and then transferred to nitrocellulose membrane. After washing, the nitrocellulose membranes were blocked for 30 min with 5% milk in TBS-T buffer (0.05% Tween 20 [pH 7.5], 150 mM NaCl, and 20 mM Tris-HCl) or for 1 h with 5% BSA (Sigma) in TBS-T. The membranes were incubated with primary antibodies overnight, at 4°C. Primary antibodies were diluted (1:1000) in milk-TBS-T or in BSA-TBS-T. After several washing steps, the membranes were incubated with secondary antibodies for 2 h at room temperature. Eventually, membranes were mixed with ECL reagent (Millipore) and signals were detected by a LI-COR C-DiGit blot scanner. The following primary antibodies were used for western blot experiments: K48-linkage specific polyubiquitin antibody (4289 S, Cell Signaling Technologies); anti-Egd2 antibodies described previously (Panasenko et al. 2006); anti-Flag M2 antibody (F1804 Sigma-Aldrich); anti-RPS6 antibody (ab40820); anti-RPS6 (2217 Cell Signaling Technologies); anti-RPL11 (PA5-27468) (Thermo Fisher Scientific); anti-Pol II (CTD4H8) (Santa Cruz Biotechnology). Goat anti-rabbit and rabbit anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies (Dako) were used in our experiments.
dSTORM measurements
Super-resolution dSTORM measurements were performed on a custom-made inverted microscope based on a Nikon Eclipse Ti-E frame. EPI-fluorescence illumination was applied at an excitation wavelength of 647 nm (2RU-VFL-P-300-647-B1, Pmax = 300 mW, MPB Communications Ltd). The laser intensity was set to 2–4 kW cm−2 on the sample plane and controlled via an acousto-optic tunable filter. An additional laser (405 nm, Pmax = 60 mW; Nichia) was used for reactivation. A filter set from Semrock (Di03-R405/488/561/635-t1-25 × 36 BrightLine quad-edge super-resolution/TIRF dichroic beamsplitter, and FF01-446/523/600/677-25 BrightLine quad-band bandpass filter, and an additional AHF 690/70 H emission filter) was inserted into the microscope to spectrally separate the excitation and emission lights. The images of individual fluorescent dye molecules were captured by an Andor iXon3 897 BV EMCCD camera (512 × 512 pixels with 16 µm pixel size) with the following acquisition parameters: exposure time = 25 msec; EM gain = 100; temperature = −75°C. Typically, 20,000–30,000 frames were captured from a single ROI. During the measurement, the Nikon Perfect Focus System kept the sample in focus. High-resolution images were reconstructed with rainstorm localization software (Rees et al. 2013). The astigmatic 3D method was applied to determine the axial position of the fluorescent dye molecules. In this arrangement, a cylindrical lens (f = 4000 mm) placed into the detector path introduces astigmatism, and the ellipticity value of the distorted PSFs provides information for the generation of 3D dSTORM images (Huang et al. 2008). Mechanical drift introduced by either the mechanical movement or thermal effects was analyzed and reduced by means of an autocorrelation-based blind drift correction algorithm. dSTORM experiments were conducted in a GLOX switching buffer (van de Linde et al. 2011) and the sample was mounted onto a microscope slide. The imaging buffer is an aqueous solution diluted in PBS containing an enzymatic oxygen scavenging system GluOx (2000 U mL−1 glucose-oxidase [Sigma-Aldrich], 40,000 U mL−1 catalase [Sigma-Aldrich], 25 mM potassium chloride [Sigma-Aldrich], 22 mM tris[hydroxymethyl]aminomethane [Sigma-Aldrich], 4 mM tris [2-carboxyethyl] phosphine [TCEP] [Sigma-Aldrich]) with 4% (w/v) glucose (Sigma-Aldrich) and 100 mM β-mercaptoethylamine (MEA) (Sigma-Aldrich). The final pH was set to 7.4.
Cluster analysis of super-resolved images
A density-based spatial cluster analysis (DBSCAN) was used for cluster recognition. This algorithm requires two input parameters: a minimum number of points that form a cluster (Ncore) and the maximum distance between two adjacent points (ε) (Ester et al. 1996). Ncore and ε were set to 8 and 24 nm during the calculations, which was optimal for the separation of adjacent RNCs. Quantitative characteristics of RNC clusters, such as their area or the distance of the closest neighboring clusters (NND) were evaluated by means of in-built Matlab functions (convhull and pdist2). The epitope number was also estimated by a DBSCAN-based method described in a previous publication (Varga et al. 2019).
Transmission electron microscopy (TEM)
For TEM imaging, 105 A549 cells were seeded onto 0.4 µm pore polyester membrane inserts (Corning) placed in a six-well plate. Cells were left to grow until the following day when they were treated with 1,6-hexanediol in 0.5% v/v final concentration and carefully washed and fixed in 4% glutaraldehyde in PBS for 2 h, and subsequently embedded in gelatin (2% gelatin in PBS). The obtained specimen was sliced to 1–2 mm cubes, which were further embedded in epoxy (Epon 812, EMS, PA 19440) by a routine TEM sample preparation protocol. Semithin sections of 1 µm were prepared to identify the cell monolayer. Blocks were trimmed, and thin sections of 70 nm were obtained and stained with uranyl and lead solutions. In three independent measurements, ribosomes in ring orientation were counted on at least 1500 µm3 cell space for each condition. At least three cells/biological replicates were tested. Images were captured by a Jeol 1400Plus Electron Microscope.
Colony forming assay
A total of 6 × 105 cells/flask were seeded into T25 cell culture flasks (Biologix) and left to grow for 24 h. After 24 h of growth, the cells were either left completely untreated, or were treated or not with 1,6-hexanediol in 0.5% v/v final concentration, and then samples were exposed to 1 Gy irradiation delivered with a Primus Linear Accelerator (Siemens Healthcare GmbH). After 30 min of incubation, another 1 Gy irradiation was applied on a subset of samples. On the next day, cells were trypsinized, suspended in medium, and counted. From each sample, 700 cells/well were seeded into six-well plates in three replicates and left to grow for 1 wk. Then colonies were fixed in 70% methanol and 30% acetone solution and stained with 0.5% crystal violet dissolved in 25% methanol.
NGS sequencing
RNA integrity and quantity extracted from MCF7 ribosome pellets were determined by capillary gel electrophoresis using an Agilent RNA 6000 Nano Kit in an Agilent 2100 Bioanalyzer instrument. Poly(A) selected, indexed RNA sequencing libraries (two biological replicates per treatment condition) were generated using the TruSeq RNA Sample Prep Kit v2 (Illumina), following the protocol provided by the manufacturer. Purified sequencing libraries were validated and quantitated using the Agilent DNA 1000 Kit in an Agilent 2100 Bioanalyzer instrument. Sequencing libraries were pooled, denatured, and sequenced in technical triplicates in an Illumina MiSeq instrument using the MiSeq Reagent Nano Kit v2-500 and MiSeq Reagent Kit v3-150 (Illumina), generating 2 × 75 bp paired-end sequences.
FASTQ sequence files were generated by GenerateFASTQ 1.1.0.64 application on Illumina BaseSpace. Adapters and low-quality sequences were trimmed with TrimGalore, then reads were aligned to the Homo sapiens reference genome (GrCh38) using HISAT2. Gene-specific read counts were determined with the summarizeOverlaps function of the Bioconductor R package using the GrCh38.104 transcriptome annotation.
Analysis of sequenced data
All protein sequences for human were downloaded from UniProt. Protein disorder data were retrieved from the AlphaFold database (Jumper et al. 2021). Permutation tests on the association between amino-terminal protein disorder and enrichment in the EDTA-pellet were calculated in R (R Core Team 2020). We have recovered more than nine million reads from pellet and EDTA-pellet libraries and identified more than 21,000 transcripts in pellet libraries and more than 150,000 transcripts in the EDTA-pellet libraries on average. In cases where there were no reads in EDTA-pellets from a transcript, we gave an artificial read score of 0.001 to be able to calculate the ±EDTA read ratios in pellets genome-wide for all the identified ribosome-associated mRNAs.
DATA DEPOSITION
Sequencing data are available at NCBI SRA (Sequence Read Archive) under the following accession number: PRJNA877600. Other data sets analyzed in the current study are available from the corresponding author upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to Dr. Balázs Vedelek and Dr. Zsuzsa Sarkadi for valuable discussions. We thank Jawad Iqbal, Elvira Czvik, Zita Kóra, and Edina Pataki for technical assistance. We are grateful to Blanka Léhy for the graphical abstract. This work was supported by grants GINOP-2.3.2-15-2016-00020 and GINOP-2.3.2-15-2016-00038, as well as by NKFI-K 142961 (Z.V.), ÚNKP-21-5-595-SZTE (Z.V.), and ÚNKP-20-5-SZTE-655 (M.K.) from the Hungarian National Research, Development and Innovation Office. Further support was provided by the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/902/19 for Z.V. and BO/00878/19/8 for M.K.). Superresolution dSTORM experiments and their evaluation were funded by the Hungarian National Research, Development and Innovation Office (TKP2021-NVA-19), Hungarian Brain Research Program (2017-1.2.1-NKP-2017-00002) awarded to M.E., and NTP-NFTÖ-20-B-0354 awarded to D.V., as well as grant 31003A_172999 from the Swiss National Science Foundation awarded to M.A.C.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079755.123.
- Received June 21, 2023.
- Accepted June 26, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Orsolya Németh-Szatmári is the first author of this paper, “Phase-separated ribosome-nascent chain complexes in genotoxic stress response.” She is a PhD student in the Translational Control Research Group at the Department of Biochemical and Molecular Biology at the University of Szeged, led by assistant professor, Dr. Zoltán Villányi. The focus of their research is a newly discovered cytoplasmic membraneless organelle, the assemblysomes. Assemblysomes contain mRNAs and ribosomes with nascent proteins sticking out of them paused in translation.
What are the major results described in your paper and how do they impact this branch of the field?
We conclude that assemblysomes represent a type of phase-separated granule distinct from other known granules. Assemblysomes contain ribosomes paused in elongation on mRNAs encoding cotranslationally assembling protein complex subunits. Our bioinformatic prediction of proteins likely to be translated in assemblysomes identified components of the DNA damage response in yeast. We present evidence that the NCAs characterized in yeast may also be relevant for the DNA damage response in human cells, and thus have a conserved functional role. In theory, destroying condensates including NCAs responsible for translational repression of stress-responsive gene products might serve as a solution for sensitizing chemo- or radioresistant tumors in the future.
What led you to study RNA or this aspect of RNA science?
RNA is one of life's most important molecules. I started working in this field of science by chance, but I feel lucky, because it is an extremely rapidly developing area that carries a lot of excitement and great importance.
If you were able to give one piece of advice to your younger self, what would that be?
Maybe to be more patient and believe in myself.
What are your subsequent near- or long-term career plans?
Our long-term goals include mapping the relationships between the structures we examine and the resistances that appear in tumors.








