
Folding of the hammerhead ribozyme: Pyrrolo-cytosine fluorescence separates core folding from global folding and reveals a pH-dependent conformational change
- Department of Microbiology and Molecular Genetics, University of Vermont, Burlington, Vermont 05405, USA
Abstract
The catalytic activity of the hammerhead ribozyme is limited by its ability to fold into the native tertiary structure. Analysis of folding has been hampered by a lack of assays that can independently monitor the environment of nucleobases throughout the ribozyme–substrate complex in real time. Here, we report the development and application of a new folding assay in which we use pyrrolo-cytosine (pyC) fluorescence to (1) probe active-site formation, (2) examine the ability of peripheral ribozyme domains to support native folding, (3) identify a pH-dependent conformational change within the ribozyme, and (4) explore its influence on the equilibrium between the folded and unfolded core of the hammerhead ribozyme. We conclude that the natural ribozyme folds in two distinct noncooperative steps and the pH-dependent correlation between core folding and activity is linked to formation of the G8-C3 base pair.
Keywords
INTRODUCTION
The hammerhead ribozyme is a small, self-cleaving RNA motif, first discovered in viroids and satellite RNAs of plant viruses (Prody et al. 1986; Forster and Symons 1987). It catalyzes a specific phosphodiester bond isomerization reaction in the course of the rolling-circle replication of these respective RNA species and has been implicated in RNA processing reactions in vertebrate satellite RNA and mRNA (Chartrand et al. 1995; Ferbeyre et al. 1998). The minimal hammerhead may be the simplest self-processing RNA and consists of a conserved core of about 15 mostly invariant core residues at a three-helix junction (Ruffner et al. 1990). However, optimal activity requires the presence of peripheral sequences that interact via tertiary contacts (De la Pena et al. 2003; Khvorova et al. 2003). In the crystal structures of the Schistosoma mansoni hammerhead ribozyme, an interaction between a peripheral Stem II loop and a Stem I bulge is proposed to lead to structural organization of the catalytic core (Martick and Scott 2006). Additionally, structures obtained in the presence of manganese ions, together with molecular dynamics simulations, have provided insights into the location and potential functions of divalent metal ions and solvent in the hammerhead ribozyme catalysis (Lee et al. 2008; Martick et al. 2008).
Fluorescence resonance energy transfer (FRET) (Penedo et al. 2004) and electron paramagnetic resonance (EPR) (Kim et al. 2005) studies have shown that global folding occurs at much lower magnesium ion (Mg2+) concentrations in the extended construct compared with the minimal ribozyme. Peripheral loops act as auxiliary elements that support ribozyme function, presumably by promoting global folding under physiological conditions, as indicated by an apparent single-step folding pathway (Penedo et al. 2004). The minimal ribozyme folds in two distinct steps, as demonstrated by various studies (Hammann and Lilley 2002; Hampel and Burke 2003). Recent work performed with time-resolved FRET (tr-FRET) and single-molecule FRET (sm-FRET) techniques to monitor the global structure and dynamics of three hammerheads based on the Avocado Sunblotch Viroid sequence invoke a linkage between RNA structural dynamics and function. This work suggests that loop–loop interactions in extended hammerhead ribozymes and Mg2+ ions that bind to minimal ribozymes may generally allow more frequent access to a catalytically relevant conformation(s), rather than simply locking the ribozyme into a single active state (McDowell et al. 2010). Strikingly, both minimal and natural hammerhead ribozyme catalysis requires significantly higher Mg2+ concentrations than those required for tertiary folding (Rueda et al. 2003). The identity of the divalent metal ion has little effect on global folding, whereas it has a very large effect on the cleavage kinetics (Roychowdhury-Saha and Burke 2006; Boots et al. 2008).
Folding and catalytic activity are related at several levels in these RNA species. To date, elucidating of the folding pathways has been hampered by the lack of assays that can independently monitor the environment of nucleobases throughout the ribozyme–substrate complex in real time without structural ambiguity. Here, we describe a fast kinetic folding analysis approach using the highly sensitive fluorescent cytosine analog pyrrolo-cytosine (pyC) (Berry et al. 2004). pyC forms a Watson-Crick-like base pair with guanosine and was shown to maintain all structural and thermodynamic features of a standard guanosine–cytosine base pair (Dash et al. 2004; Thompson and Miyake 2005; Tinsley and Walter 2006; Zhang and Wadkins 2009). Its incorporation at multiple positions in the extended hammerhead ribozyme was previously shown not to interfere with the function of the ribozyme (Lambert et al. 2006). The combination of mutational substitution, kinetics, and pyC folding analysis has allowed us to experimentally access multiple stages of hammerhead ribozyme folding, to determine the pH dependence of folding, and to establish how G8-C3/C17 equilibrium may be functionally linked to the core folding event.
RESULTS
Distinct folding of catalytic core and auxiliary structural elements
Using a native hammerhead ribozyme motif derived from S. mansoni sequences (Fig. 1A), folding was analyzed by stopped-flow fluorescence spectroscopy. Ribozyme–substrate complexes in which the fluorophore pyC (Fig. 1B) was introduced at two positions within the core (pyC3 and pyC7, respectively), one at the base of Stem I (pyC1.1) and one in a peripheral loop (pyC1.9) (Fig. 1A), were examined. These substitutions had no significant effect on the catalytic activity of the complex at any pH studied or variant used (Supplemental Fig. S1A–D). The secondary structure formation of the ground-state ribozyme–substrate complex in the presence and absence of pyC was justified by use of hydroxyl footprinting mapping as previously established and described (Hampel and Burke 2003; Lambert et al. 2006). Furthermore, mobility-shift gel assays were conducted to test the homogeneity of ribozyme–substrate complexes carrying the fluorophore at different positions, as well as for complexes carrying point mutations. The yield and the stability of the binary complex were monitored in the absence and presence of Mg2+ and at all studied pHs (Supplemental Fig. S4).
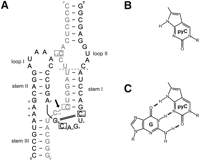
(A) Secondary structure of the Schistosoma mansoni hammerhead ribozyme construct used in this study. This representation emphasizes the global geometry as seen in the first crystal structure of the S. mansoni ribozyme (Martick and Scott 2006). Enzyme strand (black); the substrate (gray); and positions of nucleotides replaced with the fluorescent analog (boxed). Outlined letters (C17, G8, and G12) indicate core nucleotides essential for catalysis, and the cleavage site is marked with a black arrow; the canonical 8-3 Watson-Crick base pair is shown as a black double line. Thick black and gray lines indicate backbone continuity, where the sequence has been separated for diagrammatic clarity. Base numbering is according to Hertel (Hertel et al. 1992). The dotted line in the region of Loop 1 shows the truncation position of the substrate and ribozyme strands to form the minimal form of the ribozyme lacking the tertiary interaction. (B) Structure of pyrrolo-cytosine (pyC) and (C) pyrrolo-cytosine–guanosine base pair (pyC-G).
Upon induction of folding by mixing with Mg2+ (final concentrations 0.05–100 mM), significant fluorescence changes were observed in all cases. For pyC3, pyC1.1, and pyC1.9, we observed a decrease of fluorescence (pyC3: F/F0 = 0.92; pyC1.1: F/F0 = 0.82; pyC1.9: F/F0 = 0.58), and for pyC7, we observed an increase in fluorescence of F/F0 equal to 1.43 (Fig. 2A).

Folding of S. mansoni hammerhead ribozyme as observed by pyC fluorescence changes in stopped-flow fluorimeter. (A) Time courses of hammerhead ribozyme–substrate (noncleavable) complex folding upon exposure to 10 mM Mg2+, in the presence of 50 mM Tris-HCl and 100 mM NaCl (pH 7.0) at 25°C, as reported at positions in the Loop II (pyC1.9), Stem I (pyC1.1), and core region (pyC3 and pyC7). The displayed curves are averages of at least four measurements. The smooth lines represent the fit (see Materials and Methods). (B) kobs (per minute) for folding plotted as a function of Mg2+ concentration. The data have been fitted to a two-state binding model yielding a Hill coefficient equal ∼1 for all analyzed variants and an apparent dissociation constant [Mg2+]1/2: 0.12 (±0.01) mM for pyC1.9 (black diamonds); 0.14 (±0.03) mM for pyC1.1 (open diamonds); 0.8 (±0.2) mM for pyC3 (open circles); and 1.0 (±0.03) mM for pyC7 (black circles). (C) An example of time courses of folding monitored in the core of the ribozyme at position pyC7 with increasing Mg2+ concentration. Please note earlier amplitude than observed rate constant saturation with increasing Mg2+ concentration. (D) Comparison of Mg2+ dependence of the apparent kobs for pyC1.9 and pyC7 (black diamonds and circles) with Mg2+ dependence of their relative fluorescence amplitudes (gray diamonds and circles, respectively). (Left y-axis) kobs; (right y-axis) normalized relative amplitudes. The fluorescence amplitudes were normalized so that the maximal fluorescence amplitude observed in each experiment was valued at 1. The apparent dissociation constant established from changing fluorescence amplitudes was [Mg2+]1/2: 0.09 (±0.01) mM for pyC1.9 (gray diamonds) and 0.11 (±0.03) mM for pyC7 (gray circles).
The strong fluorescence decrease for pyC1.9, located in the loop region, indicates that in the absence of Mg2+, the loops must be in a different arrangement from in the fully folded state. The conformational change leading to the loop–loop interaction leads to either exposure of the pyC residue to the solvent or brings the C1.9 close to guanosine residues, which were shown to quench pyC (Hardman et al. 2008). Indeed, in the crystal structure of the native hammerhead (Supplemental Fig. S2A), C1.9 is located in the groove open to solvent and is forming close interactions with guanosine B1 (Martick and Scott 2006). The photo-induced electron transfer from proximal guanosines to pyC appears to induce much stronger quenching than exposure to solvent and reactive oxygen species (to be published elsewhere). Similar effects were observed for 2-aminopurine or fluorescein incorporated into RNA or DNA (Walter and Burke 1997; Kelley and Barton 1999).
The fluorescence decrease for the core residue pyC3 and pyC1.1, as well as the fluorescence increase for position pyC7, agrees well with the residues transferring from ground to active conformation as represented by the crystal structures of the minimal and natural ribozymes. The structure of the minimal ribozyme is generally assumed to be close to the ground state (Scott et al. 1996), and the S. mansoni ribozyme is assumed to be close to the active state (Martick and Scott 2006). In the ground state, C3 forms one hydrogen bond with C17, which is stabilized by stacking interactions, while in the active state, C3 forms a Watson-Crick base pair with G8. Since it has previously been shown that base-pairing of pyC with guanosine strongly lowers its quantum yield (Tinsley and Walter 2006), the observed decrease indicates a transition from ground to active state. For the C1.1, the base-pairing with G2.1 occurs in both ground and active states; therefore, the expected overall amplitude of signal change is small and depends only on the transition between stacking interactions with C3 in the ground state to the stacking interaction with the G8-C3 base pair in the active state. Congruently, the ground-state C7 is engaged in multiple interactions and strong base-stacking with G8 inside the ground-state core of the hammerhead ribozyme, whereas in the active state, C7 makes only a loose stacking interaction with C3 and is primarily exposed to the solvent. Hence, as anticipated for the ground-to-active-state transition, pyC7 responds with a strong fluorescence increase.
At moderate Mg2+ concentrations (<20 mM), time courses of folding were accurately fit to a single-exponential rate equation with a maximal observed rate of kobs = 2.7 × 103 min−1 at pH 7.0 (Fig. 2A). At higher Mg2+ concentrations (>20 mM), the folding appeared to be biphasic, and only the faster, dominant apparent rate of folding was taken into consideration.
When fluorophores in one of the peripheral loops (pyC1.9) and at the junction of Stem I and the core (pyC1.1) were monitored, folding was well described by a two-state model induced by non-cooperative Mg2+ binding with a Hill coefficient of ∼1 and an apparent dissociation constant of [Mg2+]1/2 = 0.12 (±0.01) mM and 0.14 (±0.03) mM for pyC1.9 and pyC1.1, respectively. The maximum velocity at saturated Mg2+ concentrations reached kobs = 2.9 × 103 min−1 (Fig. 2B). In contrast, analysis of complexes with fluorophores within the core (pyC3 and pyC7) showed an order-of-magnitude-higher concentration of Mg2+ ([Mg2+]1/2 = 1.0 [± 0.1 mM]) to be required for the reaction to reach saturation and the same maximum velocity of kobs = 2.7 × 103 min−1 (Fig. 2B). Together, these observations suggest that the stem regions closed by peripheral loops and the core fold independently of one another, reaching the same velocity only at saturating magnesium ion concentrations. Equivalent velocities of global versus core folding at saturated Mg2+ concentrations indicate that overall global folding, if inhibited, might become the rate-limiting step.
The equilibrium Mg2+-induced folding of the ribozyme–substrate complex was analyzed not only by the dependence of the observed rate constants on Mg2+ concentration, but also by the dependence of the relative amplitudes of the fluorescence change on the concentration of Mg2+. The fluorescence amplitude and associated kinetic rate changes with Mg2+ concentration for Stem I (pyC1.1) and Loop II (pyC1.9) showed an exact correlation (Fig. 2D). Again, there was no apparent cooperativity in binding, and fitting yielded a [Mg2+]1/2 = 0.08 (±0.01) mM, suggesting a two-state global folding model for the hammerhead ribozyme. Interestingly, the fluorescence amplitude changes as a function of Mg2+ concentration for core position pyC3 (data not shown) and pyC7 (Fig. 2C,D) did not correlate with Mg2+-dependent changes of the related rate constants of folding. The increase in the amplitude of fluorescence change reached saturation at ∼1 mM Mg2+; however, the rate of the fluorescence change kept increasing and saturated first at ∼10 mM Mg2+ (Fig. 2C,D). The dependence of fluorescence amplitude versus Mg2+ concentration for pyC3 and pyC7 was one order of magnitude lower ([Mg2+]1/2 = 0.12 mM [±0.03]) than that observed for their relative rates of folding and similar to that acquired for pyC1.1 and pyC1.9, assigned as global folding. This suggests that the presence of the loop–loop interaction does not influence core folding, but rather increases the fraction of the molecules able to fold the core into a transiently kinetic intermediate.
pH-dependent conformational change
The cleavage activity of the naturally occurring hammerhead ribozyme shows a log-linear pH dependence in the presence of 10 mM Mg2+ (Canny et al. 2004). A similar pH dependence has been previously observed for the minimal hammerhead (Clouet-D'Orval and Uhlenbeck 1996). However, in the presence of submillimolar concentrations of Mg2+, cleavage rates deviate from log-linear above pH 7.5, suggesting a pH-dependent conformational change, resulting in less efficient cleavage at high pH (Canny et al. 2004). To test this possibility, we measured the pH dependence of folding using hammerheads with pyC at positions in both the core and auxiliary elements. Fluorescence of pyC was previously shown to be insensitive to pH over the broad range used, pH 5.0–9.0 (Tinsley and Walter 2006). Examination of complexes with fluorophores in the peripheral loop (pyC1.9) and Stem I (pyC1.1) showed no significant change in folding rate as a function of pH (Fig. 3). In contrast, at the same Mg2+ concentration of 10 mM, complexes with fluorophores in the catalytic core (pyC3 and pyC7) showed a bell-shaped curve, with a 10-fold increase in the folding rate from pH 5.5 to 7.0, followed by a sharp decrease in kobs between pH 7.0 and 8.5 (Fig. 3). The rates of core folding at higher pH approached the rates of cleavage, indicating that core folding might become the rate-limiting step under these conditions. Fitting the data in Figure 3 to the Henderson–Hasselbalch equation yielded two apparent pKa values of 6.3 (±0.2) and 7.4 (±0.1), consistent with a model in which two protonation–deprotonation events take place within or adjacent to the active site. Furthermore, they suggest that the two pKa values observed in activity-based studies (Han and Burke 2005) may, at least in part, coincide with active-site conformational changes.
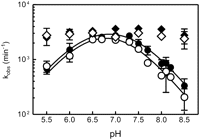
pH dependence of fluorescence-detected folding of the S. mansoni hammerhead ribozyme. The folding experiments at different pH values were performed in a stopped-flow fluorimeter by addition of Mg2+ (10 mM) to a pre-annealed complex of ribozyme and noncleavable substrate (1 μM). The folding reactions were carried out at 25°C in the presence of 100 mM NaCl and 50 mM MES (pH 5–6), MOPS (pH 6.5–7.5), HEPES (pH 7.0–8.0), TRIS (pH 7.5–8.5), TAPS (pH 8.2–8.9), and CHES (pH 8.5–9.0). The folding traces were fitted as described in Materials and Methods. Extracted, log apparent rate constants kobs (per minute) were plotted as a function of pH. The differences in the apparent rate of folding for loop pyC1.9 (black diamonds) and Stem I pyC1.1 (open diamonds) at different pH values did not exceed 10%. The apparent rate of folding for core residues pyC7 (black circles) and pyC3 (open circles) shows at least a one order of magnitude lower rate of folding at high and low pHs as compared with pH 7.0. The apparent bell-shaped curve could be fitted to a Henderson–Hasselbalch-type equation (see Materials and Methods) yielding two apparent pKa values of 6.3 (±0.2) and 7.4 (±0.1).
pH-dependent conformational change—mutational analysis
The pH-induced folding changes at positions C7 and C3 are expected to be dependent on the secondary and tertiary structure of the core. Comparison of the ground-state and the active-state hammerhead crystal structures indicates that both residues change their position during the conformational transition and in the active state both stack beneath each other (Scott et al. 1996; Martick and Scott 2006). Hence, it is reasonable to predict that fluorescence changes of both pyC7 and pyC3 might monitor the same pH-dependent folding event within the core, leading to the active-site formation. The equilibrium of the transition between both states of hammerhead folding was shown to be affected by G8-C3 base-pair formation (Przybilski and Hammann 2007a,b; Nelson and Uhlenbeck 2008). Since our earlier mutational analysis of G8 demonstrated effects both on cleavage activity and the pH profile of the reaction (Han and Burke 2005), we substituted the G8-C3 base pair with pKa-altering nucleobase analogs and examined simultaneously the pH dependence of core folding and cleavage activity.
Substitution of G8 with inosine (I8) (Fig. 4A) would be expected to result in a base pair with C3 in Watson-Crick geometry, but with only two hydrogen bonds. As shown in Figure 4B, the pH-dependent folding of the I8-C3 core appears quite similar to that of G8-C3; however, at higher pH values, the maximum velocity of folding is slower, and the decrease with pH is steeper than for G8-C3 (Table 3, below). The rate of cleavage for both G8-C3 and I8-C3 increases with pH in a log-linear manner and approaches 214 min−1 at pH 8.5 for G8-C3, while I8-C3 levels off at pH 7.5–8.0 with a maximum velocity of only ∼50 min−1 (Fig. 4C; Table 1). Fitting the cleavage data to a Henderson–Hasselbalch equation yields an apparent pKa of 8.4 (±0.1) for the G8-C3 and a slightly lower value of 8.0 (±0.2) for I8-C3 (Table 3, below). Although the difference between the pKas is small, these results appear to be consistent with the direction of the pKa difference between the two nucleobases in solution (9.4 for G and 8.5 for I, respectively) (Brown 1971).
Biochemical and fluorescence characterization of S. mansoni hammerhead ribozyme and G8-C3 variants
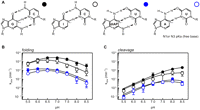
Comparison of pH dependence of cleavage and folding for the S. mansoni hammerhead ribozyme and its G8-C3 base pair Watson-Crick analogs. (A) Structure of the G8-C3 Watson-Crick base pair (black circle) and analog pairs of I8-C3 (open, black circles), diAP8-U3 (blue circles), and A8-U3 (open, blue circles). The respective pKa values of N1 of purines and N3 of pyrimidines are highlighted. (B) pH-dependent folding as monitored by fluorescence change of core pyC7 of G8-C3 and analog base pair variants. The folding data were acquired, fitted, and plotted as described for G8-C3 folding in Figure 3 and Materials and Methods. Each of the analyzed variants yielded two apparent pKas of folding: 6.1 (±0.3) and 6.8 (±0.2) for I8-C3; 5.5 (±0.1) and 7.1 (±0.2) for diAP8-U3; 5.9 (±0.1) and 6.9 (±0.1) for A8-U3 as compared with 6.3 (±0.2) and 7.4 (±0.1) for G8-C3. (C) pH-dependent cleavage assays were performed under single turnover conditions with the same buffer, salt, and Mg2+ concentrations as described for folding experiments in Figure 3. The resulting apparent rate constants for cleavage were plotted as a function of pH and fitted to the Henderson–Hasselbalch equation yielding one apparent pKa value of 8.4 (±0.2) for G8-C3 and 8.0 (±0.2) for I8-C3. For diAP8-U3 and A8-U3, the pH dependence of cleavage resembles a bell-shaped profile, and curve fitting yielded two pKas. The best fit was obtained when the second pKa was constrained to >8.5 and then the fit yielded the upright pKa of 7.5 (±0.3) for diAP8-U3, 7.0 (±0.2) for A8-U3. For the clarity of the graphs, we have displayed only every 0.5 pH unit datum. For the very low and high pH conditions, the cleavage and folding were performed in smaller increments than displayed. (Instead of 0.5 pH unit, 0.2 increments were tested, starting at pH 5 and ending at pH 9, respectively. pH 9 and 5 were not included in the fit.) The values are summarized in Tables 1–3.
To further probe the folding versus activity dependence of the G8-C3 base pair on pH, we examined two additional base pairs with reversed pKa: low on purine and high on pyrimidine. We decided on pairs of adenosine (A, pKa 4.0) and 2,6-diaminopurine (diAP, pKa 5.1) combined with uracil (U, pKa 9.6) (Fig. 4A). The diAP-U base pair forms three hydrogen bonds in a base pair analogous to the Watson-Crick G-C (Kyogoku et al. 1967; Jorgensen and Pranata 1990), and A-U is structurally and thermodynamically analogous to the Watson-Crick base pair of I-C, containing two hydrogen bonds (Fig. 4A). Since both diAP and A have low pKa as compared with G (Brown 1971), we would expect the folding to be pH-independent at high pH values, but only if the protonation of the purine is functionally relevant to folding. With the restoration of an isosteric base pair containing three hydrogen bonds for diAP8-U3, we anticipated restoring the reaction rate to that of the wild-type construct.
The pH-dependent folding of diAP8-U3 and A8-U3 variants monitored by pyC7 displayed a similarity to the unmodified ribozyme fluorescence increase at low pH and 50% lower at high pH. Both the rates of folding (Fig. 4B) and the associated amplitudes (Table 1) of fluorescence change for each variant are nearly pH-independent up to pH 7.0 and linearly decrease as a function of pH. The rates of folding are on average 10–20 times slower than that of the unmodified ribozyme and reach a minimum at pH 8.5 of 4.4 min−1 (±0.5) for diAP8-U3 and 3.5 min−1 (±0.4) for A8-U3 (Fig. 4B; Table 1). The pH-dependent cleavage of A8-U3 and diAP8-U3 at low pH is on average five times slower from that of G8-C3 and I8-C3. The difference increases to ∼50-fold at high pH. Both variants reach a maximum rate of cleavage at pH 8.0: 11 min−1 (±1) for diAP8-U3 and 5.8 min−1 (±0.8) for A8-U3). At pH 8.5, both variants display a decrease in the cleavage rate that parallels that of folding (Fig. 4B,C; Table 1).
Given that the pH profile of cleavage for the diAP8-U3 and A8-U3 variants is limited at high pH by the rate of folding, it is difficult to retrieve an overall single pKa of the cleavage reaction for the diAP8-U3 and A8-U3 variants. However, since the activity at low and neutral pH is log-linear and extends well beyond the predicted pKa for diAP and A (∼5), these data suggest that it is not the identity and the pKa of the N1 of the purine at position 8 that is important for ribozyme catalysis, but rather the stability of the interaction with pyrimidine at position 3. Moreover, these data suggest that introduction of uracil at position 3 destabilizes the active fold or, alternatively, stabilizes the ground state.
Rescue of deleterious G8-C3 mutations at low pH
We previously showed for the minimal hammerhead ribozyme that introduction of diAP at position 8 (diAP8-C3) strongly inhibited and altered the pH-dependent profile of cleavage, reducing the rate by more than three orders of magnitude (Han and Burke 2005). Although the diAP8-C3 (Fig. 5A) mutation in the native ribozyme has a surprisingly smaller impact on activity, it does have an effect on the pH profile of folding and activity (Fig. 5B,C). Folding analysis of the diAP8-C3 variant shows that the maximum velocity of 38 min−1 (±3) is reached at pH 6.5 (Fig. 5B; Table 1), and the amplitude of the fluorescence is similar to that of the wild type (Table 1). The rates of folding and the apparent amplitudes significantly decrease with higher pH and reach a minimum at pH 8.5 (Fig. 5B; Table 1). Although the fluorescence change is small at high pH, an average of five to 10 transients gave a signal-to-noise ratio that was sufficient for reliable single exponential fitting. The cleavage rate for diAP8-C3 increases over the range of pH 5.5–6.5, with rates less than two times slower than those of the unmodified ribozyme, levels off at pH 7.5, and then decreases as the pH is raised (Fig. 5C). The bell-shaped pH profile for the diAP8-C3 cleavage reaction could be fitted with pKa values of 6.7 (±0.4) and 8.1 (±0.3) (Fig. 5C; Table 3, below). Structurally, diAP has been shown to form either an unstable one-hydrogen-bond wobble-like base pair with cytosine at neutral pH or, strengthened by N1 protonation of diAP, a two-hydrogen-bond base pair (Strobel et al. 1994), thermodynamically equivalent to a two-hydrogen-bond A-U base pair. In fact, diAP8-C3 folds and cleaves at moderate pH with the same rate and to the same extent (Fig. 5B,C) as Watson-Crick A8-U3 (Fig. 4A).
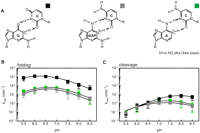
The pH dependence of cleavage and folding for wobble-like 8-3 base-pair variants. (A) G8-U3 (black squares), diAP8-C3 (gray squares), and A8-C3 (green squares) (B) pH-dependent folding as monitored by the fluorescence change of pyC7 of wobble-like 8-3 base-pair analogs. The data were acquired, fitted, and plotted as described for G8-C3 folding in Figure 4. Folding of the analyzed variants yielded a bell-shaped pH profile, and fit resulted in two apparent pKas of folding of 5.5 (±0.3) and 6.9 (±0.2) for G8-U3; 6.1 (±0.6) and 6.9 (±0.3) for diAP8-C3; 6.2 (±0.4) and 6.7 (±0.5) for A8-C3. (C) pH-dependent cleavage assays were performed as described for 8-3 analogs in Figure 4. pH dependence of cleavage for each of the analyzed variants yielded a bell-shaped profile, and curve fitting revealed two apparent pKas of 7.9 (±0.4) and >8.5 for G8-U3; 6.7 (±0.4) and 8.1 (±0.3) for diAP8-C3; 6.9 (±0.2) and 8.1 (±0.3) for A8-C3.
To test the hypothesis that the rescue of the diAP8 mutation may be due to the formation of a protonated wobble-like base pair depending on the pKa of the purine, we examined the A8-C3 and G8-U3 variants (Fig. 5A). Protonated adenosine has been shown to form a stable two-hydrogen-bond wobble-like pair with cytosine, isosteric to diAP+-C and G-U wobble pairs (Doudna et al. 1989). Our results (Fig. 5B,C) show that the ribozyme with the A8-C3 mutation has pH-dependent levels of activity and folding similar to those of the diAP8-C3 pair (Fig. 5B,C; Table 1). Since protonated wobble-like 8-3 base pairs were able to rescue the activity to a level similar to that of the unmodified ribozyme, we further expected the G8-U3 (Fig. 5A) mutant to behave similarly to the wild type. Interestingly, G8-U3 (Fig. 5B,C; Table 1) behaves rather similarly to the Watson-Crick diAP8-U3 and A8-U3 variants, indicating that its activity and folding may be affected by the pyrimidine in those pairs, U3.
Double G8 and G12 mutations in minimal versus native hammerhead ribozyme
Our previously published studies demonstrated that a simultaneous substitution of the two important nucleobases G8 and G12 with 2,6-diaminopurine shifts the pH optimum of the cleavage reaction from >8.5 (linear) to ∼6.8 (bell shaped) in two different hammerhead ribozymes (Han and Burke 2005). On the basis of pH-activity studies of targeted variants and zero-length photo-cross-linking analysis, we proposed that G8 and G12 are catalytic nucleotides within the hammerhead's active site (Han and Burke 2005; Heckman et al. 2005; Lambert et al. 2006). Subsequent crystallographic studies of Scott and colleagues (Martick and Scott 2006; Chi et al. 2008; Martick et al. 2008) are broadly consistent with this hypothesis. However, in order to interpret simultaneously the role of G12 in folding and catalysis and to determine its communication with the critical G8 residue, the pH-dependent profile of cleavage activity and folding of both single and double mutations of the respective positions were measured.
Core folding of the unmodified minimal ribozyme occurred at an overall one-order-of-magnitude-slower rate compared with the natural ribozyme. In contrast to the natural ribozyme, where folding was clearly pH-dependent (bell shaped), there are only minor differences between the rate of folding at low, neutral, and high pH for the minimal ribozyme (Fig. 6A,C; Table 2). Further differences between minimal and natural ribozymes could be observed for the single diAP8 variant. In the minimal ribozyme, this mutation showed more than five orders of magnitude inhibition of folding, whereas the same mutation in the natural ribozyme led to a decrease in the rate of folding of no more than two orders of magnitude (Fig. 6A,C; Table 2). Interestingly, the rate of folding for single diAP12 variant was only slightly influenced. For the minimal ribozyme, the difference was mostly manifested at low and high pH (fivefold decrease), whereas for the natural ribozyme, the decrease in the rate of folding was on average only twofold lower at each pH studied (Fig. 6A,C; Table 2).
Biochemical and fluorescence characterization of minimal (min) and S. mansoni (Sm) hammerhead ribozyme with G8-G12 variants
Estimation of pKa values of pH-dependent folding and cleavage for minimal (min) and natural S. mansoni (Sm) ribozymes
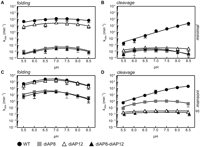
Comparison of pH-dependent core folding and cleavage of unmodified G8 (closed, black circle), diAP8 (closed, gray squares), diAP12 (open, black triangle), and double diAP8-diAP12 (black triangle) in the background of both minimal (A,B) and natural (C,D) S. mansoni hammerhead ribozyme. The pH-dependent core folding (pyC7) was performed as described in Figure 3. Folding of the analyzed minimal ribozyme and variants yielded an almost-flat pH profile, where the differences between high, moderate, and low pH did not exceed twofold, and fit resulted in two apparent pKas of folding that are rather approximations of ∼5.5 (±1.7) and 8.5 (±0.9) for G8-G12; ∼6.0 (±1.5) and 8.0 (±0.9) for diAP12; 6.2 (±1.4) and 8.0 (±0.9) for diAP8; and ∼6.2 (±1.3) and 8.0 (±1.4) for diAP8-diAP12. The same experiments performed in the background of the natural ribozyme yielded pKas of 6.3 (±0.2) and 7.4 (±0.1) for G8-G12; 6.5 (±0.4) and 7.4 (±0.5) for diAP12; 6.1 (± 0.6) and 6.9 (±0.3) for diAP8; 6.2 (±0.3) and 6.9 (±0.5) for diAP8-diAP12. The pH-dependent cleavage reactions of minimal (B) and natural (D) ribozymes were performed as described in Figure 4. The pH-dependent cleavage for unmodified minimal ribozyme is log linear with pKa higher than 9. Cleavage of the minimal ribozyme single and double G8-G12 variants yielded bell-shaped pH profiles and resulted in two apparent pKas: 5.7 (±0.4) and 8.5 (±0.3) for diAP12; 6.1 (±0.3) and 7.7 (±0.3) for diAP8; 5.9 (±0.2) and 7.7 (±0.6) for diAP8-diAP12. The same pH profiles for natural ribozyme yielded a log-linear activity increase for unmodified ribozyme with a slight leveling off at pH 8.5, yielding an apparent pKa of 8.4 (±0.2). The single diAP8 mutation showed a bell-shaped profile of cleavage, which resulted in apparent pKas of 6.7 (±0.4) and 8.1 (±0.3). Both single diAP12 and double diAP8-diAP12 variants showed too small differences in cleavage across the analyzed pH (twofold) to obtain reliable fit; consequently, the pKas were assigned to be <5.5 and >8.5. For clarity of the graph, the cleavage values for diAP8-diAP12 were incremented by 0.05 pH unit.
As shown in Figure 6, B and D, the minimal ribozyme exhibited an average of a 10–30-fold decrease in cleavage activity as compared with natural ribozyme at each pH studied. Single diAP12 substitutions showed a comparable decrease in activity in both minimal and natural ribozymes as compared with unmodified versions: one order of magnitude at low pH, two to three orders at neutral, and nearly four orders of magnitude at high pH, respectively (Fig. 6B,D; Table 2). The difference among the cleavage activities at low, neutral, and basic pH for both ribozymes did not exceed twofold; therefore, the pH profile is nearly level (Fig. 6B). Interestingly, a single diAP8 substitution showed differences in the cleavage decrease between both ribozymes. For the minimal ribozyme, this mutation caused decrease in cleavage rate of one order of magnitude at low pH, three orders at neutral pH, to more than four orders of magnitude at pH 8.5 (Fig. 6B; Table 2). In contrast, in the native hammerhead, the same substitution caused a decrease in cleavage, ranging from only less than one order of magnitude at low and neutral pH to two orders of magnitude at basic pH (Fig. 6D; Table 2). Furthermore, the cleavage rates of single diAP8 variant in the minimal ribozyme somewhat parallel those of folding at each pH studied, while those for the natural ribozyme are only similar to folding rates at pH 8 and 8.5 (Fig. 6A–D).
A comparison of the cleavage and folding rates of the minimal and native double diAP8-diAP12 ribozyme variants showed that the pH dependence of cleavage activity is restricted in both of these ribozymes by different residues (Fig. 6A–D). In the minimal ribozyme, the pH-dependent cleavage activity of diAP8-diAP12 is comparable to rates of both cleavage and folding of the single diAP8 variant, whereas for the native ribozyme, the activity of the double diAP8-diAP12 ribozyme is analogous only to the rate of cleavage of a single diAP12 variant (Fig. 6C,D). Together, these results are consistent with a model in which the pH-dependent activity of the double diAP8-diAP12 variant in the minimal ribozyme is folding-limited, whereas the pH-dependent activity of the same variant in the native ribozyme derives from the shift of the pKa of the N1 proton or/and the exchange of the O6 carbonyl to the amino group of the G12 residue.
DISCUSSION
The fluorescence of pyC provides an attractive tool for the analysis of reaction states and their dynamics in nucleic acids at specific sites. Our real-time fluorescence assay permitted us to follow topologically the kinetics of both global and local conformational changes in the folding pathway of the hammerhead ribozyme. For the first time, we were able to show that the native hammerhead ribozyme, like its more extensively studied minimal counterpart, follows not a single, but at least a two-step folding pathway with at least two distinct Mg2+ dependencies for core and global folding, each of which is one order of magnitude lower than those observed for the minimal hammerhead version (Penedo et al. 2004; Kim et al. 2005). The reason why the two previous FRET and SMD (single molecule detection) studies on the natural hammerhead ribozyme folding (Penedo et al. 2004; Boots et al. 2008; McDowell et al. 2010) failed to identify the two-step folding is because under physiological conditions, both global and core folding are very fast (in the millisecond range) and divided on the time scale by only a few seconds. When Mg2+ is fully saturating, the observed rate constant of the core folding tends toward the intrinsic rate constant of the global transition. Therefore, FRET, sensing only relatively large, global structural changes in terms of the Stem I–Stem II junction distances does not have the capability to detect the fast internal changes in the folding of the core of the natural hammerhead. In the minimal ribozyme, the kinetic and spatial differences of core and global folding are much larger; hence, it was previously possible to dissect them (Bassi et al. 1997; Hammann and Lilley 2002).
Our data on the Mg2+ dependence of the folding show no cooperativity between any sites of the ribozyme studied. This is typical for diffuse binding of Mg2+ by nucleic acids and suggests that charge screening by an ionic atmosphere is required to allow the closer approach of the different parts of the ribozyme (Krakauer 1971; Sander and Ts'o 1971). The rate of cleavage of extended and minimal hammerheads increases with magnesium ion concentration, often without evidence of saturation, and species with higher rates of cleavage show weaker affinity for divalent ions (Clouet-d'Orval and Uhlenbeck 1997; Rueda et al. 2003; Canny et al. 2004). The reason for this behavior is not explainable by a straightforward approach, and it was proposed that the low affinity could partially be due to one or more Mg2+, further increasing the rate with which active conformations occur (Rueda et al. 2003). Our results argue against this model, because the fluorescence data show clear saturation in the observable rate of folding induced by the core Mg2+ binding identified by pyC7 and pyC3, and the fact that the core of the minimal version of the ribozyme folds with rates far exceeding the rate of cleavage. However, we cannot exclude binding of another Mg2+ ion to the active site without inducing conformational change, since our experimental approach does not measure the direct binding of ions, but rather the conformational consequences of this binding. Whether there is indeed a synergy between catalytic and structural Mg2+ and whether the Mg2+ with a 1 mM core folding affinity has a physiological and/or catalytic role remain to be determined and are currently being pursued.
The Mg2+ dependence of the steady-state relative fluorescence amplitudes of core and global folding are identical, even though each site requires a distinct concentration of Mg2+ to reach the maximum velocity of folding. The amplitude of the fluorescence variation depends on the degree to which the conformational change affects the environment of the fluorophore and provides equilibrium steady-state information about the population of molecules undergoing the transition upon addition of a ligand. Alternatively, the observed transient kinetic rate constants provide information about the mechanism of the folding events (Johnson 1992). The discrepancy between the amplitude and velocity of magnesium dependence of core folding suggests that the mechanisms of the core and global folding are different. Furthermore, it suggests that global folding in the presence of the auxiliary elements sets the population of molecules with the requisite energy to reach the transition state. Even though the separate folding events fit well to the Hill equation, the equation cannot rule out more complicated, multistate reactions, while combined. On one hand, Figure 2B shows that the folding is a rather two-state event, although those particular events can be approximated as well as two sequential, more-or-less independent folding reactions. On another hand, the amplitude versus velocity comparison of pyC7 data in Figure 2D suggests that the folding is not approximated by a two-state reaction and there must be intermediate state(s). However, without more detailed temperature-dependent folding studies and core folding analysis in the background of a cleavable substrate, dissecting transient kinetic step of the active core falling back to the ground state conformation, we are unable to devise the simplest, most probably three-state model that could account for all of the data. The clear separation between global and core folding shows explicitly that the tertiary elements do not have a direct role in the core folding mechanism leading to the catalytic activity. Taken together, our results support the hypothesis that was previously introduced by Uhlenbeck's group, that tertiary interactions may have evolved to have kinetic values of global folding that adjust to the value needed for its physiological function (Nelson et al. 2005; Nelson and Uhlenbeck 2006, 2008). Our preliminary data on pyC folding, in the background of different tertiary arrangements in various natural hammerheads (IA Buskiewicz, unpubl.), suggest that global folding might become, under certain conditions, a limiting step for the core to undergo conformational change. As shown for the S. mansoni ribozyme in this study, the tertiary interaction only controls the population of core molecules with the requisite energy to reach the transition state. In conjunction with our current folding data, this supports the notion why the tertiary elements are not phylogenetically conserved (Flores et al. 2001) and can be replaced by other stabilizing tertiary elements (Saksmerprome et al. 2004). The rapid conformational switching between active and inactive states could be a critical property in the evolution of hammerheads and will defiantly help to understand the folding mechanism of the hammerheads with pseudoknot secondary motifs recently identified (Perreault et al. 2011).
The log-linear dependence of the cleavage rate on pH is generally accepted as evidence that the chemical step is rate-limiting in kinetically well-behaved hammerhead ribozyme sequences. However, at physiological Mg2+ concentrations, the log-linear pH profile levels off at high pHs (Canny et al. 2004), indicating that folding might become a rate-limiting step, as suggested by crystallographic analysis of cleavage in the crystal lattice (Murray et al. 2002). Indeed, our fluorescence folding analysis indicates that the pH-dependent conformational change occurs locally in the core of the ribozyme and that this confined core change happens not only at high, but also at low pHs. Several models could explain the observed pH rate dependence of the folding. The simplest explanation is that the bell-shaped pH profile of folding could be interpreted by well-known acid and alkaline denaturation (Bevilacqua et al. 2005) of the G8-C3 Watson-Crick base pair, because replacement of either nucleotide of this base pair influences both the pH dependence of activity and core folding. This is the least speculative hypothesis, because it does not invoke protonation of any nucleobase. However, acid–alkaline denaturation cannot explain why pH profiles of folding for G8-C3 and for diAP8-U3 or A8-U3 are different. In contrast to G8-C3, ribozymes with diAP8-U3 and A8-U3 show no pH dependence in rate of folding between pH 5 and 7. An alternative explanation of the pH dependence of folding would be the presence of an ionizable group at the same or different site within the ribozyme, responsible for weakening of the G8–C3 interaction at low or/and high pHs and strengthening the ground-level interaction of either G8 or C3. In fact, the relative effect of different mutations at positions 8 and 3 was shown to depend on the identity of residue 17 in both the minimal and natural ribozymes (Nelson and Uhlenbeck 2008). This synergistic effect was explained by transient pairing between residues 17 and 3, which stabilize an inactive ground conformation. The cleavage site nucleotide (N17) is a relatively variable nucleotide in the core of the hammerhead ribozyme. It is commonly a C or an A, bases that are frequently protonated at close to neutral pHs in various RNAs (Bevilacqua et al. 2005), seldom a U, and never a G (Flores et al. 2001; Carbonell et al. 2006). In the ground state, the N6 of C17 interacts with N3 of C3, forming a wobble-like base pair via a single hydrogen bond interaction (Supplemental Fig. S3A). In the active state, C17 interacts via O2 with N6 of A13. Interestingly, a site-specific N3 protonation of C17 or C3 could cause formation of a hydrogen bond to O2 of C3. This would transform the weak, one-hydrogen bond C17-C3 pair to a double bond C+17-C3 or C17-C+3 pair, which was shown to be thermodynamically equivalent to a G-U wobble base pair (SantaLucia et al. 1991; Masquida and Westhof 2000). The formation of C-C+ base pairs was previously shown in DNA (Gray et al. 1984; Edwards et al. 1990) and RNA (Brahms et al. 1967; Guschlbauer 1975). Interestingly, large changes in thermodynamic parameters were previously observed for loops holding C–C interaction upon changing the pH, because the Tm was increased by 12°C, and ΔG becomes more favorable when the pH is lowered from pH 7.0 to 5.5 (SantaLucia et al. 1991). If protonation of C17 or C3 should occur in the ground state, then the active-state formation would be destabilized and could explain the observable decrease in the rate constant and amplitude of folding at low pH as shown in Figure 3. The possible protonation of the C17–C3 interaction could also explain the lack of pH dependence of folding for U3 variants. C17–U3 interaction in the ground state can form stable double-bond Watson-Crick like base pairs (Supplemental Fig. S3B). If C17, protonated at N3, approached U3, no interaction could occur and the equilibrium would shift toward the active state (Supplemental Fig. S3B). In fact, all U3 mutants are stable at low pH in both folding and cleavage. Although the above proposed hypothesis of C17-C3 protonation explains the pH dependence of folding for the U3 variants at low pHs, it cannot explain why the three-hydrogen-bond base pair of diAP8-U3 does not fully restore the unmodified ribozyme activity. The diAP-U base pair has been previously shown to form a stable base pair in DNA and RNA duplexes (Howard et al. 1966; Muraoka et al. 1980; Howard and Miles 1984). As the terminal pair in an RNA duplex, diAP-U was shown to be 0.5 kcal/mol more stable than A-U, but 1.2 kcal/mol less stable than G-C, which also forms three hydrogen bonds. The lower stability of a diAP-U pair has been attributed to repulsive electrostatic interactions between the juxtaposed functional groups of the base pair. Taken together, this suggests that lower strength of the diAP-U base pair and possibly more favorable interaction of the U3 with C17 in the ground state would explain why with restoring the three-hydrogen-bond base pairs, we do not restore wild-type-like activity.
Introduction of the loop–loop interaction within the ribozyme was shown to induce a large semi-rigid bend that flexes the catalytic core in a defined direction (Martick and Scott 2006). Wobble-like mutants at the 8-3 position in the minimal ribozyme (G8-U3 and diAP8-C3) reduce the catalytic rates by more than three orders of magnitude (Fu and McLaughlin 1992; Tuschl et al. 1993; Han and Burke 2005). In the native ribozyme, those mutations are less than one order of magnitude affecting both the folding and the activity. The wobble geometry of the 8-3 base pair would introduce only a 1 Å displacement of the 2′-OH of G8, and the thermodynamic stability of the G8-U3 pair is only slightly weaker than that of a canonical G-C pair (Masquida and Westhof 2000), supporting the notions that the loop–loop interaction shifts the equilibrium of the core folding to the more locked conformation and small, otherwise deleterious distortions are not restricting this transition. The question whether the opening of the G8-C3 base pair leading to proton exchange is of a local fluctuation or occurs during a long-range distortion of the core, when an exchange of an N1-proton with the solvent is easily possible, will need to be further carefully studied. The greater-than-usual flexibility leading to the proton shuffle is right now only supported by formation of protonated wobble pairs: diAP8-C3 and A8-C3, with the same geometry as a G-U (Strobel et al. 1994; Sashital et al. 2004), which rescue the activity of the ribozyme in a way similar to that observed for G-C+ wobble in the Tetrahymena ribozyme (Knitt et al. 1994). To date, it is one of the few examples that demonstrate that, under certain circumstances, RNA tertiary structure can directly influence secondary structure (Knitt et al. 1994; Sashital et al. 2004).
A comparison of the cleavage and folding rates of the minimal and native double diAP8-diAP12 ribozyme variants showed that the pH dependence of cleavage activity is restricted in both of these ribozymes by different residues (Fig. 6A–D). In the minimal ribozyme, the pH-dependent cleavage activity of diAP8-diAP12 is comparable to rates of both cleavage and folding of the single diAP8 variant, whereas for the native ribozyme, the activity of the double diAP8-diAP12 ribozyme is analogous only to the rate of cleavage of a single diAP12 variant (Fig. 6C,D). Together, these results are consistent with a model in which the pH-dependent cleavage of the double diAP8-diAP12 variant of the minimal ribozyme is folding-limited, whereas the pH-dependent activity of the same variant in the background of the native ribozyme originates from the shift of the pKa of the N1 proton and/or the exchange of the O6 carbonyl to the amino group of G12, the residue. The pH-independent core folding of the minimal version of the S. mansoni ribozyme resembles rather global folding of the natural version, further supporting our above observation that the global folding, even much faster than the apparent rates of cleavage, is a limiting factor.
As presented here, combining time-resolved folding analysis with sequence and nucleotide analog variants provides a strategy to both establish relationships and to distinguish between kinetically equivalent mechanisms that commonly confound mechanistic analyses of acid–base catalysis in the ribozyme field. The number of similarities and differences between the two kinetic folding phases determined in the present study, particularly with regard to core folding, suggests that we are able to observe functionally significant changes in the environment of the core residues and that these changes are not part of, but only dependent on, global alteration of the hammerhead ribozyme–substrate complex. Our newly established site-specific folding assay will allow in the future, precise analysis of other site-specific mutations in the core of the hammerhead and other ribozymes, to answer questions of their role in the catalytic mechanism from both the chemical and folding point of view. The most intriguing question will be to answer the folding of G12 variants, which do not show pH dependence of cleavage or folding of newly identified ribozymes with pseudoknot motives.
MATERIALS AND METHODS
Chemical synthesis
All oligonucleotides were generated by solid-phase synthesis using nucleotide phosphoramidiates purchased from Glen Research or ChemGenes, and then deprotected and purified by denaturating polyacrylamide gel electrophoresis and HPLC, as described previously (Butcher and Burke 1994; Han and Burke 2005). Oligonucleotides containing pyrrolo-cytosine were synthesized in a similar way as described previously (Heckman et al. 2005), but deprotection was carried out under milder conditions (ammonium hydroxide for 12 h at room temperature instead of methylamine for 10 min at 60°C).
Ribozyme cleavage assays
Catalytic activities of the hammerhead ribozyme and its variants were measured under single-turnover conditions as described
previously (Han and Burke 2005). Cleavage reactions were carried out in 50 mM MES (pH 5–6), MOPS (pH 6.5–7.5), HEPES (pH 7.0–8.0), TRIS (pH 7.5–8.5), TAPS
(pH 8.2–8.9), and CHES (pH 8.5–9.0) buffers, containing additionally 100 mM NaCl and 10 mM MgCl2. Complexes of ribozyme (2 μM) and 5′-32P-end-labeled ribozyme substrate (<5 nM) were prepared by heat denaturation for 5 min at 75°C and pre-incubated for 15 min
at 25°C for equilibration and folding in the absence of Mg2+ and presence of 0.1 mM EDTA. The cleavage reactions were initiated by adding MgCl2 solution to a final concentration of 10 mM and performed at 25°C. For slow kinetic reactions (pH 5.5–7.0) 2-μL aliquots were
removed at each time point and quenched with 10 volumes of loading solution (90% formamide, 50 mM EDTA, 0.005% each of xylene
cyanol and bromophenol blue) and immediately frozen. For fast kinetic reactions (pH 7.5–8.5), cleavage reactions were performed
in a rapid quench flow apparatus (Kintek RQF-3). Reactions were initiated by rapidly mixing (2 msec) equal volumes (∼15 μL)
of pre-assembled complexes with Mg2+ ions. Fast kinetic reactions were quenched with 90 μL of the formamide-loading buffer and frozen immediately. Samples corresponding
to each reaction time point were collected independently, electrophoresed on denaturating 20% polyacrylamide, 8 M urea gel,
and quantified using a Bio-Rad Molecular Imager FX system. The fraction cleaved Ft at time t was fit using GraphPad Prism 5 software either to a single-exponential equation at low and neutral pH (pH 5.5–7.0):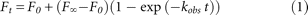 or to a biexponential equation at high pH (pH 7.5–8.5), where the two cleavage rates were (the second observed rate was ∼2%
of the total amplitude for pH 7.5 and 6%–8% for pH 8.0–8.5)
or to a biexponential equation at high pH (pH 7.5–8.5), where the two cleavage rates were (the second observed rate was ∼2%
of the total amplitude for pH 7.5 and 6%–8% for pH 8.0–8.5)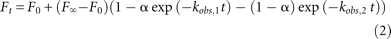 where F0 is the zero-point correction, F∞ is the estimated plateau value at infinite time, α is the fraction of the cleaved population with a rate constant of kobs,1, and (1 − α) is the fraction cleaved with a rate constant of kobs,2. Uncertainties were calculated from both curve fitting and from analysis of at least three-times-repeated independent measurements
for each variant studied. Furthermore, we tested three independent ribozyme and substrate preparations.
where F0 is the zero-point correction, F∞ is the estimated plateau value at infinite time, α is the fraction of the cleaved population with a rate constant of kobs,1, and (1 − α) is the fraction cleaved with a rate constant of kobs,2. Uncertainties were calculated from both curve fitting and from analysis of at least three-times-repeated independent measurements
for each variant studied. Furthermore, we tested three independent ribozyme and substrate preparations.
For pH-dependent cleavage analysis, variants showing one titration group were fit to the Henderson–Hasselbalch equation: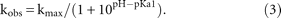 Variants displaying two titratable groups were fit with
Variants displaying two titratable groups were fit with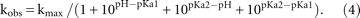
For the very low and high pH conditions, the cleavage and folding assays were performed in smaller increments than displayed. (Instead of 0.5 pH unit, 0.2 increments were tested, starting at pH 5 and ending at pH 9, respectively. The pH 9 and 5 were not included in the fit.)
Rapid folding kinetics
Fluorescence stopped-flow measurements were performed on an SX-18MV spectrometer (Applied Photophysics). Hammerhead ribozymes
and variants containing pyrrolo-cytosine at different positions were excited at 350 nm and measured after passing a KV500
cut-off filter (Schott). Hammerhead ribozyme–substrate (1–1.5 μM) complexes with C17 on the substrate strand substituted with
2′-deoxycytosine were pre-equilibrated under the same conditions as described for cleavage. The folding reaction was initiated
by rapid mixing (dead time 2 msec) of equal volumes (47 μL) of each hammerhead ribozyme–substrate strand complex and Mg2+ (final concentration 10 mM) at 25°C. Measurements of folding were performed in parallel in four different time ranges of
1000 data points each, and at least six shots were averaged per folding curve. Each folding experiment was performed at least
three times with independently assembled ribozyme–substrate complexes. The reproducibility of the rate constant and amplitudes
in the independent experiments was better than ±5% and ±10%, respectively; within one experiment, the reproducibility from
shot to shot was ±2% for both parameters. The complex formation and stability were monitored by native gel electrophoresis
at each pH (data not shown). The contribution of the C17 to 2′-deoxy-C17 substitution was checked at pHs from 5.5 to 7.0,
where cleavage is four orders of magnitude slower than folding, but no significant changes in folding were observed (data
not shown). Folding data were fit using the single exponential function described above for cleavage analysis for moderate
magnesium concentrations (up to 20 mM); at higher magnesium concentrations, two rates were observed, where the second rate
did not exceed 5% of the total amplitude of fluorescence change. For the pH-dependent analysis of the folding, only the first
dominant rate was considered. The pH dependence of the folding was fit as above described for the pH-dependent cleavage reactions
with the Henderson–Hasselbalch equation. Plots of folding rate versus magnesium ion concentration were fit to a linear version
of the Hill equation, representing a two-state model: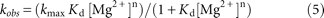 where [Mg2+] is the magnesium ion concentration, kobs is the observed rate constant for the folding at a given magnesium concentration, kmax is kobs at the saturating magnesium ions concentration, n is the Hill constant representing the lower limit of the number of magnesium ions taken up by RNA, and Kd is the apparent dissociation constant for ion binding to RNA.
where [Mg2+] is the magnesium ion concentration, kobs is the observed rate constant for the folding at a given magnesium concentration, kmax is kobs at the saturating magnesium ions concentration, n is the Hill constant representing the lower limit of the number of magnesium ions taken up by RNA, and Kd is the apparent dissociation constant for ion binding to RNA.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Professor David Draper from Johns Hopkins University and Professor Dan Herschlag from Stanford University for insightful discussions. Furthermore, we thank Dr. Joyce Heckman from the University of Vermont for help with preparing this manuscript. This work was supported by the US National Institutes of Health, award no. AI044186.
Footnotes
-
↵1 Corresponding author.
E-mail ibuskiew{at}uvm.edu.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.030999.111.
- Received October 19, 2011.
- Accepted November 9, 2011.
- Copyright © 2012 RNA Society








