
An energetically beneficial leader–linker interaction abolishes ligand-binding cooperativity in glycine riboswitches
Abstract
Comprised of two aptamers connected by a short nucleotide linker, the glycine riboswitch was the first example of naturally occurring RNA elements reported to bind small organic molecules cooperatively. Earlier works have shown binding of glycine to the second aptamer allows tertiary interactions to be made between the two aptamers, which facilitates binding of a separate glycine molecule to the first aptamer, leading to glycine-binding cooperativity. Prompted by a distinctive protection pattern in the linker region of a minimal glycine riboswitch construct, we have identified a highly conserved (>90%) leader–linker duplex involving leader nucleotides upstream of the previously reported consensus glycine riboswitch sequences. In >50% of the glycine riboswitches, the leader–linker interaction forms a kink-turn motif. Characterization of three glycine ribsowitches showed that the leader–linker interaction improved the glycine-binding affinities by 4.5- to 86-fold. In-line probing and native gel assays with two aptamers in trans suggested synergistic action between glycine-binding and interaptamer interaction during global folding of the glycine riboswitch. Mutational analysis showed that there appeared to be no ligand-binding cooperativity in the glycine riboswitch when the leader–linker interaction is present, and the previously measured cooperativity is simply an artifact of a truncated construct missing the leader sequence.
Keywords
INTRODUCTION
Riboswitches are cis-acting mRNA elements that regulate gene expression by direct binding to small molecule metabolites (Breaker 2011). Upon binding of the biologically important metabolites, riboswitches undergo conformational changes that may affect the structure of the associated expression platforms and control gene expression through transcriptional termination, translational initiation, or alternative splicing. Many riboswitches have been computationally discovered and experimentally verified (Roth and Breaker 2009; Breaker 2011). Most of the currently reported 24 classes of riboswitches are found in the 5′ UTRs of bacterial genes, though TPP riboswitches are also present in plants and fungi (Cheah et al. 2007; Wachter et al. 2007). They vary widely in their structures and functional mechanisms, discriminatively sensing a plethora of metabolites (Roth and Breaker 2009; Breaker 2010, 2011).
Glycine riboswitches are among the earliest discovered riboswitches (Mandal et al. 2004). Using BLASTN searches followed by iterative secondary structure refinement and extension with the SEQUENCESNIFFER program, Breaker and coworkers identified a gcvT motif that resides upstream of bacterial genes expressing enzymes involved in the metabolism of glycine (Barrick et al. 2004; Mandal et al. 2004). The gcvT motif, termed the glycine riboswitch, forms an on-switch when bound to glycine, controlling the expression of enzymes that catalyze the initial reactions for the use of glycine as an energy source. These enzymes are components of the glycine cleavage system, which breaks down glycine and produces carbon dioxide, ammonia, and 5-10-methylenetetrahydrofolate in the presence of tetrahydrofolate (Fujiwara et al. 1984). 5-10-methylenetetrahydrofolate serves as a source of the methyl groups for methionine and 2′-deoxythymidine biosynthesis (Birdsall et al. 1996). In addition, 5-10-methylenetetrahydrofolate can also serve as the substrate in the reversible conversion of glycine to serine. Combined together, glycine and serine account for up to 15% of carbon assimilated by E. coli growing on glucose (Pizer and Potochny 1964). Beside their direct roles in protein synthesis, glycine and serine serve as precursors for purines, heme groups, cysteine, phosphatidylserine, enterochelin, and tryptophan (Ravnikar and Somerville 1987; Tripp et al. 2009). Thus, the glycine riboswitch self-regulates by biofeedback an important metabolite that serves as a precursor to one of the main biosynthetic trunks in bacteria (Tripp et al. 2009).
Interestingly, glycine riboswitches contain two tandem glycine-sensing domains and purportedly use, uniquely, ligand-binding cooperativity. The most biochemically characterized construct, Vibrio cholerae (VC) glycine riboswitch, was reported to bind glycine with a Hill coefficient of 1.5–1.6 (Mandal et al. 2004; Kwon and Strobel 2008; Erion and Strobel 2011), the largest among all reported glycine riboswitches. Deleterious mutations in each aptamer of the VC glycine riboswitch also greatly reduced the glycine-binding affinity in the other aptamer, providing direct evidence of the ligand-binding cooperativity of the tandem riboswitch (Mandal et al. 2004; Erion and Strobel 2011). As the first example of naturally occurring RNA elements reported to bind small organic molecules cooperatively (Mandal et al. 2004; Trausch et al. 2011), glycine riboswitches have inspired many biochemical and biophysical studies, including nucleotide analog interference mapping (NAIM) to investigate the chemical basis of the cooperativity (Kwon and Strobel 2008), and small-angle X-ray scattering (SAXS) to dissect the roles of metal ions and ligand binding in structural transitions (Lipfert et al. 2007, 2010). Recent efforts have resulted in the proposal of the asymmetrical model of glycine-binding cooperativity (Erion and Strobel 2011), as well as crystal structures of the single aptamer VCII with an extended P1 helix (Huang et al. 2010) and the tandem glycine riboswitch from Fusobacterium nucleatum (Butler et al. 2011).
In working with the tandem VC glycine riboswitch, we have found our minimal construct exhibited a distinctive protection pattern in the linker region compared with literature results. This led to the discovery of a surprising leader–linker interaction involving nucleotides upstream of the reported consensus glycine riboswitch sequences. We have defined the energetic benefit of the leader–linker interaction through in-line probing of the tandem riboswitches and further characterized its effect on glycine binding and interaptamer interaction using biochemical assays with two aptamers in trans. Our mutational analysis surprisingly showed that interaptamer inhibition does not exist in the full-length VC glycine riboswitch. Together, our data suggest that glycine binding and interaptamer interaction synergize to fold the global structure of the glycine riboswitch in which its two aptamers do not bind glycine cooperatively.
RESULTS
Discovery of the leader–linker interaction
As RNA tertiary structures are generally more flexible than proteins, to reduce terminal entropy and increase crystallizability of RNAs, crystallographers generally remove the single-stranded flanking regions from their RNA constructs. In an attempt to crystallize the VC tandem glycine riboswitch, we have constructed the minimal sequence VCIIIs (nt 7–225) (Fig. 1A). The overall glycine-binding characteristics of VCIIIs (Kd = 47 ± 14 μM) (Table 1; Supplemental Fig. S1) are very similar to VCIII (nt 1–225, Kd = 59 ± 12 μM) (Table 1). In the linker region (nt 135–137), VCIIIs remained unstructured independent of glycine concentration (Supplemental Fig. S1), while VCIII exhibited local structure flexibility with increasing glycine concentration (Mandal et al. 2004). This suggested that its 5′ leader sequence may influence the structure of the linker region. One simple scenario is direct duplex formation between the leader and the linker region, which proved correct upon retrieval of sequences from the NCBI database and manual comparison of the leader with the linker sequences. Thus, a leader–linker duplex (termed P0) beyond the published consensus glycine sequence was unearthed. Supported by Mfold (Zuker 2003), the P0 helix contains 3–7 bp and is 96% conserved (53 out of 55) in the tandem glycine riboswitch sequences reported by Breaker and coworkers (Mandal et al. 2004). In addition, the P0 helix is present in 91% of the 22 tandem glycine riboswitch seed sequences in Rfam (Gardner et al. 2011). Further analysis showed that in 40% of these glycine riboswitches, P0 helices are connected to P1 through simple internal loops/bulges (Supplemental Fig. S2). In the rest (>50%) of the glycine riboswitch sequences (Fig. 1B), however, P0 helices are connected to P1 through kink-turn motifs (Klein et al. 2001; Goody et al. 2004; Liu and Lilley 2007; Cruz and Westhof 2011). Three characteristics show that these motifs fit the pattern of the consensus kink-turns derived from the ribosome kink-turns: (1) The C stems most often end with two C-G/G-C pairs; (2) the internal loop follows the RNN(N) consensus rule; (3) the NC stems usually start with two sheared G-A/A-G base pairs. Interestingly, the canonical base pair adjacent to the internal loop (-1b/-1n pair) has a strong preference for G-C, just the opposite to the C-G base-pair preference in the ribosome, indicating a possible deviation from the consensus kink-turn motif, as the G-C pair may not support the type I A-minor interaction as well as C-G (Klein et al. 2001).
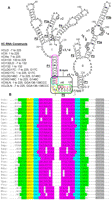
(A) Secondary structure of the full-length VC glycine riboswitch. r2–r7; *a–*b indicate the representative glycine-induced perturbation regions in the in-line probing assay and the hydroxyl radical footprinting assay of VCIII, respectively; the corresponding nucleotides are circled. (I, II) Aptamer I and aptamer II, respectively. Green, yellow, cyan, purple, and gray regions indicate C stem (P0 helix), internal loop, NC stem, P1 helix of aptamer I, and P1 helix of aptamer II, respectively. The kink-turn motif is shown in a black box. Red nucleotides indicate previously defined linker sequence. (B) Alignment of the kink-turn motifs formed between leader and linker regions in 31 of 55 tandem glycine riboswitch sequences reported by Breaker and coworkers (Mandal et al. 2004). Abbreviations of the names of the organisms are taken from the same article.
Binding affinities and Hill coefficient values of glycine riboswitches and mutantsa
The global structures of the full-length glycine riboswitches are similar to those of the truncated constructs
To evaluate the global structures of the full-length glycine riboswitches, we have constructed both full-length and truncated versions of the well-studied glycine riboswitches from three organisms: Vibrio cholerae (VCLD [nt −7 to 225] and VCIII), Fusobacterium nucleatum (FNLD [nt −11 to 158], and FNIII [nt 1–158]), and Bacillus subtilis BSLD (nt 1–202) and BSIII (nt 13–202). These RNA sequences are downstream from the transcription start sites predicted by Prokaryotic Promoter Analysis Using Sequence Alignment Kernel software (Gordon et al. 2003). Interestingly, the Bacillus subtilis glycine riboswitch previously reported (Mandal et al. 2004) contains the leader sequence, although the leader–linker interaction was not recognized. In-line probing assays map the local structure changes by measuring the flexibility of the phosphodiester bond to achieve in-line geometry for nonenzymatic cleavage through internal transphosphorylation (Soukup and Breaker 1999; Regulski and Breaker 2008). Under standard in-line probing conditions (50 mM Tris-Hcl at pH 8.3, 20 mM MgCl2, 100 mM KCl, 25°C), at saturating glycine concentrations, the full-length glycine riboswitches exhibit the same protection patterns as the truncated ones outside of the linker region in all three VC, FN, and BS glycine riboswitches (Fig. 2A; Supplemental Figs. S3–S5). To compare the regions directly, we calculated the relative band intensities of the major cleavage bands and known glycine-perturbed regions. Results showed that these bands have similar relative intensities within any given construct in the in-line probing assay of glycine riboswitches with or without leader sequences (Table 2). In the linker region, the full-length glycine riboswitches are protected from spontaneous cleavage, while the truncated glycine riboswitches show cleavage bands, consistent with a leader–linker interaction. Therefore, in-line probing assays showed that, except for the linker regions, the full-length glycine riboswitch folds into the same structure as the truncated constructs at high magnesium and glycine concentrations.
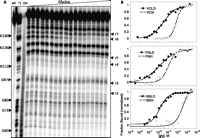
In-line probing of full-length and truncated glycine riboswitches. (A) Representative in-line probing gel image of BSLD. NR, T1, and OH represent no reaction, partial digestion with RNase T1, and partial digestion with alkali, respectively. r2–r7 indicate regions defined in Table 2. (B) Representative semi-log plots of the normalized fraction of RNA bound versus glycine concentration. Shown from top to bottom: VCLD/VCIII (r3); FNLD/FNIII (r6); BSLD/BSIII (r6).
Comparison of glycine riboswitches' protection patterns with and without the leadera
As in-line probing assays may detect structure formation with heavy focus on secondary structures, to compare tertiary structures of the glycine riboswitches, we used hydroxyl radical footprinting (HRFP), one of the most frequently used methods for studying RNA folding (Latham and Cech 1989; Sclavi et al. 1998; Brenowitz et al. 2002; Wakeman and Winkler 2009). HRFP is sequence independent and measures the perturbation of solvent accessibility of local tertiary structures during conformational change by hydroxyl radical-triggered RNA backbone cleavage. Similar to the in-line probing results, HRFP experiments showed that, at saturating glycine concentrations, the full-length glycine riboswitches exhibit the same protection patterns as the truncated ones outside of the linker region in all three VC, FN, and BS glycine riboswitches (Supplemental Figs. S6–S8).
Combining in-line probing and HRFP experiments, results showed that full-length glycine riboswitches fold into the same global structure as the truncated version at saturating magnesium and glycine concentrations.
Energetic benefit of the leader–linker interaction
Unexpected tertiary structures involving flanking sequences of functional RNAs have been reported before and are functionally beneficial. For instance, the loop/bulge interaction involving the 5′-flanking sequence of the hammerhead ribozyme increases the catalytic rate substantially (Khvorova et al. 2003; Martick and Scott 2006). To understand the functional benefit of the leader–linker interaction, we obtained the ligand-binding affinities of the full-length glycine riboswitches through in-line probing assays at standard conditions, similar to that reported previously for glycine riboswitches (Mandal et al. 2004; Kwon and Strobel 2008; Erion and Strobel 2011) (Fig. 2; Supplemental Figs. S3–S5). In the linker regions, the full-length glycine riboswitches showed constant protection with or without glycine, suggesting that the leader–linker interaction is formed independent of glycine. Other glycine-perturbed regions, including both aptamer I and aptamer II regions, showed similar glycine-binding affinities for the full-length constructs within experimental error. The affinities calculated from all of the major glycine-perturbed regions over at least two duplicated experiments were averaged together and tabulated in Table 1. Results showed that glycine-binding affinities of the full-length constructs have been increased by 4.5- to 86-fold compared with the truncated ones. This corresponds to a free-energy benefit of 0.9–2.7 kcal/mol under standard in-line probing conditions. Interestingly, all three full-length riboswitches showed similar glycine-binding affinities with Kds of 5.0 ± 4.0 μM, 3.1 ± 2.5 μM, and 3.6 ± 3.4 μM for VCLD, FNLD, and BSLD, respectively, while the truncated constructs showed much more varied glycine-binding affinities. We have also obtained glycine-binding affinities with HRFP, which gave similar values for VCIII and FNIII. However, we could not obtain any visible glycine perturbation for the full-length constructs using HRFP. At the corresponding regions, where glycine induces protection in the truncated glycine riboswitches, full-length constructs exhibit protection with or without glycine, precluding visible perturbation. Typically, hydroxyl radical footprints are observed for the interior portion of closely packed helical segments for structured RNAs. We inspected the protected regions of FNIII in the published crystal structure (Butler et al. 2011) in PyMol (http://www.pymol.org) and found that these regions are mostly buried between the two aptamers. For instance, the sugar moieties (HRFP susceptible) of nt 63–67, 134–136, and 117–119 are buried by the α-α′ interaptamer interaction; sugar moieties of nt 85–86 are partially buried by the β-β′ interaptamer interaction, and the packing against flipped base A111, which is very important for the formation of the glycine-binding pocket; nt 110–112 directly participate in the glycine-binding pocket. The HRFP protection in these regions of FNLD in the absence of glycine suggests that the full-length constructs promote the interaptamer interaction, form more compact and organized pockets for glycine binding, and render any possible further compaction by the glycine binding undetectable through HRFP experiments.
Mutation in the linker region selectively reduces the ligand-binding affinities of the full-length, but not the truncated glycine riboswitches
The full-length glycine riboswitches exhibit significantly higher ligand-binding affinities compared with the truncated ones. To link the energetic benefit directly to the leader–linker interaction, we focused on the VC glycine riboswitch, which was studied most extensively with biochemical analysis. Incorporating a GGA136–138CCC mutation to prevent the formation of the P0 helix, we prepared VCIIILN and VCLDLN, the linker mutants in the truncated and full-length VC glycine riboswitches, respectively. Similar to the wild-type constructs, the linker mutants also showed similar glycine-binding affinities in both aptamer I and aptamer II regions within experimental error. The glycine-binding affinities were calculated as the average affinities of all of the major regions over at least two duplicated experiments. Results showed that VCLDLN binds glycine with a Kd of 61 ± 11 μM (Fig. 3A; Table 1), very similar to that of VCIII, 59 ± 12 μM, losing the entire energetic benefit (12-fold) of the leader–linker interaction observed in VCLD. This affinity loss could be potentially caused by a loss of an unidentified linker interaction with regions other than the 5′ leader. To control for this possibility, we obtained the glycine-binding affinity of VCIIILN. Unlike VCLDLN, the linker mutation in VCIIILN did not reduce the glycine-binding affinity compared with VCIII. These results are consistent with the model that leader–linker interaction contributes solely to the energetic benefits in VCLD. Interestingly, there is about a fourfold increase in the glycine-binding affinity of VCIIILN compared with VCIII (Fig. 3B; Table 1). We noticed that both VCIII and VCIIILN contain three guanosines at the 5′, with two artificial guanosines added for improved transcription efficiency. Suggested by Mfold (Zuker 2003), this GGG sequence could potentially pair with CCC136–138 in VCIIILN and form an imperfect kink-turn motif (only two nucleotides in the internal loop). Thus, the unexpected fourfold increase of glycine-binding affinity of VCIIILN versus VCIII is also consistent with the functional role of the leader–linker interaction.
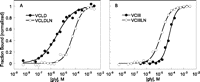
Comparison of the ligand binding of the linker mutants with wild-type VC glycine riboswitches. (A,B) The representative semi-log plots of the normalized fraction of RNA bound versus glycine concentration for r3 region (A121–G123) of VCLD with VCLDLN, and VCIII with VCIIILN, respectively.
Leader–linker interaction greatly enhances the interaptamer interaction
HRFP experiments showed that full-length glycine riboswitches promote the interaptamer interaction and form more compact structures in the absence of glycine. To observe the effect of the leader–linker interaction on interaptamer complexation directly, we designed the individual aptamers of the VC glycine riboswitch and analyzed the interaptamer binding with native gel assays. We have prepared two different aptamer I constructs, with (VCI132LD, nt −7 to 132) and without the 5′ leader (VCI132, nt 1–132), and one aptamer II construct with the entire linker region (VCII133, nt 133–225). This design prevents self-association of aptamer I through leader–linker interaction by placing the entire linker region on aptamer II and precludes interaptamer duplex formation in P1 regions by avoiding any overlapping bases between aptamers, thereby allowing direct measurement of the possible enhancement of the interaptamer complex formation through leader–linker interaction. We 5′ 32P-labeled VCII133 and incubated it with unlabeled VCI132LD or VCI132 at concentrations ranging from 0 to ∼100 μM under native conditions (10 mM MgCl2, 5 mM glycine). The mixture was then resolved via native gel based on a polyacrylamide coelectrophoresis (PACE) method (Cilley and Williamson 1999; Kwon and Strobel 2008) with radiolabeled VCLD as a size marker (Fig. 4A). VCII133 showed small fractions of probable self-complexation independent of aptamer I concentration. With increasing amounts of VCI132, a new complex with increasing intensity was formed and comigrated with VCLD, suggesting that it is a reconstituted complex between VCII133 and VCI132. The intensity of the reconstituted complex reached 16% with 25 μM VCI132 and dropped abruptly to 9% at ∼100 μM VCI132, presumably due to possible dimerization of VCI132 at such a high concentration. With increasing amounts of VCI132LD, similar complexes with increasing intensity were formed and comigrated with VCLD. Interestingly, on a well-resolved PACE gel, we observed two closely migrating bands for the VCI132LD/VCII133 complex. We reasoned that without covalent linkage between two aptamers, the kink-turn motif formed between leader and linker may assume two different conformations or dock into the rest of the glycine riboswitch in two different geometries. The reconstituted complexes of VCI132LD/VCII133 were formed more readily and reached 52% with 50 μM VCI132LD before dropping abruptly to 44% at 100 μM VCI132LD, similar to that observed for VCI132. Plotting complex formation versus aptamer I concentration (excluding the data at ∼100 μM aptamer I) gives Kd values of 0.66 ± 0.16 μM and ≥6.3 μM (Fig. 4B) for VCI132LD and VCI132, respectively (the Kd for VCI132 is estimated for lack of plateau at high aptamer I concentration). This shows that the leader–linker interaction enhanced the interaptamer interaction by ≥10-fold under native conditions, consistent with the energetic benefit obtained previously.

(A) Native gel image of 32P-labeled VCII133 titrated with aptamer I constructs in 10 mM MgCl2 and 5 mM glycine. (Lane 1) 32P-labeled VCLD; (lane 2) 32P-labeled VCII133; (lanes 3–8) 32P-labeled VCII133 with 0.2–97 μM VCI132; (lanes 9–21) 32P-labeled VCII133 with 0.2–100 μM VCI132LD. (B) Semi-log plots of unnormalized fraction of VCII133 bound versus aptamer I concentration.
Glycine-binding properties of individual aptamers
To better dissect the roles of interaptamer interaction and glycine binding on native glycine riboswitch folding in cis or in trans, we set out to analyze the glycine-binding properties of the individual aptamers with in-line probing experiments. Neither VCI132 nor VCI132LD showed glycine-dependent modulation (data not shown), consistent with previous reports that aptamer I may not be capable of independent glycine binding, at least within the limits of the in-line probing method (Erion and Strobel 2011). In addition to the aforementioned two, and two other aptamer I constructs reported previously (Erion and Strobel 2011), VCI139 (nt 1–139) and VCI150 (nt 1–150), we have also tested VCI145 (nt 1–145), VCI145LD (nt −7 to 145), VCI151 (nt 1–151), VCI151LD (nt −7 to 151), VCI166 (nt 1–166), VCI177 (nt 1–177), and VCI189 (nt 1–189), covering half of aptamer II. In-line probing experiments showed that none of these constructs bind glycine (data not shown), not even with the 5′ leader on some of the constructs (VCI145LD and VCI151LD). These experiments definitively showed that aptamer I does not bind glycine independently. Interestingly, aptamer II construct VCII133 does not bind glycine properly either (Kd of 2.0 ± 0.1 mM) (Table 1), although VCII122 (nt 122–225) binds glycine with a Kd of 4.4 ± 1.9 μM, consistent with previous reports (Mandal et al. 2004; Erion and Strobel 2011). Further analysis showed that other VC aptamer II constructs shorter than VCII122, such as VCII127 (nt 127–225) and VCII130 (nt 130–225) also do not bind glycine (data not shown). An aptamer II construct with blunt-end P1 helix VCII140m1 (nt 140–225, with CC140–141GG and GG224–225CC mutation) binds glycine with a Kd of 1.4 ± 0.2 mM, 300-fold weaker than VCII122, although it has been reported that a VC aptamer II construct with a wobble pair mutation and extended P1 helix binds glycine with a Kd of 3.5 ± 1.5 μM at 20 mM MgCl2, analyzed by ITC experiment (Huang et al. 2010). These results strongly suggest that wild-type VC aptamer II sequences also need part of aptamer I structural elements to bind glycine properly. VCII122 contains 11 nucleotides from aptamer I, and it may happen to mimic the interaptamer interaction needed for aptamer II to bind glycine properly or assume an alternative conformation that allowed proper glycine binding. Further evidence supporting that individual aptamers need structural elements on the other aptamer comes from the FN glycine riboswitch. The corresponding constructs of VCI145, VCI151, and VCII122 on FN glycine riboswitch are FNI84 (nt 1–84), FNI90 (nt 1–90), and FNII61 (nt 61–158), none of which bind glycine in the in-line probing experiments (data not shown).
Leader–linker interaction greatly enhances the glycine-binding affinities of the individual aptamers
Via native gel assays, we have also tried to analyze the complex formation between aptamer I and aptamer II without glycine. However, no complex was visible (without glycine added in the gel) when VCII133 was incubated with either VCI132 or VCI132LD in the absence of glycine (data not shown). The previous section showed that without proper interaptamer interaction, an individual aptamer does not bind glycine properly. This suggests that glycine binding and interaptamer interaction (assisted by leader–linker interaction) have to function synergistically in global glycine riboswitch folding. The PACE gel results showed that saturating glycine helps aptamers interact with each other in trans and form the interaptamer complex. We then sought to investigate whether a saturating concentration of an individual aptamer will enhance glycine binding on the other aptamer and what role the leader–linker interaction might play. We carried out the in-line probing experiment using radiolabeled VCII133 incubated with 15 μM VCI132 in the presence of a range of glycine concentrations. Results showed that VCI132 greatly enhances the glycine binding of VCII133 from 2 mM to a Kd of 191 ± 44 μM (Fig. 5A; Table 1). When 15 μM VCI132LD was added to the in-line probing assay, VCII133 showed glycine binding with a Kd of 7.4 ± 1.0 μM (Fig. 5A; Table 1) enhanced 26-fold further from that with VCI132, restoring the glycine affinity almost completely to that of the full-length tandem riboswitch. This enhancement is slightly better than the ∼12-fold enhancement of VCLD vs. VCIII, suggesting that the leader–linker interaction benefits more in trans, which is consistent with its role in holding the two aptamers together. We also carried out the in-line probing experiment using radiolabeled VCI132 incubated with 15 μM VCII133 in the presence of a range of glycine concentrations. Results showed that VCII133 rescues the glycine binding of VCI132 from completely nondetectable to an apparent Kd of 8.1 ± 2.5 mM (Fig. 5B; Table 1). A similar in-line probing experiment was carried out with radiolabeled VCI132LD as well. Results showed that VCII133 also rescues the glycine binding of VCI132LD from completely nondetectable to a Kd of 190 ± 41 μM (Fig. 5B; Table 1), enhanced ∼40-fold further from that with VCI132. This affinity is substantially smaller than that of VCLD, unlike the glycine-binding affinity of VCII133 rescued by VCI132LD. A possible explanation is that aptamer I requires the interaction between the P3b hairpin of aptamer II and the P1 helix of aptamer I (Kwon and Strobel 2008; Huang et al. 2010; Erion and Strobel 2011), which is right next to the break point in our in trans construct design. As evidenced by the two bands of the reconstituted complexes in native gel assays (Fig. 4A), in this noncovalently linked version, kink-turn leader–linker interaction in the VC glycine riboswitch may not be formed perfectly, leading to imperfect interaction between the P3b hairpin of aptamer II and the P1 helix of aptamer I, and exhibiting only partial rescue of the glycine-binding affinity.

Representative semi-log plots of aptamers rescuing each other in trans in-line probing assays. (A) r7 of VCII133 rescued by 15 μM of aptamer I constructs. (B) r2 of VCI132LD and VCI132 rescued by 15 μM VCII133.
Combining the results of the trans in-line probing assays, we have demonstrated that interaptamer interactions can enhance the glycine-binding affinities of the individual aptamers, while the leader–linker interaction provides an additional 26- to 40-fold enhancement.
Full-length glycine riboswitch does not exhibit glycine-binding cooperativity
We have shown that glycine binding and interaptamer interaction functions synergistically in glycine riboswitch folding through trans assays with native gel and in-line probing experiments. There are two glycine-binding sites and multiple interaptamer interactions, which may or may not take place concurrently. The leader–linker interaction forms in the absence of glycine, as indicated by the constant protection in the linker region in the in-line probing assay and the lack of glycine perturbation in the HRFP experiments of the full-length glycine riboswitches. This seems to suggest that not all interactions are formed at the same time. One specific question is whether the two glycine molecules bind cooperatively. One crude measure is to use Hill coefficients of the glycine binding in the double aptamer riboswitches. Indeed, our results showed that the truncated VC and FN glycine riboswitches have Hill coefficient values larger than one, exhibiting reasonable apparent cooperativity, although smaller than reported (Mandal et al. 2004; Kwon and Strobel 2008). Two linker mutants, VCLDLN and VCIIILN, which contain no leader–linker interaction, showed Hill coefficient values larger than one as well, exhibiting similar cooperativity to that of the truncated construct, VCIII. However, all three full-length glycine riboswitches showed Hill coefficient mean values less than or close to one, and the upper limits of these values are all close to one, exhibiting no glycine-binding cooperativity. We recognize that Hill coefficient values are extremely sensitive to the accidental error of the data points, and therefore may not be the definitive measure for glycine-binding cooperativity. The unequivocal evidence of the glycine-binding cooperativity of the truncated construct VCIII is from mutation analysis. Breaker and coworkers reported that deleterious mutations in each aptamer greatly reduced the glycine-binding affinities of the other aptamer, demonstrating interaptamer inhibition and cooperativity between two glycine-binding sites (Mandal et al. 2004). Our results showed that VCIIIG17C (nt 1–225, with G17C mutation) not only completely abolished glycine binding in the aptamer I regions, but also reduced the aptamer II glycine-binding affinity to 1.1 ± 0.1 mM, while VCIIIG146C binds glycine with a Kd of 1.9 ± 0.3 mM in both aptamers, comparable to the previous reports (Mandal et al. 2004; Erion and Strobel 2011). These experiments are consistent with the cooperative binding in the two glycine-binding sites of the truncated glycine riboswitch VCIII. To assess the glycine-binding cooperativity of VCLD, we therefore constructed VCLDG17C (nt −7 to 225, with G17C mutation) and carried out in-line probing experiments. Results showed that most of the aptamer I regions modulated in VCIII and VCLD do not show glycine modulation in VCLDG17C, such as nt 63–64, 70–72, 74 (r2), 78–80, and 83 (Fig. 6A). However, much to our surprise, unlike VCIIIG17C, the aptamer II regions including nt 146 (r5), 170–172 (r6), 177–178 (r7), and 207–208 showed strong glycine modulation with a Kd of 2.8 ± 0.4 μM, retaining full glycine-binding capacity of VCLD in these regions (Figs. 6A,C; Table 1). Interestingly, A121–G123 (r3) is also perturbed with a similar glycine-binding affinity, consistent with our finding that aptamer II needs structural elements from aptamer I to bind glycine. We have also mutated aptamer II and constructed VCLDG146C (nt −7 to 225, with G146C mutation). In-line probing experiments showed that most of the aptamer II regions modulated in VCIII and VCLD do not show glycine modulation in VCLDG146C, such as nt 170–172 (r6), 177–178 (r7), and 207–208 (Fig. 6B). However, again unexpectedly, unlike VCIIIG146C, the aptamer I regions including nt 70–72, 74 (r2), 78–80, and 121–123 (r3) showed great glycine modulation with a Kd of 12 ± 2 μM, retaining most of the glycine-binding capability of VCLD in these regions (Fig. 6B,D; Table 1). Interestingly, nt 146 is also perturbed with a similar glycine-binding affinity, consistent with our finding that aptamer I needs structural elements from aptamer II to bind glycine. The in-line probing results of VCLDG17C and VCLDG146C showed that an individual glycine-binding site may retain its full or majority binding capacity, even though the other site is not occupied, abolishing interaptamer inhibition observed in VCIII, and strongly suggesting that VCLD does not bind two glycine molecules cooperatively.

(A,B) The in-line probing gel images of VCLDG17C and VCLDG146C, respectively. NR, T1, and OH represent no reaction, partial digestion with RNase T1, and partial digestion with alkali, respectively. r2–r7 indicate regions defined in Table 2. (C,D) The representative semi-log plots of the normalized fraction of RNA bound versus glycine concentration for VCLDG17C (r3, r5, and r6) and VCLDG146C (r2, r3, and r5), respectively.
DISCUSSION
Prompted by the different protection patterns in the linker regions of two different truncated glycine riboswitches (VCIII and VCIIIs), we have identified a leader–linker interaction involving nucleotides upstream of the current published consensus glycine riboswitch sequences. This leader–linker interaction forms a P0 helix with 3–7 bp and is greatly conserved (>90%) in tandem glycine riboswitches. In more than half of the glycine riboswitches, the P0 helix is connected to the P1 helix through a kink-turn motif. These kink-turn motifs fit largely to the consensus kink-turns derived from the ribosome kink-turns, except that the base pair adjacent to the internal loop has a strong preference for G–C, just the opposite to the C–G base-pair preference in the ribosome kink-turns. The kink-turn motif is not unprecedented in riboswitches; it has been reported in SAM-I riboswitches connecting to the four-way junction motif (Montange and Batey 2006) as well as in the class II c-di-GMP (Lee et al. 2010) and lysine riboswitches (Blouin and Lafontaine 2007). This SAM-I kink-turn motif follows the exact consensus derived from the ribosome kink-turns and is stabilized by RNA tertiary structure (Schroeder et al. 2011), different from those stabilized by metal ions (Liu and Lilley 2007) or protein binding (Turner et al. 2005; Turner and Lilley 2008). It will be interesting to investigate which strategy is used in the glycine riboswitch to stabilize the kink-turn motif located at the 5′ boundary of the regulatory RNA element.
We have shown that full-length glycine riboswitches bound glycine 4.5- to 86-fold tighter than the truncated versions under saturating magnesium concentrations (Lipfert et al. 2007). Interestingly, three full-length glycine riboswitches, VCLD, FNLD, and BSLD, showed glycine-binding affinities very similar to each other, unlike their truncated versions, raising the question of whether glycine riboswitches have convergently evolved to bind glycine at a cellularly specific value for optimum metabolism.
We have demonstrated direct binding between two aptamers in trans without any overlapping bases using native gel assays. The leader–linker interaction enhanced the interaptamer interaction by at least 10-fold with the riboswitches saturated with glycine. As interaptamer complexation cannot be detected by native gel assay in the absence of glycine, these results suggest that glycine binding enhances the interaptamer interaction. Conversely, aptamers also greatly enhanced the glycine-binding ability of each other with the leader–linker interaction contributing an additional 26- to 40-fold improvement. These data suggest that interaptamer interaction and glycine binding complement each other. One prominent property of the truncated glycine riboswitches is their glycine-binding cooperativity. Surprisingly, the Hill coefficients of the wild-type full-length glycine riboswitches, and more definitively, the mutational analysis with VCLDG17C and VCLDG146C, showed that there is no ligand-binding cooperativity between two glycine-binding sites in the full-length glycine riboswitches.
Together, our results support a new global folding model of the tandem glycine riboswitches. Independent of glycine, the leader–linker interaction can be formed in the presence of magnesium ions and leads to a more compact structure that enhances glycine binding and other interaptamer interactions, such as the α-α′, β-β′, and γ-γ′ interaptamer interactions reported previously (Huang et al. 2010; Butler et al. 2011; Erion and Strobel 2011). The glycine binding at either site acts synergistically with interaptamer interactions to achieve global folding of the tandem glycine riboswitch, while the two glycine-binding sites do not operate cooperatively.
Our biochemical results focused on the VC glycine riboswitch and strongly supported the lack of glycine-binding cooperativity in the full-length VC glycine riboswitch. Although we cannot rule out possible ligand-binding cooperativity in other full-length glycine riboswitches, since the truncated construct VCIII is the most cooperative glycine riboswitch reported, our finding is likely to be general. The lack of glycine-binding cooperativity in the full-length glycine riboswitches may have been manifested in the great variability of the reported cooperativity values with Hill coefficients ranging from 1.1 to 1.6 (Mandal et al. 2004; Lipfert et al. 2007; Kwon and Strobel 2008; Tripp et al. 2009; Erion and Strobel 2011).
One interesting question remains: Without the advantage of cooperative and more “digital” response to the cellular glycine concentration, why do glycine-sensing RNAs evolve into the tandem arrangement? One possibility is that as our data pointed out, glycine sensing may not be achieved efficiently with a single aptamer, given glycine is the smallest organic molecule sensed by RNAs. Individual aptamers need structural elements from other aptamers to achieve efficient glycine binding; the only reported functional wild-type single aptamer construct, VCII122, uses part of the aptamer I sequence to mimic an efficient glycine-binding strategy. Another possibility is that through tandem arrangement, the glycine riboswitch uses the leader–linker interaction to promote the interaptamer interactions and preorganize the glycine-binding pockets. Three experimental results support the more organized and compacted structure formation in the absence of glycine: (1) Protection of the regions important for interaptamer interaction or the glycine-binding pocket is independent of glycine in HRFP of the full-length glycine riboswitches (Supplemental Figs. S6–S8); (2) the longer VCLD runs slightly faster than VCIII in the absence of glycine on native gel, while running comparably or slightly slower than VCIII in the presence of glycine (Supplemental Fig. S9); (3) two antibody fragments (Fabs) selected against VCIII (Ravindran et al. 2011) bind VCLD with similar affinities, but higher percentage binding, at saturating Fab concentrations, presumably because VCLD is less flexible and retained better during the washing step in the filter-binding assay (data not shown). The more compact and organized structure of full-length glycine riboswitches may have advantages in glycine sensing under physiological conditions in vivo if fast on rate is desired, similar to other riboswitches under kinetic control (Wickiser et al. 2005; Smith and Strobel 2011; Trausch et al. 2011).
MATERIALS AND METHODS
Preparation of the RNAs
The glycine riboswitches VCIII, FNIII, BSIII, and single aptamers were generated by annealing DNA oligonucleotides (Integrated DNA technologies) and amplified by PCR with an EcoRI restriction site and T7 promoter sequence engineered at the 5′ terminus, and EarI and HindIII sites engineered at the 3′ terminus. Double-digested riboswitch DNA inserts and pUC19 vector were ligated together to generate glycine riboswitch plasmids. The full-length glycine riboswitches and mutants were constructed by a similar method or QuikChange site-directed mutagenesis (Stratagene) from the corresponding wild-type plasmids and proper DNA oligonucleotide primers. The plasmids were transformed into JM109 cells, amplified and purified with a Qiagen plasmid preparation kit, and their sequences confirmed by sequencing (Genewiz Inc). The resulting plasmids were then linearized with EarI and transcribed by T7 RNA polymerase. The RNA transcripts were purified by 6%–8% denaturing PAGE, electroeluted, PCA extracted, ethanol precipitated, and redissolved in TE buffer.
In-line probing assay and quantification
RNA transcripts were dephosphorylated with Shrimp alkaline phosphatase (Fermentas Life Sciences) and 5′-32P-labeled with [γ32P]ATP (Perkin Elmer) and T4 polynucleotide kinase (New England Biolabs). For transcripts with the 5′ end buried by or too close to the duplex region, to improve the labeling yield, the transcript was first annealed to a short DNA oligonucleotide to free the 5′ end before dephosphorylation and kinase reactions. In-line probing assays were carried out similar to the reported procedure (Soukup and Breaker 1999). Briefly, ∼50 kcpm 32P-labeled RNA transcript was incubated at 25°C for 48 h in 50 mM Tris-HCl (pH 8.3), 20 mM MgCl2, 100 mM Kcl, under various glycine concentrations. For the trans in-line probing assay, 15 μM unlabeled proper aptamer was also added to the reaction. Spontaneously cleaved RNA fragments were resolved by 8% denaturing PAGE, dried, exposed to PhosphorImager screens, and quantified by ImageQuant (Molecular Dynamics). For VC glycine riboswitches and mutants, the following regions are quantified if contained on the constructs and well resolved: U74 (r2), A121–G123 (r3), G136–G137 (r4), G146 (r5), G170–A172 (r6), C177–A178 (r7), and U207–C208. To control for loading differences, the band intensities were normalized to nonvariable bands (U94 and U156–U161). For FN glycine riboswitches, the following regions are quantified if contained on the constructs and well resolved: G35, U46 (r2), G61 (r3), G85 (r5), G110–A112 (r6), and C117–A119 (r7). Band intensities were normalized to nonvariable bands (U23 and C90). For BSLD and BSIII, the following regions are quantified: G60, U83 (r2), C88–A89, A151–A153 (r6), and A158–G160 (r7). Band intensities were normalized to nonvariable bands (G136–U138). The Hill coefficients and dissociation constants were obtained by fitting each plot using the following equation in KaleidaGraph software: fraction bound = m1 × [gly]n/(Kdn + [gly]n) + m2.
Hydroxyl radical footprinting
For footprinting, 5′ end-labeled RNA were dissolved in 10 mM Na-MOPS (pH 7.0). A total of 1 μL of this RNA solution (∼100 kcpm) was incubated with 1 μL of 500 mM Na-MOPS (pH 7), 1 μL of 100 mM MgCl2, 1 μL of 10x stock glycine solution at various concentrations, and 5 μL of water for 20 min at 50°C. A 10x footprinting reagent was prepared separately that contained 1 mM Fe(NH4)2(SO4)2, 1.25 mM EDTA (pH 8), and 60 mM sodium ascorbate. To each sample, 1 μL of this reagent was added to initiate the footprinting reaction. The reaction proceeded for 1 h at room temperature and was quenched by the addition of 5 μL of thiourea stop solution (9 M urea, 300 mM thiourea, 0.1% each xylene cyanol and bromophenol blue). The footprinting reactions were fractionated with 8% denaturing PAGE. The gels were dried, exposed to PhosphorImager screens, and quantified by ImageQuant (Molecular Dynamics). For VCIII, the protections around G146 and G123 are quantified and normalized to nonvariable band C108. For FNIII, the protections around G85–A86 and U65–C66 are quantified and normalized to nonvariable band G72. For BSIII, the protection around C88 is quantified and normalized to nonvariable band G75. The Hill coefficient and dissociation constant were obtained by fitting each plot using the following equation in KaleidaGraph software: fraction bound = m1 × [gly]n/(Kdn + [gly]n) + m2.
Native gel analysis of interaptamer complexation
Native gel electrophoresis was carried out similar to the procedure described previously (Cilley and Williamson 1999; Erion and Strobel 2011). A total of 1 μL of 5′ end-labeled VCII133 (∼5 kcpm) was incubated with 1 μL of 2x incubation buffer (180 mM Tris-borate at pH 8.3, 10 mM glycine, 20 mM MgCl2) for 20 min at room temperature. In a separate Eppendorf tube, 1 μL of 4x stock unlabeled VCI132 (or VCI132LD) solution at various concentrations was incubated with 1 μL of 2x incubation buffer for 20 min at room temperature. The solution was combined together and incubated for an additional 20 min at room temperature. After homogenizing with 1 uL of 5x native loading dye (50% glycerol, 0.1% xylene cyanol), the reaction mixture was resolved via a 6% native PAGE in TB buffer containing 10 mM glycine and 5 mM glycine. Electrophoresis was carried out with TB buffer containing 10 mM MgCl2 at 4°C for 4 h under ∼12.5 V/per cm of gel height. The gels were dried, exposed to PhosphorImager screens, and quantified by ImageQuant (Molecular Dynamics). The dissociation constant was obtained by fitting each plot using the following equation in KaleidaGraph software: fraction bound = m1 × [RNA]/(Kd + [RNA]) + m2.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
NOTE ADDED IN PROOF
R. Das and colleagues (Kladwang et al. 2012) recently report computational and biochemical evidence for an interaction between the 5′ leader and internal linker in glycine riboswitches from Fusobacterium nucleatum.
ACKNOWLEDGMENTS
We thank Mohammed Elsayed, Bryan Brinda, Waise Quarni, Cody Ott, Aditi Ramcharitar, Wei Zhang, Sean Holmes, and Yassmeen Abdel-Aty for making some of the constructs, and the Ye lab members, especially Breena Stoner, for helpful discussions. We also thank Dr. Piccirilli (University of Chicago) for imaging supplies. E.S. dedicates this work to the memory of D.C. Avrett. This work is supported by the start-up fund provided to J-D.Y. from the University of Central Florida.
Footnotes
-
↵1 Corresponding author.
E-mail yejingdong{at}gmail.com.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.031286.111.
- Received November 9, 2011.
- Accepted December 14, 2011.
- Copyright © 2012 RNA Society








