
Processing and decay of 6S-1 and 6S-2 RNAs in Bacillus subtilis
- Jana Christin Wiegard1,4,
- Katrin Damm1,4,
- Marcus Lechner2,
- Clemens Thölken2,
- Saravuth Ngo3,
- Harald Putzer3 and
- Roland K. Hartmann1
- 1Philipps-Universität Marburg, Institut für Pharmazeutische Chemie, D-35037 Marburg, Germany
- 2Philipps-Universität Marburg, Center for Synthetic Microbiology (SYNMIKRO), Bioinformatics Core Facility, D-35032 Marburg, Germany
- 3Expression Génétique Microbienne, CNRS, Université Paris Cité, Institut de Biologie Physico-Chimique, 75005 Paris, France
- Corresponding author: roland.hartmann{at}staff.uni-marburg.de
Abstract
Noncoding 6S RNAs regulate transcription by binding to the active site of bacterial RNA polymerase holoenzymes. Processing and decay of 6S-1 and 6S-2 RNA were investigated in Bacillus subtilis by northern blot and RNA-seq analyses using different RNase knockout strains, as well as by in vitro processing assays. For both 6S RNA paralogs, we identified a key—but mechanistically different—role of RNase J1. RNase J1 catalyzes 5′-end maturation of 6S-1 RNA, yet relatively inefficient and possibly via the enzyme's “sliding endonuclease” activity. 5′-end maturation has no detectable effect on 6S-1 RNA function, but rather regulates its decay: The generated 5′-monophosphate on matured 6S-1 RNA propels endonucleolytic cleavage in its apical loop region. The major 6S-2 RNA degradation pathway is initiated by endonucleolytic cleavage in the 5′-central bubble to trigger 5′-to-3′-exoribonucleolytic degradation of the downstream fragment by RNase J1. The four 3′-exonucleases of B. subtilis—RNase R, PNPase, YhaM, and particularly RNase PH—are involved in 3′-end trimming of both 6S RNAs, degradation of 6S-1 RNA fragments, and decay of abortive transcripts (so-called product RNAs, ∼14 nt in length) synthesized on 6S-1 RNA during outgrowth from stationary phase. In the case of the growth-retarded RNase Y deletion strain, we were unable to infer a specific role of RNase Y in 6S RNA decay. Yet, a participation of RNase Y in 6S RNA decay still remains possible, as evidence for such a function may have been obscured by overlapping substrate specificities of RNase Y, RNase J1, and RNase J2.
Keywords
- 6S RNA
- noncoding RNA
- pRNA
- RNA degradation
- endoribonuclease
- RNase Y
- RNase J1
- RNase PH
- exoribonucleases
- Bacillus subtilis
INTRODUCTION
Bacterial 6S RNAs are noncoding RNAs (ncRNAs) with a size of ∼200 nt that interact with the active site of housekeeping RNA polymerase (RNAP) holoenzymes to globally regulate transcription (Steuten et al. 2014; Wassarman 2018). 6S RNAs adopt a rod-shaped structure composed of a largely single-stranded central region, termed central bubble (CB), that is flanked by two irregular helical stems. This architecture, recognized by bacterial housekeeping RNAP holoenzymes, mimics an open B-form DNA promoter (Wassarman and Storz 2000; Barrick et al. 2005; Trotochaud and Wassarman 2005; Chen et al. 2017).
In most of the studied bacteria, 6S RNA levels were reported to increase toward the stationary growth phase, but different expression profiles were reported as well (for review, see Elkina et al. 2017). So far, relatively little is known regarding the adaptation of 6S RNA levels in response to environmental cues such as stresses. One example was reported for the phototrophic cyanobacterium Prochlorococcus MED4, where induction of cell cycle synchronization by light–dark cycles resulted in decreasing 6S RNA levels during night periods (Axmann et al. 2007). Another example is the α-proteobacterium Bradyrhizobium japonicum that is capable of nitrogen fixation in symbiosis with plants. Here, 6S RNA levels increased when the bacterium formed bacteroids in root nodules relative to free-living cells (Madhugiri et al. 2012).
Interestingly, and unlike most other bacteria, the genomes of Bacillus subtilis and closely related Firmicutes encode two paralogous 6S RNAs, termed 6S-1 (gene bsrA) and 6S-2 (gene bsrB) RNA (Ando et al. 2002; Suzuma et al. 2002; Trotochaud and Wassarman 2005; Beckmann et al. 2011). 6S-1 RNA, considered to be the canonical 6S RNA, reaches the highest cellular levels from late exponential to stationary phase, where its levels exceed those of 6S-2 RNA (Hoch et al. 2015). The levels of B. subtilis 6S-2 RNA were reported to peak in exponential phase (Ando et al. 2002; Barrick et al. 2005; Beckmann et al. 2011; Thüring et al. 2021) or to remain quite constant throughout growth (Trotochaud and Wassarman 2005; Cavanagh et al. 2012). These divergent observations may suggest that 6S-2 RNA expression is sensitive to strain background or culture conditions, but so far, potential adaptations of 6S-1 and 6S-2 RNA levels in response to changes in environmental conditions have not been scrutinized.
The function of canonical 6S RNAs, such as Escherichia coli 6S RNA or B. subtilis 6S-1 RNA, is thought to reprogram transcription toward stationary phase to cope with nutrient deprivation (Trotochaud and Wassarman 2004; Steuten et al. 2014) and to keep RNAP in a state where the enzyme can be instantly released from sequestration upon supply of fresh nutrients. Release is then triggered by elevated cellular NTP levels, allowing RNAP to transcribe so-called “product RNAs” (pRNAs) of extended length (≥13 nt in the case of E. coli 6S RNA and B. subtilis 6S-1 RNA) on 6S RNA as template. Such extended pRNAs form a stable hybrid helix with the template 6S RNA and persistently rearrange its structure to induce dissociation from RNAP. As a result, RNAP becomes available again for transcription initiation at chromosomal DNA promoters and the released 6S RNA undergoes accelerated degradation (Wassarman and Saecker 2006; Wurm et al. 2010; Beckmann et al. 2011, 2012; Panchapakesan and Unrau 2012; Cabrera-Ostertag et al. 2013). Reported phenotypes of B. subtilis 6S-1 RNA gene knockouts include growth to lower optical density during extended stationary phase (Hoch et al. 2015; Thüring et al. 2021) and delayed outgrowth from stationary phase (Cavanagh et al. 2012).
The block of RNAP by 6S-2 RNA is also reversed by pRNA synthesis, although pRNA synthesis appears less efficient on 6S-2 versus 6S-1 RNA (Cabrera-Ostertag et al. 2013; Hoch et al. 2016). Regarding the function of the second 6S-2 RNA in B. subtilis, 6S RNA gene deletions in the NCIB 3610 wild-type strain were revealing (Thüring et al. 2021). The Δ6S-2 derivative strain showed derepressed biofilm formation, retarded swarming activity and accelerated spore formation. Remarkably, the NCIB 3610 Δ6S-1&2 double deletion strain displayed prolonged lag phases of growth under oxidative, high salt and alkaline stress conditions, in addition to decelerated spore formation (Thüring et al. 2021). The finding that the phenotype of the NCIB 3610 double knockout strain is not the sum of the single 6S RNA knockouts suggests a complex intertwining of transcriptional regulation by 6S-1 and 6S-2 RNAs in B. subtilis.
The primary sequence conservation of 6S RNAs across the bacterial superkingdom is remarkably low, as evident from the current Rfam consensus secondary structure (https://rfam.org/family/RF00013). The secondary structure of B. subtilis 6S-1 RNA can be predicted with good confidence by algorithms such as RNAfold and was confirmed experimentally in solution (Beckmann et al. 2012). For the paralogous 6S-2 RNA, RNAfold predicts alternative near-isoenergetic structures that deviate from the typical 6S RNA secondary structure, and only a sequence/structure alignment of 6S-2 RNA homologs from other Firmicutes allowed the prediction of a consensus secondary structure (Burenina et al. 2014) that conforms to the overall shape of the Rfam model. Nonetheless, the identification of 6S RNA candidates based on the Rfam model often remains tentative and requires experimental verification. A genuine 6S RNA should bind to RNAP holoenzymes in a sigma-dependent manner, should serve as a template for pRNA transcription and should refold and dissociate from RNAP upon the synthesis of longer pRNAs.
In general, the functionality and steady-state level of transcripts depend on RNA synthesis, processing, and decay. Surprisingly, RNA turnover differs in the well-studied model organisms E. coli and B. subtilis due to the involvement of different sets of RNases (Condon 2003; Lehnik-Habrink et al. 2012). One global player in B. subtilis RNA degradation is RNase Y, a membrane-associated endonuclease (Shahbabian et al. 2009; Lehnik-Habrink et al. 2011a,b). Acting as a functional counterpart of RNase E in E. coli, RNase Y was reported to initiate RNA degradation by cleavage of preferably 5′-monophosphorylated substrates preferentially in A/U-rich single-stranded regions (Shahbabian et al. 2009; Lehnik-Habrink et al. 2011b; Durand et al. 2012a; Laalami et al. 2013). Based on bacterial two-hybrid studies and in vivo crosslinking experiments, it has been postulated that RNase Y binds to the exoribonucleases RNase J1 and PNPase as well as to the helicase CshA and to two glycolytic enzymes (enolase and phosphofructokinase) to form an RNA degradosome (Commichau et al. 2009; Lehnik-Habrink et al. 2010, 2011a). The formation of such RNA degradosome complexes in B. subtilis is still a matter of discussion, as they could not be isolated in the absence of crosslinking reagents (Laalami et al. 2014; Redder 2018).
Another key enzyme in B. subtilis is RNase J1 that can form a heterotetramer with its paralog RNase J2, an inefficient 5′-exoribonuclease (Mathy et al. 2010). RNase J1, the first known bacterial 5′-to-3′-exoribonuclease (Mathy et al. 2007), acts as a dual function nuclease with endo- and exonucleolytic activity modes (Even et al. 2005; Li de la Sierra-Gallay et al. 2008; Newman et al. 2011). For acting as a 5′-to-3′-exoribonuclease, the enzyme prefers single-stranded 5′-extremities with monophosphorylated or 5′-OH ends generated by a preceding endonucleolytic cleavage (Deikus et al. 2008; Yao and Bechhofer 2010) or by a pyrophosphohydrolase reaction (Richards et al. 2011). Gene knockouts of RNase J1 and Y lead to retarded growth and altered cell morphologies, make cells hypersensitive to a wide range of antibiotics, and cause major defects in sporulation and competence development programs (Figaro et al. 2013).
In B. subtilis, three major pathways of RNA transcript turnover have been described: (a) the RNase Y-dependent pathway, (b) 5′-directed RNase J1-dependent degradation, and (c) an alternative RNase J1-dependent pathway involving internal cleavage by RNase J1. The major pathway is initiated by endonucleolytic RNase Y cleavage, followed by degradation of the downstream product via the 5′-to-3′-exoribonucleolytic activity of RNase J1 (Yao and Bechhofer 2010; Durand et al. 2012a; Lehnik-Habrink et al. 2012). Upstream fragments are degraded by one or more of the four known 3′-exoribonucleases (PNPase, RNase R, RNase PH, and YhaM) (Craven et al. 1992; Luttinger et al. 1996; Mitra et al. 1996; Oussenko and Bechhofer 2000; Oussenko et al. 2002), of which polynucleotide phosphorylase (PNPase) is considered to be the major 3′-exonuclease (Deutscher and Reuven 1991; Oussenko et al. 2005; Liu et al. 2014). Alternatively (pathway b above), the pyrophosphohydrolase RppH generates a 5′-monophosphorylated RNA substrate that is degraded in the 5′-to-3′-direction by RNase J1 (Richards et al. 2011; Durand et al. 2012a). Only a few targets are known at which J1 initiates degradation by endonucleolytic cleavage (pathway c above) (Even et al. 2005; Deikus and Bechhofer 2009; Laalami et al. 2014). Upstream and downstream cleavage fragments are subsequently degraded in the same manner as described above for the major RNase Y-dependent pathway.
In this work, we investigated the decay of B. subtilis 6S-1/2 RNAs and 6S-1 RNA-encoded pRNAs using a set of different strains with knockouts for RNase Y, RNases J1 and J2, RppH, RNase III, RNase PH, or all four known 3′-exonucleases (ΔRNase R, ΔPNPase, ΔRNase PH, and ΔYhaM). Degradation products were analyzed by northern blot and RNA-seq experiments. The possible roles of RNases J1 and Y in 6S RNA decay were further investigated by in vitro cleavage assays.
RESULTS
Northern blot analysis of 6S-1 RNA processing and decay
We analyzed 6S-1 RNA processing and decay by northern blot experiments using probes against the full-length RNA, its 5′- and 3′-half molecules, to each quarter of the RNA and to the 5′-leader of precursor 6S-1 (pre-6S-1) RNA (for details, see Table 1; Fig. 1).
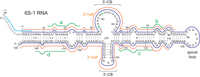
Antisense probes used for the detection of B. subtilis 6S-1 RNA and fragments thereof in northern blot experiments. The probes are illustrated as colored lines along the secondary structure of 6S-1 RNA; sky blue line: LNA/DNA mixmer probe to specifically detect 6S-1 RNA precursor molecules (probe pre-6S-1); dark blue line: antisense 6S-1 RNA (probe 6S-1_mature); orange lines: RNAs antisense to roughly the 5′-half (5′, probe 6S-1_5′-half) or 3′-half (3′, probe 6S-1_3′-half) of 6S-1 RNA; green lines: LNA/DNA mixmer probes (#a–d, probes 6S-1_a–d) for the detection of 6S-1 RNA fragments derived from the first to the fourth quarter of the RNA. 5′-CB, 3′-CB: 5′- and 3′-parts of the CB. For further details, see Materials and Methods.
Northern blot probes specific for 6S-1 RNA, 6S-2 RNA, or 5S rRNA
For the experiment shown in Figure 2A, obtained with probe 6S-1_mature, total RNA was isolated from stationary phase cultures of the parental (W168, termed wt in the following) and derivative strains with RNase gene knockouts. The Δrny strain reproducibly showed an intermediate stagnation phase at ∼0.4–0.8 OD600 (Supplemental Fig. S1), termed stat1 phase in the following, that was initially considered as the stationary phase and used for RNA isolation. The northern blot (Fig. 2A) revealed (i) low signal intensities for RNA samples from Δrny bacteria in stat1 phase (lane 2), (ii) accumulation of pre-6S-1 RNA in the RNase J1 knockout (ΔrnjA) strain (lane 3), and (iii) reduced amounts of 6S-1 RNA-derived fragments (termed “long” and “shorter” fragments) in ΔrnjA bacteria (lane 3) and particularly in Δrny stat1 phase cells (lane 2; see also Supplemental Fig. S2B). Furthermore, the signal patterns for the RNase PH knockout (Δrph) and the quadruple mutant with deletion of all four known B. subtilis 3′-exonucleases, Δ[rph_rnr_yhaM_pnp] (termed strain Δ4exo in the following) suggested the presence of 3′-extended pre-6S-1 RNA molecules (Fig. 2A, lanes 4,5).
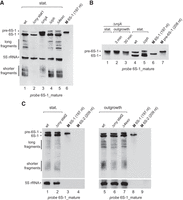
Northern blot analysis of 6S-1 RNA processing and decay in the B. subtilis wt (W168) strain and RNase knockout derivative strains. (A) Total RNAs isolated from stationary (stat.) phase cells of the wt (lane 1), ΔrnjA (ΔRNase J1, lane 3), Δrph (ΔRNase PH, lane 4) strain, and the quadruple mutant Δ4exo (lane 5) with knockouts of rph, rnr (RNase R), yhaM (YhaM), and pnp (polynucleotide phosphorylase, PNPase). In the case of Δrny (ΔRNase Y, lane 2) bacteria, RNA was not isolated at the final stationary phase, but at an intermediate stagnation phase at 0.4–0.8 OD600 (Supplemental Fig. S1), termed stat1 phase. RNAs were separated by 10% denaturing PAGE. Lane 6: a 197 nt T7 transcript of 6S-1 RNA loaded as size marker (M). (B) Northern blot analysis after extended electrophoresis to resolve the region between precursor and mature 6S-1 RNA. Lanes 1–3: total RNA isolated from ΔrnjA bacteria in stationary (stat.) phase (lane 1) or after 3 min (lane 2) and 30 min (lane 3) of outgrowth (1:5 dilution of stationary cells in fresh LB medium); lanes 4,5: total RNA isolated from stationary phase cells of the wt and Δrph strains; lanes 6,7: T7 transcripts of mature (197 nt) and precursor (208 nt) 6S-1 RNA used as size markers (for details, see Supplemental Methods). The shown blots are representative of 10 (panel A) or five (panel B) individual blots using two to four independent RNA preparations. (C) 6S-1 RNA northern blot analysis (12% denaturing PAGE) using total RNA from Δrny bacteria in the final stationary (stat2) phase (lane 2) or 30 min after induction of outgrowth (lane 6). RNA preparations from wt and Δ4exo strains harvested at the corresponding growth stages were loaded for comparison (lanes 1,5,7). The 6S-2 RNA marker transcripts (lanes 4,9) were loaded to control for probe specificity. The shown blots are representative of at least three individual blots performed with independent RNA preparations. For the specificity of probe 6S-1_mature, see Supplemental Figure S2A. 5S rRNA was used as loading control in panels A and C. 6S-1 RNA and 5S rRNA were probed simultaneously (panel A) or successively (panel C) using full-length probes complementary to 6S-1 RNA and 5S rRNA (Table 1; Fig. 1); we confirmed in preceding northern blot experiments that the 5S RNA probe only gives rise to a single 5S rRNA-specific signal (indicated at the left margin) under the applied conditions.
To better resolve the region of precursor and mature 6S-1 RNA, we hybridized a blot after extended gel electrophoresis (Fig. 2B), also using total RNA from stationary cells of the ΔrnjA (lane 1), wt (lane 4), and Δrph strain (lane 5), and additionally RNA from the ΔrnjA strain 3 and 30 min after induction of outgrowth from stationary phase (lanes 2,3). Enhanced resolution of the region comprising pre- and mature 6S-1 RNA verified that 5′-end maturation of 6S-1 RNA is abolished in the ΔrnjA strain independent of growth phase; instead, pre-6S-1 RNA bands with partial shortening of the 11 nt 5′-leader appeared (Fig. 2B, lanes 1–3 vs. lane 4), indicating that another RNase acts on the 5′-leader with low efficiency in the absence of RNase J1. The extended gel run also suggested minor 3′-extensions of pre- and also mature 6S-1 RNA in the Δrph strain (Fig. 2B, compare lanes 4 and 5).
We then noticed during cultivation experiments that the Δrny strain resumes growth after its stat1 stagnation phase and grows to final cell densities comparable to those for the other strains (Supplemental Fig. S1). We thus analyzed the total RNA isolated in this final stationary phase (termed stat2 phase) and compared the northern blot signal pattern with that of the wt strain (Fig. 2C, lanes 1,2). Furthermore, total RNA from stat2 Δrny bacteria isolated 30 min after induction of outgrowth (lane 6) was analyzed as well, using corresponding RNA preparations from the wt and Δ4exo strain as controls (lanes 5,7). This revealed not only very similar 6S-1 RNA signal patterns for wt stationary and Δrny stat2 cells (Fig. 2C, lanes 1,2), but this similarity basically persisted when such cultures were subjected to outgrowth (Fig. 2C, lanes 5,6). The results suggested that RNase Y has either no direct role in 6S-1 RNA decay or its role can be taken on by other RNases.
RNA-seq analysis of total RNA from the wt and RNase knockout strains
We performed two independent RNA-seq experiments for the wt (W168) and RNase knockout strains (stationary phase, see Supplemental Fig. S1), with a time lag between the two and using moderately different protocols for library construction. In the first RNA-seq experiment, the ratios of 6S-1 RNA fragment reads to full-length 6S-1 RNA reads (precursor and mature) were higher than in the second RNA-seq experiment. This is illustrated in Supplemental Figure S3 for 6S-1 RNA reads in the libraries of the wt strain. Conformity between the libraries of the first and second replicates was evident at the (pre-)6S-1 RNA end regions (Supplemental Fig. S3A,B, top panels) and for 3′-ends in the nt 88–95 region (middle panels). Read ends in the latter region, suggesting endonucleolytic cleavage of 6S-1 RNA in its apical loop region, are consistent with the “shorter fragments” observed in northern blots (Fig. 2A). Overall, RNA-seq libraries derived from stationary cells of wt, Δrph and Δ4exo bacteria contained far more reads mapping to the 6S-1 than to the 6S-2 locus (Table 2), in line with 6S-1 RNA levels exceeding those of 6S-2 RNA in stationary phase (Beckmann et al. 2011; Hoch et al. 2015). For the wt strain, 5′-end coverage verified the transcription start site (at nt −11) and the mature 5′-terminus of 6S-1 RNA (Fig. 3A, gray bars). The percentage of 5′-ends mapping to position −11 was largely increased in the ΔrnjA strain (Fig. 3A, light green bars), thus independently confirming the block of 6S-1 RNA 5′-end maturation in the ΔrnjA strain. Also, new 5′-ends appeared between positions +1 and −11 in this strain (Fig. 3A, light green bars) that were absent from the wt libraries, in line with the northern blot results (Fig. 2B, lanes 1–3 vs. lane 4) and indicating RNase J1-independent cleavages within the 5′-leader. A possible candidate for this activity might be RNase J2 in its endo- or 5′-exonucleolytic mode (Mathy et al. 2010; Laalami and Putzer 2011). RNA-seq further identified increased percentages of molecules with the mature 3′-end extended by 5–9 nt in the Δrph and Δ4exo strains (Fig. 3B,C; percentages given below the bars relative to wt). Although basically in line with the northern blot signals of slightly retarded mobility in Figure 2B (lane 5), these 3′-extensions were more evident from the northern blot than RNA-seq data, for unknown reasons.
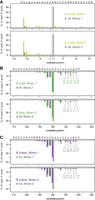
Representative 6S-1 RNA 5′- and 3′-end read profiles for the indicated strains and libraries, given as the percentage of all mapped 5′- or 3′-ends. (A) 5′-end read distribution in the region between position +1 and −15, illustrated by bars pointing upward, and shown for libraries 1 and 2 of the ΔrnjA strain (light green bars) and the wt strain (gray bars). (B,C) 3′-end read distribution in the region covering nt 180 and 199, illustrated by bars pointing downward. Nucleotide sequences are given above each graph, with capital letters indicating nucleotides of the mature 6S-1 RNA and lower case letters nucleotides present in precursor molecules. The percentages of 3′-ends at positions 195–199 are given below the bars for the Δrph (B, green numbers) and Δ4exo (C, purple numbers) libraries, and underneath for the corresponding wt library (gray numbers in B and C).
RNA-seq read numbers for 6S-1 and 6S-2 RNA
Northern blot analysis of 6S-1 RNA using region-specific probes
Signal intensities of the so-called “shorter fragments” were reduced in RNA pools from ΔrnjA bacteria relative to wt, Δrph, and Δ4exo bacteria (Fig. 2A). This observation suggested that RNase J1-catalyzed removal of the 5′-triphosphorylated leader (either directly by the “sliding endonuclease” activity of RNase J1 or following 5′-triphosphate [5′-ppp] removal by a pyrophosphohydrolase, see below) generates mature 6S-1 RNA with a 5′-monophosphate (5′-p), which in turn favors cleavage by an endonuclease to generate the “shorter fragments” that are roughly half molecules derived from cleavage in the apical loop region of 6S-1 RNA (see below).
Northern blots with probe 6S-1_5′-half showed that signals in the area of the “shorter fragments” were partly retarded in mobility when using RNA from the ΔrnjA relative to the wt strain (Fig. 4A, lane 2 vs. lane 1). These shifted signals were assigned to 5′-half molecules extended by 5′-leader nucleotides. Usage of a 5′-leader-specific probe further confirmed the enrichment of pre-6S-1 RNA in the ΔrnjA strain (Fig. 4B, lane 2 vs. lanes 1,3) and the presence of 5′-half molecules (“shorter fragments”) including the 5′-leader. The latter were also observed in wt and Δrph bacteria, but with much lower intensity (Fig. 4B, lanes 1,3 vs. lane 2). This suggests that some endonucleolytic cleavage of 6S-1 RNA (around the apical loop region; see Fig. 1) also occurs before 5′-leader removal by RNase J1. The 5′-leader-specific probe seemed to also detect some of the pre-6S-1 RNAs with 5′-ends between positions −11 and +1 accumulating in the ΔrnjA strain (Fig. 4B, lane 2, broadened pre-6S-1 RNA band). Assuming that these shortened pre-6S-1 RNA carried 5′-monophosphate ends, they may have contributed to the signal for the “shorter fragments” in Figure 4B (lane 2) through facilitating cleavage by an endoribonuclease in the apical loop region.
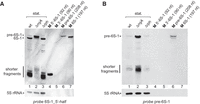
(A) Northern blot analysis using probe 6S-1_5′-half to determine the identity of the shorter 6S-1 RNA fragments. Lanes 1–3: as lanes 1,3,4 in Figure 2A. In addition to the 208- and 197-nt transcripts (lanes 6,7), we further loaded T7 transcripts mimicking the 5′- and 3′-halves of 6S-1 RNA (lanes 4,5; for details, see Supplemental Methods). (B) As in panel A, but using probe pre-6S-1 to specifically detect 6S-1 RNA species containing the 5′-precursor segment (11 nt; see Fig. 1). The shown blots are representative of four (panel A) and seven (panel B) individual blots using two to four independent RNA preparations.
We further analyzed the nature of the shorter decay intermediates using total RNA from the wt strain and probes 6S-1_5′-half and 6S-1_3′-half, unveiling that both 5′- and 3′-half fragments (∼90 nt in length), contribute to the signals in this area of the blot (Fig. 5A, compare lanes 5 and 9 with the size markers in lanes 6 and 11, respectively). This finding was confirmed using probes 6S-1_a–d (Fig. 1) specific for the individual quarters of 6S-1 RNA (Fig 5B, C). Probe 6S-1_d in particular also detected even smaller fragments (Fig. 5C, lane 13; compare with size markers in lanes 14 and 15) that can be assigned to cleavage in the 3′-central bubble (3′-CB, ∼position 140) and further downstream (∼position 160). Cleavage in the nt 160 region is consistent with 3′-ends mapping to positions 158 and 162–164 in basically all of our RNA-seq libraries, particularly in those of the first RNA-seq experiment enriched for 6S-1 RNA fragments (see https://synmikro.pages.uni-marburg.de/ag-lechner/6sdecay/). Likewise, the so-called long fragments of 6S-1 RNA (three major bands, Fig. 2A, lanes 1,4,5) were detectable with probes 6S-1_a–c, but essentially not with probe 6S-1_d (Fig. 5B,C), indicating that these are primarily fragments lacking at least the 3′-terminal 20 nt of 6S-1 RNA. This observation would also be in line with cleavage in the nt 160 region. We thus conclude that a fraction of 6S-1 RNA molecules is initially cleaved in the 158/162–164 region by a yet unidentified endonuclease, representing an alternative decay pathway.

Northern blot analysis of 6S-1 RNA processing and decay in the parental B. subtilis W168 strain (wt) using probes against different portions of 6S-1 RNA. (A) Hybridization with probes 6S-1_mature, 6S-1_5′-half and 6S-1_3′-half. (B) Hybridization with probe 6S-1_a. (C) Hybridization with probes 6S-1_b to d; in the blot on the right, 12% (instead of 10%) denaturing PAGE was used to resolve fragments as short as 25 nt. The shown blots in panels A–C are representative of two individual blots performed with independent RNA preparations. For details on the probes, see Figure 1 and Table 1. For T7 transcripts used as size markers and to control probe specificity, see Supplemental Methods; M 3′-6S-1 (25 nt) and M 3′-6S-1 (50 nt) are DNA oligonucleotides identical in sequence to the last 25 or 50 nt of 6S-1 RNA (see Fig. 1).
Testing for an involvement of other nucleolytic activities in 6S-1 RNA decay
We further examined a possible role of the pyrophosphohydrolase RppH as well as RNases J2 and III, the latter being a double-strand RNA-specific endonuclease (Mitra and Bechhofer 1994). The ΔrppH strain was included as the enzyme converts 5′-ppp to 5′-p ends, which we hypothesized may favor 5′-leader removal by RNase J1. However, for all three knockout strains, northern blot analysis of RNA from stationary phase cells provided no evidence for changes in the 6S-1 RNA fragment pattern relative to the wt strain (Supplemental Fig. S4A), arguing against specific roles of the three enzymes in 6S-1 RNA decay.
The RNase III deletion (Δrnc) strain was additionally tested under outgrowth conditions, where a burst of pRNA synthesis takes place and leads to the release of 6S-1 RNA:pRNA complexes from RNAP (Beckmann et al. 2011, 2012). We considered the possibility that the released 6S-1 RNA:pRNA complex with its ∼14 bp long hybrid helix might be a substrate for RNase III. However, the fragmentation pattern of 6S-1 RNA was essentially unchanged in the Δrnc strain under outgrowth as well as stationary phase conditions (Supplemental Fig. S4B).
The assignable maturation and degradation activities of 6S-1 RNA based on northern blot analysis and consistent with 5′- and 3′-ends mapped in RNA-seq experiments are illustrated in the context of the 6S-1 RNA secondary structure in Figure 6.
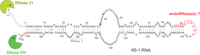
Summary of main findings obtained on 6S-1 RNA processing and decay in this study, illustrated in the context of the 6S-1 RNA secondary structure. The 5′-end and 3′-end nucleotides representing mature 6S-1 RNA, as identified in the 6S-1 RNA-seq libraries, are marked in blue (5′-end) or red (3′-ends). The read 3′-ends at nt 86–96 corresponding to the 5′-half molecules (shorter fragments) of 6S-1 RNA are highlighted in red as well. RNase activities inferred in this study to be involved in the processing and decay of 6S-1 RNA are indicated near the structural elements where hydrolysis takes place.
Cleavage of pre-6S-1 RNA by RNase J1 in vitro
To shed light on the mechanism of pre-6S-1 RNA maturation by RNase J1, we performed in vitro processing assays. Pre-6S-1 RNA with two extra G residues at the 5′-end for efficient T7 transcription was prepared with a 5′-32P-monophosphate or 5′-[γ-32P]-triphosphate end. Incubation of the first substrate with RNase J1 yielded one major product migrating at the position of a mononucleotide, as inferred from comparison with a nuclease P1 ladder of a 13-meric mimic of the 5′-leader (Fig. 7, compare lanes 2 and 5). In contrast, only little amounts of mononucleotide accumulated in the lane with the substrate carrying a 5′-triphosphate end (lane 4). Using the 5′-monophosphorylated pre-6S-1 RNA with its native 5′-A terminus (lacking the two extra G residues) also yielded the main mononucleotide product in comparable amounts as obtained with the 5′-monophosphorylated 6S-1 RNA extended by two G residues at the 5′-end (Fig. 7, compare lanes 11 and 2), excluding that the two extra G residues had produced an artificial result. The absence of prominent bands derived from internal cleavage of the leader in lanes 2,11 indicates that RNase J1 primarily removes the 5′-leader in its 5′-to-3′-exonucleolytic mode when pre-6S-1 RNA carries a 5′-monophosphate, in line with previous data (Li de la Sierra-Gallay et al. 2008). As 5′-leader removal from pre-6S-1 RNA is not very efficient in vivo (Beckmann et al. 2011), it remains a possibility that the bulk of intracellular pre-6S-1 RNA retains its 5′-triphosphate and RNase J1 (or J2) slowly removes the 5′-triphosphorylated leader in its endonucleolytic mode. Alternatively, a rate-limiting, inefficient pyrophosphohydrolase reaction that removes the γ/β-phosphates from the pre-6S-1 primary transcript in vivo may precede RNase J1 cleavage. As RppH could be experimentally ruled out (Supplemental Fig. S4A), in line with B. subtilis RppH requiring a G residue at the second position from the 5′-end (A in pre-6S-1 RNA; Fig. 1), an alternative pyrophosphohydrolase needs to be invoked in such a case. Indeed, there is evidence for the presence of another phosphate-removing enzyme in B. subtilis that is relatively insensitive to sequence variation at the 5′-terminal three positions and that was estimated to be responsible for 30% of the cellular pyrophosphohydrolase activity in B. subtilis (Hsieh et al. 2013).
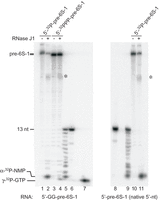
In vitro RNase J1 cleavage of precursor 6S-1 RNA using either mono- or triphosphorylated substrates. Lanes 1,2: monophosphorylated, 5′-32P-endlabeled pre-6S-1 RNA carrying two additional G residues at the 5′-end for efficient T7 transcription, incubated with (lane 2) or without (lane 1) RNase J1; lanes 3,4: as lanes 1,2, but using the same RNA with a triphosphorylated (γ-32P) 5′-end; lanes 5,6,8,9: a 13 nt long synthetic RNA oligonucleotide (5′-GGAAGUUAAAUAA-3′) corresponding to the 5′-leader of the pre-6S-1 RNA analyzed in lanes 1–4, incubated with (lanes 5,9) or without nuclease P1 (lanes 6,8); lane 7: γ-32P-GTP; lanes 10,11: as lanes 1,2, but using monophosphorylated, 5′-32P-endlabeled pre-6S-1 RNA starting with the native 5′-terminal A residue (i.e., lacking the two additional G residues). Asterisks next to lanes 4,11 indicate additional larger cleavage products. For more details, see Materials and Methods.
Cleavage of 6S-1 RNA by RNase Y in vitro
The Δrny strain is severely defective in RNA metabolism, as inferred from its slow growth and the intermediate stat1 phase growth arrest (Supplemental Fig. S1). Northern blot analysis of the Δrny strain (stat1 phase) using probes 6S-1_a and 6S-1_d detected faint signals in the ∼nt 90 region, indicating that inefficient cleavage in the apical loop region can occur in the absence of RNase Y (Supplemental Fig. S2C, lanes 2,7). With probe 6S-1_d, an aberrant signal with an estimated length of ∼80 nt appeared in the Δrny strain (Supplemental Fig. S2C, lane 7). This would correspond to a 3′-fragment originating from cleavage around position 110. Altogether, these observations may suggest that RNase Y is the main activity cleaving in the apical loop region in wt cells. However, normal levels of these “shorter fragments” in the Δrny strain in stat2 phase created doubts about this interpretation (see below). To shed more light on this issue, we investigated 6S-1 RNA cleavage by purified RNase Y in vitro. This was analyzed without and with the addition of 8 mM Mg2+ (Supplemental Fig. S5A, compare lanes 1–11 with 12–22). The analysis revealed cleavage primarily in the 5′-CB (∼nt 40–52), the most unstructured region of the molecule (Supplemental Fig. S5A, lanes 3,14). In the absence of Mg2+, RNase Y also showed enhanced cleavage in the region around ∼nt 145 of the 3′-CB. In the case of a direct key role of RNase Y in 6S-1 RNA decay, we would have expected prominent cleavages in the apical loop region. However, only very weak cleavages close to background levels were observed in this region in the presence of 8 mM Mg2+ (around position 95; Supplemental Fig. S5A, lane 14). Upon addition of a 10-fold molar excess of σA-RNAP over 6S-1 RNA, RNase Y cleavage patterns remained unchanged, but signal intensities decreased, assignable to partial protection of 6S-1 RNA from cleavage by RNase Y when bound to RNAP (Supplemental Fig. S5A, compare lanes 5 and 3, as well as 16 and 14). Annealing of an LNA mimic of a 6S-1 pRNA 14-mer reduced the overall signal intensity of RNase Y cleavages in the 5′-CB and narrowed cleavage down to the ∼nt 50 ± 4 region (Supplemental Fig. S5A, lanes 9,20); in addition, new cleavages appeared, tentatively assigned to the region of approximately nt 160–170 that refolds upon pRNA annealing (Supplemental Fig. S5B). Altogether, these results did not provide evidence for a role of RNase Y in endonucleolytic cleavage of the apical loop region in 6S-1 RNA.
6S-1 RNA—synthesis and decay of pRNAs
A key function of 6S-1 RNA is to serve as template for pRNA synthesis. We therefore asked if the removal of the 5′-leader from pre-6S-1 RNA is a prerequisite for pRNA transcription. For this purpose, cells were harvested 3 min after dilution of stationary phase cells into fresh LB medium (outgrowth) to extract total RNA via the TRIzol method to enrich for small RNAs (Damm et al. 2015a). The northern blot analysis revealed pRNA synthesis to occur with similar efficiency in the wt and the RNase J1 knockout strain (Fig. 8, lanes 1,2). We conclude that pRNA synthesis, and by inference 6S-1 RNA function including RNAP binding, is independent of 5′-leader removal. Increased levels of 6S-1 pRNAs were detected in RNA derived from the quadruple mutant (Fig. 8, lane 7 vs. lane 6), indicating that one or more of the 3′-to-5′-exoribonucleases are responsible for pRNA decay.

Northern blot analysis of 6S-1 pRNA synthesis and decay in B. subtilis. Endogenous pRNAs (∼14 nt), synthesized from 6S-1 RNA as template, were detected in total RNA extracts (6 µg) withdrawn from wt and ΔrnjA cells 3 min after induction of outgrowth (lanes 1,2). As specificity control, total RNA from a 6S-1 RNA knockout strain (ΔbsrA, lanes 3,9) was analyzed in parallel. A chemically synthesized pRNA 14-mer (5′-GUUCGGUCAAAACU-3′) was loaded in two different amounts (0.25 and 1 ng) as length marker (lanes 4,5,10,11). To identify RNases involved in 6S-1 pRNA decay, 10 µg of total RNA from wt cells, the quadruple mutant (Δ4exo), ΔrnjA and ΔbsrA bacteria, harvested 30 min after outgrowth, were analyzed by 10% native PAGE and northern blotting (lanes 6–9). The shown blots are representative of four individual blots performed with two to three independent RNA preparations; pRNAs were detected by probe 6S-1_pRNA.
6S-2 RNA turnover in B. subtilis
6S-1 and 6S-2 RNA have similar overall structures and both function as templates for pRNA synthesis in vitro and in vivo (Burenina et al. 2014; Hoch et al. 2016), raising the question whether their decay pathways are similar. In rich media, the W168 and related strains accumulate the highest 6S-2 RNA levels during exponential phase that gradually drop toward stationary phase (Beckmann et al. 2011). As for 6S-1 RNA, 6S-2 RNA turnover was investigated by northern blot analysis (probes illustrated in Fig. 9) and RNA-seq. In stationary phase, fragments of 6S-2 RNA accumulate in the wt strain (Fig. 10A). Northern blots revealed the accumulation of a new 6S-2 RNA-derived fragment in the RNase J1 knockout strain (Fig. 10B, “new signal” in lane 3) that was undetectable in RNA of the wt strain (lane 1). These findings indicated a central role for RNase J1 in 6S-2 RNA decay. This “new signal” went along with the accumulation of RNA-seq 5′-ends in the 5′-CB (Fig. 10D, positions 42–50) and increased the fraction of reads mapping to the 6S-2 RNA locus (Table 2). Supplemental Figure S6D illustrates how this intermediate dominated the 6S-2 RNA-seq profile, while read profiles for the other mutant strains, including the Δrny strain in stat2 phase, were similar to that of the wt strain (Supplemental Fig. S6A–C). As a northern probe specific for the 5′-end of 6S-2 RNA did not detect this new signal (Fig. 10C, lane 3), we concluded that the latter represents the 3′-product derived from cleavage in the region of nt 42–50. This indicates that endonucleolytic cleavage in the 5′-CB at nt 42–50 triggers rapid RNase J1-catalyzed degradation of the 3′-cleavage product via its 5′-to-3′-exonucleolytic activity.

Illustration of probes used in northern blot analysis of 6S-2 RNA processing and decay. The dark blue line symbolizes the full-length antisense probe; LNA/DNA mixmer probes (Table 1) were 6S-2_5′ (sky blue, complementary to nt 14–1), 6S-2_5′ short (pink, complementary to nt 10–1), 6S-2_3′ (green, complementary to nt 198–185), and 6S-2_3′ short (orange, complementary to nt 209–198).
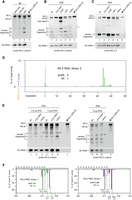
Analysis of 6S-2 RNA processing and decay. (A) Northern blot detection of 6S-2 RNA and fragments thereof (12% denaturing PAGE, probe 6S-2_mature) in total RNA preparations from wt cells harvested in early (OD600 = 1.35; at 3.2 h) and late (OD600 = 3.2; at 5.2 h) exponential as well as stationary phase (OD600 = 2.4; at 27 h). Full-length antisense 6S-2 RNA (probe 6S-2_mature, dark blue line in Fig. 9) was used as the probe. M (lane 4), T7 transcript of 6S-2 RNA, 209 nt in length (for details, see Supplemental Material). 5S rRNA served as a loading control. 6S-2s stands for 6S-2shortened, a processing product somewhat shorter than full-length 6S-2 RNA. (B) Corresponding northern blot analysis of 6S-2 RNA decay in the wt and RNase knockout derivative strains (10% denaturing PAGE). Total RNA was isolated from stationary phase cells, except for Δrny stat1 representing the intermediate stagnation phase at 0.4–0.8 OD600 (Supplemental Fig. S1). (C) As in panel B, but using the 5′-end-specific probe 6S-2_5′ (sky blue line in Fig. 9). The identity of the lower signal as fragment “c” was inferred from its migration distance relative to the full-length RNA signal (gels in panels B and C were run identically) and further confirmed in Supplemental Figure S7C. The shown blots are representative of 10 (panel B) or three (panel C) individual blots using two to five independent RNA preparations. (D) Percentage of RNA-seq 5′-end reads mapping to the 6S-2 RNA locus up to nt position 60 in ΔrnjA library 2 (green bars); gray bars indicate the values for the respective library 2 of the wt strain. (E) Northern blot analysis (12% denaturing PAGE) of 6S-2 RNA using total RNA derived from stationary cells of the wt strain, the Δrny strain (stat2, terminal stationary phase), and the Δ4exo strain using probe 6S-2_mature and different RNA concentrations as indicated above the blots. M, 209 nt T7 transcript used as marker for mature 6S-2 RNA. The swung dashes next to lanes 3, 6 and 10 mark the upshifted fragment “a” in the Δ4exo lanes. (F) RNA-seq profiles showing depletion of mature 3′-end reads (position 211) and simultaneous accumulation of 3′-end reads representing 6S-2 RNAs with their 3′-end extended by 3 nt (3′-end at position 214) in the Δrph and Δ4exo strains (green and magenta bars) relative to the library 1 wt strain (gray bars). The percentages of 3′-ends at positions 211–214 in the libraries for the wt and mutant strains are indicated below the corresponding bars in the same color code.
In the wt strain, we observed a 20- to 26-fold excess of reads mapping to the 6S-1 RNA locus relative to the 6S-2 RNA locus, which is basically consistent with, but may overestimate, the excess 6S-1 RNA levels relative to those of 6S-2 RNA in stationary phase (Beckmann et al. 2011; Hoch et al. 2015). This ratio decreases to 0.5- to 1.1-fold in the two ΔrnjA libraries (Table 2). One potential reason is that the 3′-fragment of 6S-2 RNA that massively accumulates in the ΔrnjA strain (Supplemental Fig. S6D) outcompetes other RNAs, including 6S-1 RNA, during library construction. Reduced levels of the shorter 6S-1 RNA fragments (Fig. 2A, lane 3 vs. lane 1; Fig. 4A, lane 2 vs. lane 1) and accumulation of pre-6S-1 RNA carrying a 5′-triphosphate may have contributed as well to the lowered number of reads mapping to the 6S-1 locus in the ΔrnjA strain. Moreover, the 5′-triphosphate of pre-6S-1 RNA may not have been fully converted to 5′-monophosphates for library inclusion or may have been, with its 11 nt single-stranded 5′-leader, a suboptimal substrate for adapter ligation, although this remains speculative at present.
Next, we analyzed the identity of the “shorter fragments” (Fig. 10A,B, signals “a–c”). By coelectrophoresis with size markers, the length of fragment “a” could be narrowed down to ∼115 nt, that of fragment “b” to ∼95 nt and that of “c” to ∼85 nt (Supplemental Fig. S7A,B). With the two 5′-end-specific probes, 6S-2_5′ and 6S-2_5′ short, only fragment “c” was detectable (Fig. 10C; Supplemental Fig. S7C). This identified signal “c” as a 5′-fragment and signals “a” and “b” as fragments lacking the 5′-terminal region of 6S-2 RNA. The intensity reduction for the fragment “c” signal in the Δrny strain (stat1 phase) and in the quadruple mutant (Fig. 10C, lanes 2,5) is difficult to explain, particularly for the latter, but may have indirect causes owing to the perturbation of RNA metabolism in these mutants. Degradation products “a” and “b” were confirmed as 3′ fragments using probe 6S-2_3′ (Supplemental Fig. S7C). RNA-seq identified an increased fraction of 6S-2 RNAs with 3′-ends extended by three nucleotides in the Δrph and Δ4exo strains (Fig. 10F). This correlates with northern blot signal “a” in Figure 10B (lanes 4,5, marked by the ∼symbol) shifted slightly upwards relative to the other strains (lanes 1–3; see also Fig. 10E, lanes 3,6), identifying the shifted signal “a” as fragments with unprocessed 3′-ends. We further conclude from the northern blot and RNA-seq results (Fig. 10) that 6S-2 RNA primary transcripts of 214 nt are synthesized in B. subtilis, which are trimmed at the 3′-end mainly by RNase PH. It should also be noted that the length of mature 6S-2 RNA in the wt strain is 211 nt (Fig. 9) and not 203 nt as previously proposed (Ando et al. 2002).
The weaker signal for the 5′-terminal fragment “c” in Figure 10B and C (lanes 2) observed with total RNA from Δrny bacteria in stat1 phase might have suggested that RNase Y is the major activity generating intermediate “c.” We therefore analyzed RNA from Δrny bacteria in the genuine stationary (stat2) phase. This did not reveal any significant differences in the northern blot signal pattern relative to the wt strain (Fig. 10E), arguing against a specific role of RNase Y in 6S-2 RNA degradation.
A signal migrating slightly faster than the full-length 6S-2 RNA (named 6S-2s for 6S-2shortened) was observed in the 6S-2 RNA-specific northern blots (Fig. 10A–C,E). This signal was also detected with the shorter probes 6S-2_5′ and 6S-2_5′ short covering only the 5′-terminal 14 or 10 nt of 6S-2 RNA, respectively (Figs. 10C,11A), indicating that this RNA species represents a 6S-2 RNA fragment somewhat shortened at the 3′-end. We then analyzed the 3′-portion of 6S-2s RNA by use of probes 6S-2_3′ short and 6S-2_3′ (Table 1). Unfortunately, we were unable to obtain specific signals with probe 6S-2_3′ short directed against the terminal stem of 6S-2 RNA (nt 198–209, Fig. 9) for unknown reasons. However, with probe 6S-2_3′, covering nt 185–198 (Fig. 9), we were able to detect 6S-2s RNA (Fig. 11C, lane 1). The northern blot signal was specific for 6S-2 RNA; it was not detectable in 6S-2 deletion strains (ΔbsrB and ΔbsrA/B; Fig. 11C, lanes 3,4). The signal was also obtained with the 194 nt 6S-2 RNA marker fragment covering nt 1–193 of 6S-2 RNA, but not with the 190 nt marker covering only nt 1–189 (Fig. 11C, compare lanes 6 and 7). This finding suggests that the intermediate 6S-2s has its 3′-end in the 3′-terminal loop region (nt 193–197, Fig. 9). The northern blot results are summarized in Figure 12, further illustrating the formation of fragments a–c by cleavage in the apical loop region of 6S-2 RNA. As for 6S-1 RNA, we analyzed the effects of RppH, RNase J2 and RNase III knockouts on 6S-2 RNA degradation and did not find any evidence for a role in 6S-2 RNA decay (Supplemental Fig. S4D).
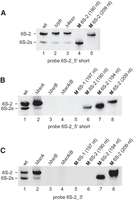
Northern blot analysis of a 6S-2 RNA degradation intermediate termed 6S-2s using the probes as indicated below each blot. (A) The presence of the 6S-2s signal is detectable (lane 1) with probe 6S-2_5′ short (see Fig. 9), indicating that 6S-2s RNA is shortened at the 3′-end; 6S-2s RNA is also detectable in the Δrph and Δ4exo strains (lanes 2,3), indicating that it is generated by endonucleolytic cleavage rather than 3′-exonucleolytic degradation of 6S-2 RNA. (B) Detection of 6S-2s RNA by probe 6S-2_5′ short in a B. subtilis PY79 6S-1 RNA deletion strain (ΔbsrA; lane 2), but not in a 6S-2 RNA deletion strain (ΔbsrB; lane 3) or a 6S-1/2 double deletion strain (ΔbsrA/B; lane 4). (C) As in panel B but using probe 6S-2_3′ (Table 1; Fig. 9). This probe detects the 194 nt 6S-2 marker RNA (lane 7) that has its 3′-end at position 193 (Fig. 9), but not the 190 nt marker RNA with the 3′-end at position 189 (lane 6). For the sequence of 6S-1 and 6S-2 marker RNAs, see the Supplemental Material.

Summary of main findings obtained on 6S-2 RNA decay in this study, illustrated in the context of the 6S-2 RNA secondary structure. The 5′-end reads accumulating in RNA-seq libraries of the ΔrnjA strain are highlighted in blue. The terminator hairpin loop inferred to be cleaved endonucleolytically to yield 6S-2s RNA is marked in orange. RNase activities inferred in this study to be involved in the decay of 6S-2 RNA are indicated near the structural elements where hydrolysis takes place. The shorter fragments “a” to “c,” representing an alternative decay pathway, are indicated by lines above and below the secondary structure.
DISCUSSION
In this work, we investigated the processing and decay pathways of the two noncoding 6S RNA paralogs in B. subtilis. In contrast to the majority of mRNAs (Hambraeus et al. 2003), both RNAs are abundant and quite stable (Beckmann et al. 2011), such that cleavage products are well detectable in northern blots of RNA derived from the “wild-type” strain W168. 6S-1 RNA is transcribed as a 5′-precursor (pre-6S-1 RNA) with 11 extra nucleotides, whereas the transcription start is the mature 5′-end in the case of 6S-2 RNA. We found that mature 6S-2 RNA is longer (211 nt) than previously reported (203 nt) (Ando et al. 2002). At least a substantial fraction of pre-6S-1 and 6S-2 RNAs carry 5′-ppp ends as inferred from differential RNA-seq data (Beckmann et al. 2011).
Our results support a model according to which 6S-1 RNA degradation is initiated by RNase J1-catalyzed removal of the single-stranded 5′-precursor segment. This generates mature 6S-1 RNA carrying a 5′-monophosphate end, which in turn stimulates an endoribonuclease to cleave in the RNA's apical loop, generating roughly 5′- and 3′-half fragments (termed “shorter fragments”) that accumulate in stationary phase. Stimulation of endonucleolytic cleavage in the apical loop region upon RNase J1-catalyzed removal of the 5′-leader can be inferred from reduced amounts of these “shorter fragments” in the ΔrnjA strain (Fig. 4A) and a lower fraction of RNA-seq reads with 3′-ends in the nt 90–100 region for the same strain (Supplemental Fig. S8). The “shorter fragments” or nicked 6S-1 RNA molecules are assumed to remain bound to RNAP and apparently evade rapid degradation in stationary phase, as inferred from their clear detectability in northern blots. During outgrowth from stationary phase, when 6S-1 RNA is released from RNAP upon pRNA synthesis, these fragments are rapidly degraded by 3′- and/or 5′-exoribonucleases (Supplemental Fig. S4C, compare lanes 1 and 3). The northern blots shown for the quadruple mutant relative to the wt strain (Supplemental Fig. S4C, compare lanes 3 and 4) are consistent with the involvement of 3′-exonucleases in their decay during outgrowth.
A potential candidate for the endoribonuclease cleaving in the apical loop region of 6S-1 RNA is endoribonuclease RNase Y acting preferably on 5′-monophosphorylated RNA substrates (Shahbabian et al. 2009). However, several observations appear to argue against this possibility: the Δrny strain showed neither substantial changes in the northern blot pattern of stationary (stat2) phase cells (Fig. 2C) nor a substantial overall reduction of reads with 3′-ends in the nt 86–96 region (Supplemental Fig. S8). Moreover, the in vitro assays for cleavage of 6S-1 RNA by RNase Y provided no evidence for prominent cleavages in this apical loop region (Supplemental Fig. S5A). Another possibility is that RNase J1 also acts as an endonuclease (see Introduction) to cleave in the apical loop region. This would be in line with a decreased fraction of reads with 3′-ends in the 86–96 nt region in the ΔrnjA strain (Supplemental Fig. S8) and an endonucleolytic J1 cleavage product observed in vitro (Fig. 7, asterisks). On the other hand, a backup RNase for apical loop cleavage has to be invoked, as we still observed reduced numbers of reads with 3′-ends in the nt 86–96 region in the ΔrnjA strain (Supplemental Fig. S8). RNase J2 may be a candidate for this backup activity, considering that the paralogous RNases J1 and J2 (70% amino acid similarity) have the same endonuclease specificity at some substrates (Even et al. 2005). Furthermore, also RNase Y, like J1 and J2, acts as a single strand-specific endonuclease. At some substrates, such as the thrS leader, all three enzymes cleave the same U-rich site, although the relative activities of RNases Y and J1 at the thrS substrate were found to be reversed in vitro versus in vivo (Laalami et al. 2021). Taken together, it cannot be excluded that RNase Y, J1, and J2 may be basically able to replace each other for the cleavage of 6S-1 RNA in the apical loop region or 6S-2 RNA in the 5′-CB. This might have obscured the identification of specific endonucleolytic cleavages for RNase Y, J1, and J2 in 6S RNA decay. The Δrny strain showed aberrant northern blot patterns for the two 6S RNAs in the intermediate growth arrest (stat1) phase. These patterns became wt-like when the strain had reached the final stationary phase (stat2). This may also be explained by overlapping Y, J1, and J2 endonuclease activities. Growth retardation and deceleration of RNA metabolism may enable Δrny bacteria to compensate the defects by making use of RNases J for processing functions that are carried out more efficiently by RNase Y in wt bacteria. In this context, it is worth mentioning that mRNA levels for RNases J1/J2 were unaffected by RNase Y depletion (Laalami et al. 2013), and depletion of RNases J1 and J2, either alone or together, did not affect mRNA levels of RNase Y (gene rny, formerly named ymdA) (Mäder et al. 2008; Durand et al. 2012a).
In vitro cleavage of pre-6S-1 RNA by RNase J1 (Fig. 7) supports 5′-to-3′-exonucleolytic removal of the 5′-leader, preferentially in the presence of a 5′-terminal monophosphate. Exonucleolytic removal would be in line with the absence of northern blot signals intermediate between the 5′-precursor and 5′-mature 6S-1 RNA, and with the very low frequency of RNA-seq reads with 5′-ends between nt −11 and +1 in all strains except for the ΔrnjA mutant strain (Fig. 3A; https://synmikro.pages.uni-marburg.de/ag-lechner/6sdecay/). It is likely that the bulk of (pre-)6S-1 RNA carries a 5′-triphosphate, as inferred from considerable levels of pre-6S-1 RNA at all growth stages except for extended stationary phase (Fig. 1B therein; Beckmann et al. 2011). This view is supported by RNA-seq experiments on total RNA from stationary phase cells: 5′-end reads representing pre-6S-1 RNA were strongly enriched upon 5′-triphosphate removal before library construction (Beckmann et al. 2011, Fig. 1E therein). This raises the question whether (pre-)6S-1 RNA processing and decay involve a pyrophosphohydrolase activity at all. As we found no evidence for an involvement of RppH, an alternative, yet unidentified pyrophosphohydrolase activity that was inferred to exist in B. subtilis (Hsieh et al. 2013) might act on pre-6S-1 RNA. However, the abundance of 5′-triphosphorylated pre-6S-1 RNA throughout growth predicts that this activity would be very low between exponential and early stationary phase and might increase only toward extended stationary phase. Alternatively, there may be no pyrophosphohydrolase acting on pre-6S-1 RNA, and RNase J1 may slowly mature 5′-triphosphorylated pre-6S-1 RNA. This would be consistent with the “sliding endonuclease” activity of RNase J enzymes that was observed at single-stranded, 5′-triphosphorylated RNA end regions creating an entry site for the 5′ exonuclease activity of RNase J1 (Taverniti et al. 2011; Liponska et al. 2018). Toward extended stationary phase, 5′-end maturation of 5′-triphosphorylated 6S-1 RNA, either by RNase J1 directly or via an inefficiently acting pyrophosphohydrolase, catches up, likely due to the down-regulation of 6S-1 RNA synthesis. Removal of the 5′-leader, generating 5′-monophosphorylated mature 6S-1 RNA, then stimulates endonucleolytic cleavage in the apical loop region, either by RNase J1 itself and/or another endoribonuclease. This model is in line with the observed correlation of increased 6S-1 RNA 5′-end maturation and the appearance of the “shorter fragments” toward extended stationary phase (Beckmann et al. 2011). Moreover, we provided evidence that 6S-1 RNA 5′-maturation is not essential for the function of 6S-1 RNA, as synthesis of 6S-1 pRNAs was indistinguishable between the wt and ΔrnjA strain (Fig. 8). Taken together, we conclude that the primary role of the 5′-triphosphorylated leader is to enhance 6S-1 RNA stability via keeping 5′-processing and decay initiation by RNase J1 at bay.
Although the processing and decay of 6S-2 RNA also involve RNases J1 and PH (Fig. 10), the mechanism is fundamentally different. The level of RNAs derived from the 6S-2 RNA locus increased in stationary phase in the RNase J1 knockout strain, mainly due to the accumulation of a 3′-fragment derived from cleavage in the 5′-CB (positions 42–50; Fig. 10; Supplemental Fig. S6D). This clearly indicates that endonucleolytic cleavage in the 5′-CB triggers rapid RNase J1-catalyzed degradation of the 3′-cleavage product via the 5′-to-3′-exonucleolytic activity of this enzyme. The profoundly altered 6S-2 RNA read profile of the ΔrnjA strain showed largely decreased read coverage for the 5′-terminal ∼45 nt of 6S-2 RNA and essentially no reads with 3′-ends derived from cleavage at nt 42–50 (Fig. 10D). Likewise, we were unable to detect 6S-2 RNA fragments of ∼45 nt in northern blots of the wt and RNase knockout strains, including the Δ4exo strain. We conclude that the fragments derived from cleavage in the 5′-CB are rapidly degraded by an unknown mechanism. As for 6S-1 RNA, we did not find evidence for endonucleolytic cleavage of 6S-2 RNA by RNase Y (Fig. 10E), but cannot exclude the involvement of RNase Y whose role may have been masked by the redundant endonuclease function of RNases Y, J1, and J2 as discussed above. Beyond the major decay pathway, we further observed cleavage of 6S-2 RNA in the apical loop region and in the loop of the putative terminator hairpin, giving rise to the shorter fragments “a–c,” where fragment “a” covers the 3′-end and fragment “c” the 5′-end, while fragment “b” was inferred to arise from cleavage of fragment “a” in the terminator hairpin loop (Fig. 12). It remains to be seen how this alternative decay pathway that, as for 6S-1 RNA, involves cleavage in the apical loop region, is interdigitated with the major 6S-2 RNA decay pathway.
Bacillus subtilis RNase J1 was recently shown to resolve stalled RNAP complexes through degrading the nascent RNA and causing the disassembly of stalled transcription complexes upon collision with RNAP (Šiková et al. 2020). qPCR validation of ChIP-seq data revealed an almost sixfold increased RNAP occupancy of the 6S-2 RNA gene (bsrB) in a ΔrnjA strain; this correlated with a ∼10-fold increase in 6S-2 RNA levels as determined by RT-qPCR (Šiková et al. 2020). These data were obtained with total RNA derived from exponentially grown cells (OD600 ∼ 0.5). The authors further showed an RNA-mediated co-pull down of RNase J1 and RNAP, particularly in stationary phase (Šiková et al. 2020), in line with J1 and RNAP colocalization on the periphery of the nucleoid as inferred from fluorescence microscopy experiments (Šiková et al. 2020). Increased association of RNase J1 and RNAP in stationary phase correlates with decreasing steady-state levels of 6S-2 RNA toward this phase (Beckmann et al. 2011). Increased 6S-2 RNA decay in stationary phase may act in concert with reduced 6S-2 RNA transcription in this nutrient-poor growth phase. The 6S-2 RNA gene (bsrB) promoter, at which transcripts are initiated with GTP (Ando et al. 2002), has some features of promoters that are sensitive to the cellular GTP concentration (Sojka et al. 2011); the concentration of GTP was reported to drop by roughly 70% upon entry into stationary phase (Lopez et al. 1979). An open question is whether RNase J1 accesses 6S-2 RNA exclusively on stalled transcription complexes at the bsrB locus, or if the enzyme also acts on free 6S-2 RNA or 6S-2 RNA bound to the active site of RNAP.
Both 6S-1 and 6S-2 RNAs, or major fractions thereof, are transcribed with a few extra nucleotides at the 3′-end that are removed primarily by the trimming activity of RNase PH (Figs. 2B, 10B). This might have a stabilizing effect on 6S RNAs, assuming that RNase PH protects the RNA from attack by other 3′-exoribonucleases that could use the 3′-overhang as a starting point for more invasive degradation. Trimming of 3′-ends by a few nucleotides (4 nt) by RNase PH or the other three 3′-exonucleases was also observed for the signal recognition particle RNA of B. subtilis (Yao et al. 2007), termed small cytoplasmic RNA (scRNA) (Struck et al. 1989). Interestingly, RNase J1 cleaves the scRNA primary transcript endonucleolytically in the 3′-region (Yao et al. 2007).
In E. coli, a major activity in the degradation of its singular 6S RNA (functionally corresponding to B. subtilis 6S-1 RNA) is the endonuclease function of RNase BN. The expression of RNase BN, which has no ortholog in B. subtilis (Condon 2003), peaks in exponential phase to decrease 6S RNA levels twofold relative to an RNase BN knockout strain (Chen et al. 2016). The enzyme essentially disappears toward stationary phase. Remarkably, RNase BN was shown in vitro to degrade 6S RNA:pRNA complexes more efficiently than naked 6S RNA. We observed the appearance of a hypersensitive RNase Y cleavage site in the nt 150–170 region of B. subtilis 6S-1 RNA upon rearranging its structure by annealing of a pRNA mimic (Supplemental Fig. S5). It is thus conceivable that also RNase Y accelerates the decay of 6S-1 RNA:pRNA complexes after their release from RNAP upon initiation of outgrowth, although clear evidence supporting this idea is lacking (Fig. 2C). Other protein interactants of 6S RNA beyond RNAP are potential factors that may additionally modulate 6S RNA decay. So far, no other 6S RNA-binding proteins than RNAP have been identified by methods like Grad-seq (Gerovac et al. 2021), but proteins to be identified that interact with 6S RNAs, be it transiently or in specific conditions, might contribute to the regulation of 6S RNA decay in vivo.
We have discovered a differential key role for RNase J1 in the processing/decay of B. subtilis 6S RNAs, but were unable to infer a direct role of RNase Y in these processes. It was recently reported that a Δrny strain developed suppressor mutations, for example, in the β, β′ subunits of RNAP and the transcription elongation factor GreA, that decreased transcription efficiency as a prerequisite for cell growth of the rny knockout strain (Benda et al. 2021). The authors concluded that RNase Y and RNAP cooperate in balancing RNA synthesis and degradation. The strong perturbation of RNA metabolism in the Δrny strain also manifested in our study in the form of an intermediate growth stagnation phase (Supplemental Fig. S1). At this stage, 6S-1/2 RNA northern blot patterns varied substantially (Figs. 2A, 10B; Supplemental Fig. S2B,C) between the Δrny strain and the wt and other mutant strains, although we are aware that this stagnation phase is not directly comparable to the growth state of the other strains at the same OD600 (0.4–0.8; Supplemental Fig. S1). Taken together, it seems reasonable to hypothesize that deceleration of RNA metabolism enables the Δrny strain to maintain viability through compensating its processing defects by utilizing minor decay pathways and alternative enzymes with overlapping substrate specificities (RNases J1 and J2).
Our study has demonstrated an important function of RNase J1 in the metabolism of both 6S RNA paralogs in B. subtilis. This raises the question of how processing and decay are accomplished in bacteria lacking RNase J1 and whether there might be a correlation between the presence of RNase J1 and two 6S RNA paralogs. As discussed above, RNase BN plays a key role in the decay of the singular 6S RNA in E. coli that lacks RNase J1 (Chen et al. 2016), and the endoribonucleases E and G are responsible for 5′-end maturation of 6S RNA precursors initiated at two different promoters in E. coli (Kim and Lee 2004; Chae et al. 2011). The occurrence of RNases J1/J2 is quite patchy among bacteria. Homologs were identified in some cyanobacteria, actinobacteria, Tenericules species, (Lacto)Bacillales, Clostridia, and α/γ/ε-proteobacteria (Kaberdin et al. 2011). Only some of those bacteria (mainly Bacilli and closely related Firmicutes) encode a second 6S RNA paralog (Wehner et al. 2014). As an example, an RNase J1 homolog was unequivocally identified in the α-proteobacterium Rhodobacter sphaeroides (Rische and Klug 2012) that expresses only one 6S RNA species (Elkina et al. 2017). It thus appears that 6S RNA processing and decay pathways have diverged substantially among bacteria, and a consistent link between the presence of RNase J1 and the expression of two 6S RNA paralogs is not evident.
MATERIALS AND METHODS
Bacterial strains and cell culture
All RNase knockout strains used here are derivatives of the B. subtilis strain W168 and were kindly provided by the laboratory of Ciaran Condon (see Table 3). The following concentrations of antibiotics were used for selection on agar plates or in precultures: 100 µg/mL spectinomycin, 20 µg/mL tetracycline, 5 µg/mL phleomycin, 5 µg/mL erythromycin, and 5 µg/mL kanamycin. To grow strain CCB396, a quadruple mutant, a low salt LB agar (10 g/L tryptone, 5 g/L sodium chloride, 5 g/L yeast extract, adjusted to pH 7.5; 15 g/L agar) containing 50 µg/mL spectinomycin, 10 µg/mL tetracycline, 1 µg/mL phleomycin, and 5 µg/mL kanamycin was used. For the control of the specificity of northern blot signals, 6S-1 and 6S-2 RNA knockout strains (ΔbsrA, ΔbsrB; derived from B. subtilis PY79) were grown in the presence of spectinomycin (100 µg/mL) or kanamycin (10 µg/mL), respectively (Table 3). Cells were grown in LB medium at 37°C and under gentle shaking (200 rpm, Aquatron waterbath shaker, Infors AG). Complete growth curves were recorded (by optical density measurements at 600 nm = OD600) using 300 mL LB medium without antibiotics after inoculation with an overnight culture (grown with antibiotics) to an OD600 of 0.05. For all strains except CCB434 (ΔrnjA) and CCB441 (Δrny), the aforementioned overnight culture was inoculated directly with a colony picked from an agar plate. Owing to the slow growth of strains CCB434 and CCB441, a preculture inoculated with a plate colony was grown for subsequent inoculation of the overnight culture. For RNA-seq, 10 OD600 of stationary cells from each strain were harvested at time points indicated in the context of the respective growth curves (Supplemental Fig. S1). To induce outgrowth, cells from extended stationary phase were diluted 1:5 in fresh prewarmed LB medium. Samples of 10 mL (for phenol extraction) or 40 mL (for TRIzol extraction) were withdrawn at different time points for RNA preparation. For stationary cells analyzed in northern blots, cell cultures were harvested at time points comparable to those used for RNA-seq experiments (Supplemental Fig. S1), where cells had already been in stationary phase for >5 h and where the optical density of cultures had already begun to decrease in many cases.
Strains used in this study
RNA preparation and sequencing
Total cellular RNA was isolated from frozen cell pellets by extracting three times with hot phenol, whereas B. subtilis 6S-1 pRNAs (∼14 nt) were extracted via the TRIzol method (Damm et al. 2015a). RNA integrity was verified on 6% denaturing polyacrylamide (PAA) gels (8 M urea) before RNA-seq was performed. Ribosomal RNA molecules were depleted from the total RNA samples using either the Ribo-Zero rRNA Removal Kit (Epicentre) for bacteria (first RNA-seq), or using an in-house protocol (Vertis Biotechnologie AG; second RNA-seq). Then, the library was enriched for RNA fractions <500 nt using the RNeasy MinElute Cleanup Kit (Qiagen; first RNA-seq), or for RNA fractions <200 nt using the RNA Monarch RNA Cleanup Kit (NEB; second RNA-seq). The RNAs were subsequently treated with tobacco acid pyrophosphatase (TAP, Epicentre; first RNA-seq) or RNA 5′ Polyphosphatase (Biozym; second RNA-seq) to convert 5′-ppp to 5′-p ends for linker ligation. Oligonucleotide adapters were ligated to the 5′- and 3′-ends of RNA molecules. First-strand cDNA synthesis was performed using Moloney murine leukemia virus (M-MLV) reverse transcriptase and a primer complementary to the 3′-adapter. The resulting cDNAs were PCR-amplified using a high-fidelity DNA polymerase. The generated cDNA was amplified by 19 PCR cycles until a DNA yield of ∼9–26 ng/µL was reached. The PCR reaction mixtures containing the cDNA libraries were purified using the Agencourt AMPure XP Kit (Beckmann Coulter Genomics), analyzed by capillary electrophoresis and pooled. Library construction was carried out at Vertis (see above). The paired-end sequencing reaction was conducted using an Illumina sequencer.
All libraries were processed including quality assessment, adapter trimming and trimming of bad quality bases at the ends, using fastp v0.23.2 (Chen et al. 2018). Trimmed reads were then mapped against the B. subtilis W168 genome (NC_000964.3, GCF_000009045.1_ASM904v1) and alignments extracted for loci of 6S-1 and 6S-2 RNA ± 100 nt. RNA-seq reads from the first and second RNA-seq experiments were mapped with bwa (Li and Durbin 2010) v0.7.17-r1188 in paired-end mode with default parameters. Only read pairs that mapped uniquely to a locus and in proper configuration (from both sides), and without mismatches after trimming, were considered for further analysis (Table 2). The region in the transcript between matching read pairs was considered fully covered in sequencing regardless of actual overlap.
Northern blotting
For the analysis of 6S-1 and 6S-2 RNAs and if not stated otherwise, 3 µg of total RNA were loaded on a 10% or 12% denaturing (8 M urea) PAA gel and northern blotting was performed as described previously (Damm et al. 2015b). If not indicated differently, digoxigenin (DIG)-labeled antisense transcripts covering the full-length mature 6S RNA or 5S rRNA (internal loading control) were used. Antisense transcripts were synthesized by T7 RNAP from linearized plasmid DNA or PCR products as templates using a DIG RNA labeling mix (Roche Diagnostics) (Damm et al. 2015b). Analysis of total RNA from B. subtilis PY79 strains with a 6S-1 RNA (ΔbsrA) or 6S-2 RNA (ΔbsrB) gene deletion, respectively (Table 3), allowed us to demonstrate that the 6S-1 and 6S-2 RNA full-length probes were specific for the respective 6S RNA and fragments thereof. The northern blot probes applied in this study, including several specific LNA/DNA mixmer probes labeled with DIG at the 5′- and 3′-end, are specified in Table 1. For pRNA detection, 6 or 10 µg of total cellular RNAs were separated by 10% native PAGE (for details, see Beckmann et al. 2010).
In vitro RNase J1 cleavage assays
RNase J1 cleavage assays were performed with 2000 Cherenkov cpm of either 5′-[α-32P]-endlabeled pre-6S-1 RNA (5′-monophosphorylated) or 5′-[γ-32P]-endlabeled pre-6S-1 RNA (triphosphorylated RNA); for details, see Supplemental Material. Samples (final volume: 20 µL) further contained 1× RNase J1 reaction buffer (20 mM HEPES pH 8.0, 8 mM MgCl2, 100 mM NaCl) and 1 µg of RNase J1. Samples were incubated for 60 min at 30°C and stopped by adding 2× RNA denaturing loading buffer (0.02% [w/v] bromophenol blue, 0.02% [w/v] xylene cyanol blue, 8 M urea, 50% [v/v] formamide, 2× TBE). Nuclease P1 cleavage of the RNA 13-mer (5′-GGAAGUUAAAUAA-3′, corresponding to the native 11 nt 5′-leader with two additional G residues at the 5′-end) was performed as follows: 2.5 µg of the RNA 13-mer were supplemented with 2000 Cherenkov cpm of 5′-32P-endlabeled 13-mer in 1× nuclease P1 buffer [40 mM NH4(OAc)2; 0.4 mM ZnSO4, pH 5.3 at room temperature, pH 5.1 at 70°C] in a volume of 10 µL. After preincubation of the sample for 1 min at 70°C, 1 U of nuclease P1 (USBiological/Biomol) was added; after 45 sec at 70°C, the reaction was stopped by adding an equal volume of 2× RNA denaturing loading buffer (see above). All samples were heated up to 98°C for 3 min, immediately followed by cooling on ice for 5 min and electrophoresis on a 25% denaturing PAA gel (polyacrylamide/bisacrylamide [24:1], 8 M urea, 1× TBE) using 1× TBE as running buffer.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Ciarán Condon (Paris, France) for providing RNase knockout strains, Torsten Hain (Gießen, Germany) for access to their Illumina sequencing platform, Simone Bach and M. Amri C. Schlüter for initial support with northern blot experiments, and Dominik Helmecke for technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (GK 1384, GK 2355) to K.D., J.C.W., and R.K.H.
Footnotes
-
↵4 Joint first authors.
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079666.123.
- Received March 22, 2023.
- Accepted June 6, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








