
Loop of fate: structural and mechanistic insights into hnRNPA1 binding to the hepatitis C virus RNA
- Ajit Kumar1,2,
- Srinivasa Penumutchu3,
- Love Panchariya4,
- Priyanka Kumari1,
- Shubham Thakur1,
- Purba Daripa1,
- Vandana Singh1,
- Arockiasamy Arulandu4,
- Souvik Maiti1,2,
- Mandar V. Deshmukh5 and
- Niyati Jain1,2
- 1CSIR-Institute of Genomics and Integrative Biology, New Delhi 110025, India
- 2Academy of Scientific and Innovative Research (AcSIR), Ghaziabad 201002, India
- 3Department of Biochemistry and Biophysics, University of Pennsylvania, Philadelphia, Pennsylvania 19104, USA
- 4Structural Biology Group, International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi 110067, India
- 5CSIR-Centre for Cellular and Molecular Biology, Hyderabad 500007, Telangana, India
- Corresponding author: niyati2002us{at}gmail.com
-
Handling editor: Britt Glaunsinger
Abstract
Hepatitis C virus (HCV) is a major global health burden, associated with chronic liver diseases, including cirrhosis and hepatocellular carcinoma. Viral replication critically depends on conserved cis-acting replication elements (CREs), such as the 5BSL3.2 stem–loop near the 3′ end of the open reading frame. This element forms a long-range kissing-loop interaction with the SL2 domain of the 3′X tail, essential for efficient genome replication. However, the role of host RNA-binding proteins (RBPs) in regulating this RNA–RNA interaction remains poorly understood. To explore this, we investigated whether the host RBP hnRNPA1 modulates HCV replication by targeting the 5BSL3.2 element. Using an integrated approach combining structural biology, biophysics, and biochemical assays, we identify the terminal loop of 5BSL3.2 as a high-affinity binding site for the tandem RNA recognition motifs (RRMs) of hnRNPA1. Our data reveal that adenine-rich residues within the loop are critical for binding specificity. Our results uncover a structural mechanism by which hnRNPA1 binding perturbs the kissing-loop interaction between 5BSL3.2 and the SL2 element of the viral 3′X-tail, which impacts viral replication. This study highlights a previously unrecognized role of hnRNPA1 in modulating viral RNA structure and suggests a novel interface for host-directed antiviral intervention.
Keywords
- hnRNPA1
- hepatitis C virus (HCV)
- cis-acting replication element (CRE)
- 5BSL3.2 stem–loop
- RNA–protein interaction
INTRODUCTION
Hepatitis C virus (HCV) is a leading cause of liver inflammation, affecting an estimated 58 million individuals globally (WHO 2022), resulting in liver cirrhosis and hepatocellular carcinoma. HCV is a positive-stranded, tiny, enveloped RNA virus that belongs to the Flaviviridae family (Choo et al. 1989; Morozov and Lagaye 2018). HCV has a ∼9.6 kb genome that encodes for a single open reading frame flanked by nontranslated regions (NTRs) at the 5′ and 3′ ends (Bartenschlager and Lohmann 2000; Rijnbrand et al. 2001). The 5′ and 3′-NTRs have multiple conserved regulatory elements that are essential in viral translation and replication. The 5′-NTR of the viral genome has an internal ribosome entry site (IRES) guiding the production of the viral polyprotein precursor, which consists of ∼3000 amino acids (Ashfaq et al. 2011). This precursor is then cleaved into 10 distinct proteins by host and viral proteases (Vieyres et al. 2014; Pirakitikulr et al. 2016). The 3′-NTR is an essential site for the initiation of viral replication (Oh et al. 2000; Friebe and Bartenschlager 2002; Yi and Lemon 2003). Other evolutionary conserved RNA secondary structures within the genome, distinct from those found in NTRs, are reported to serve as cis signals that regulate crucial events of the viral life cycle (Haekyung et al. 2004; Tuplin et al. 2004; You et al. 2004). Among these RNA regulatory elements, the cis-acting replication element (CRE) located within the 3′-end of the ORF is of particular interest.
The CRE at the 3′ terminus of the ORF (5BSL3) is a cruciform structure that consists of three stable stem–loops 5BSL3.1, 5BSL3.2, and 5BSL3.3 (Shi and Lai 2006). The mutational studies disrupting the RNA secondary structure of 5BSL3.2 implicated its critical role in viral replication (You et al. 2004). While the other two stem–loops, 5BSL3.1 and 5BSL3.3, are suggested to act as viral translational managers (Romero-López et al. 2018), the stem–loop 5BSL3.2, which is clamped between 5BSL3.1 and 5BSL3.3, is ∼50 bases long RNA. The genetic studies showed the importance of the apical loop of 5BSL3.2 that forms the kissing-loop tertiary interactions with the loop region of the stem–loop 2 in the 3′X tail (Friebe et al. 2005; You and Rice 2008). Importantly, the mutational studies within the loops established that the complementarity between the nucleotides of 5BSL3.2 and SL2 in the 3′X tail is essential for replication (Friebe et al. 2005). The apical loop and the internal bulge of the 5BSL3.2 are shown to be involved in long-range RNA–RNA interactions and modulate viral replication (Friebe et al. 2005; Diviney et al. 2008; You and Rice 2008); however, only a few studies have reported the interaction of CRE with cellular proteins. In one such study, mass spectrometry analysis identified several cellular proteins associated with the CRE that may influence HCV replication (Oakland et al. 2013; Ríos-Marco et al. 2016). Notably, most of these proteins contained RNA recognition motifs (RRMs) (Ríos-Marco et al. 2016). Among the host factors examined, heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) emerged as a key regulator of HCV replication. Knockdown of hnRNPA1 significantly increased HCV replicon levels, whereas its overexpression suppressed viral replication, suggesting that hnRNPA1 functions as a negative regulator of the HCV life cycle (Ríos-Marco et al. 2016).
hnRNPA1 is a highly abundant RNA-binding protein known to be involved in diverse RNA metabolic processes, such as alternative pre-mRNA splicing, transcription, protein translation, mRNA stability, and miRNA processing (Jean-Philippe et al. 2013; Feng et al. 2022). Human hnRNPA1 consists of an N-terminal fragment spanning 1–196 amino acids known as unwinding protein 1 (UP1) (Herrick and Alberts 1976; Williams et al. 1985) that contains two RNA recognition motifs (RRMs) and is separated by a short linker, followed by the C-terminal glycine-rich region (glycine-rich domain, 197–320 amino acids) that includes an RGG-box (197–249 amino acids) that also constitutes an RNA binding motif (Kiledjian and Dreyfuss 1992; Ghosh and Singh 2020), a prion-like domain (233–267 amino acids), and M9 shuttling sequence (268–305 amino acids) (Fig. 1A). As mentioned above, previous studies have shown hnRNPA1 to negatively regulate viral replication by associating with the CRE region of HCV (Fig. 1B; Ríos-Marco et al. 2016). However, the precise molecular mechanism underlying this regulation remains unclear. Given the established involvement of hnRNPA1 in HCV replication, we hypothesized that hnRNPA1 may bind to the 5BSL3.2 element and interfere with its long-range kissing-loop interaction with the 3′X-tail SL2 domain, thereby modulating the RNA architecture required for genome circularization and efficient replication.
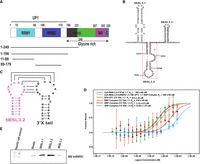
hnRNPA1 binds the 5BSL3.2 element and disrupts its kissing-loop interaction with the 3′X-tail (SL2). (A) The protein contains two RNA recognition motifs (RRMs), RRM1 (residues 15–88) and RRM2 (residues 106–179), followed by an RGG box (residues 196–233), a glycine-rich region (residues 233–267), an M9 nuclear localization/export signal (residues 267–305), and a C-terminal region (residues 305–320). The UP1 fragment includes both RRMs (residues 1–196). Truncated constructs used in this study are indicated below the domain map. (B) Secondary structure of the HCV cis-acting replication element (CRE) encompassing the 5BSL3.1, 5BSL3.2, and 5BSL3.3 stem–loop regions. The 5BSL3.2 stem–loop, important for long-range RNA–RNA interactions with the 3′X tail (SL2) region, is highlighted by a red box. Nucleotide positions are numbered according to the HCV genome. (C) Secondary structure diagram showing the long-range RNA–RNA interaction between the apical loops of the 5BSL3.2 element and the 3′X tail (SL2) region, forming a conserved kissing-loop complex essential for HCV replication. (D) Microscale Thermophoresis (MST) analysis showing the interaction between the 5BSL3.2–UP1 complex and the 3′X tail (SL2). The experiment was performed by preforming the 5BSL3.2–UP1 complex at increasing UP1:5BSL3.2 molar ratios, followed by titration with the 3′X tail. The binding affinity between the RNP complex and the 3′X tail decreases progressively with increasing amounts of UP1 relative to 5BSL3.2. (E) RNA pull-down assays reveal that hnRNPA1 is preferentially enriched with 5BSL3.2 compared to 5BSL3.1 or 5BSL3.3.
To address this, we employed a multidisciplinary approach, combining structural biology, biophysical techniques, biochemical assays, and functional studies, to reveal the molecular basis of 5BSL3.2 recognition by hnRNPA1, shedding light on its role in modulating HCV replication. Our biophysical and NMR analyses demonstrate that the terminal loop of 5BSL3.2 (9265–9308) acts as a high-affinity binding site for the tandem RRM, with adenines playing a pivotal role in this interaction. Notably, MST analyses reveal that hnRNPA1's tandem RRMs engage the terminal loop of the cis-element, thereby modulating and preventing the formation of kissing-loop interactions in a concentration-dependent manner that are crucial for HCV replication. These findings offer a novel paradigm for understanding the regulation of HCV replication, emphasizing the pivotal role of RNA-binding proteins (RBPs) in recognizing and modulating key structural elements such as the terminal loop.
RESULTS
hnRNPA1 disrupts the 5BSL3.2–3′X tail (SL2) kissing-loop interaction critical for HCV replication
The Hepatitis C virus (HCV) genome relies on a highly structured RNA architecture that mediates long-range interactions essential for its replication. One such interaction, a conserved kissing-loop pairing between the apical loop of the 5BSL3.2 stem–loop in the NS5B coding region and the SL2 hairpin within the 3′X untranslated region (UTR) (Fig. 1C), has been shown to promote genome circularization and replication. However, how host RNA-binding proteins might influence or interfere with this critical interaction remains unclear.
To explore this, we asked whether hnRNPA1, a host RNA-binding protein known to modulate RNA structure and viral replication, might interact with the 5BSL3.2 element and interfere with this critical long-range kissing-loop interaction. To address this, we performed in vitro binding assays, microscale thermophoresis (MST) experiments (MST measures binding by detecting changes in the thermophoretic movement of a fluorescently labeled molecule when its binding partner is titrated; these changes are plotted to generate a binding curve, from which the dissociation constant [KD] is calculated) (Jerabek-Willemsen et al. 2014), as detailed in the Materials and Methods section, to determine whether hnRNPA1 binding could prevent or weaken the interaction between these two RNA elements. In the absence of protein, these two RNA elements formed a complex with an apparent dissociation constant (KD) of 360 ± 60 nM. As a control, 5BSL3.2 binding to hnRNPA1 (UP1) showed a comparable affinity (KD = 399 ± 66 nM), and a similar affinity was observed for GFP-UP1 binding to the 3′X-tail RNA (KD = 414 ± 28 nM). However, when UP1 and 5BSL3.2 were present at a 1:1 molar ratio, the affinity of 5BSL3.2 for the 3′X tail decreased (KD = 618 ± 48 nM). This inhibitory effect became more pronounced as the UP1:5BSL3.2 molar ratio increased. At a 2:1 ratio, the binding affinity further declined (KD = 3.16 ± 0.5 µM), and at a 5:1 ratio, the interaction was substantially weakened (KD = 13.05 ± 1.07 µM) (Fig. 1D).
These findings indicate that specific binding of hnRNPA1 to the 5BSL3.2 loop, suggesting a potential mechanism by which hnRNPA1 may interfere with 5BSL3.2–3′X-tail kissing-loop interaction in a concentration-dependent manner. At elevated hnRNPA1 levels, its binding to 5BSL3.2 likely masks or alters the structure of the apical loop, thereby obstructing this essential RNA–RNA contact required for HCV replication. This supports a model in which hnRNPA1 regulates HCV genome architecture and replication by competitively or sterically hindering critical long-range RNA interactions.
hnRNPA1 is preferentially enriched at the 5BSL3.2 element of the HCV CRE domain
To investigate the interaction of hnRNPA1 with individual RNA elements of the HCV CRE domain, we performed RNA pull-down assays using in vitro transcribed and biotinylated 5BSL3.1, 5BSL3.2, and 5BSL3.3 RNAs. These RNAs were incubated with HepG2 cell lysates, and RNA-bound proteins were captured using streptavidin magnetic beads and analyzed by SDS-PAGE followed by western blotting with an anti-hnRNPA1 antibody.
The western blot revealed distinct hnRNPA1 binding patterns. The RNA pull-down data represent enriched fractions, resulting in stronger signals for the hnRNPA1 protein. Among the three stem–loops tested, 5BSL3.2 showed the strongest and most consistent enrichment of hnRNPA1. At the same time, 5BSL3.1 exhibited a weaker signal, and 5BSL3.3 showed minimal binding and was essentially indistinguishable from the background observed with the beads-only negative control (Fig. 1E). The input lane shows a weaker band intensity compared to the pull-down lanes, likely due to the lower total protein amount loaded for the input sample. These results indicate that hnRNPA1 preferentially recognizes the 5BSL3.2 stem–loop, which might lead to the potential disruption of long-range RNA–RNA interactions, which supports its possible role as a specific modulator of this cis-acting RNA element within the HCV genome.
The phylogenetically conserved 5BSL3.2 element folds show the sequence of the apical loop to be conserved
To investigate the evolutionary conservation of the 5BSL3.2 RNA structural element within the hepatitis C virus (HCV) genome, we performed an extensive phylogenetic analysis using isolate sequences available in the NCBI HCV database. A total of ∼1000 HCV genome sequences, representing various genotypes and geographical origins, were aligned for this purpose. The alignments served as the basis for a maximum likelihood phylogenetic analysis, allowing the construction of a tree that reflects the evolutionary relationships among these isolates, illustrating their divergence from a common ancestral sequence (Cho 2012; Yang and Rannala 2012).
The results of this analysis are depicted in the accompanying Figure 2A,B, which shows both the maximum likelihood phylogenetic tree and isolate-specific sequence logos derived from the 5BSL3.2 region. The sequence logos reveal a strikingly high degree of conservation across the aligned HCV isolates, underscoring the functional significance of this structural element in the viral life cycle. Secondary structure prediction based on the global consensus sequence of the 5BSL3.2 element yields a characteristic stem–loop conformation. This structure includes a 12 nt apical loop, an 8 nt internal bulge, and well-defined upper and lower stem regions (Fig. 2C). Notably, both the apical loop and bulge regions have been shown to play essential roles in HCV replication, as demonstrated in prior functional studies (Friebe et al. 2005). The apical loop contains the conserved 5′-UCACAGC-3′ motif, which is complementary to the loop region of the 3′X tail of the viral genome. This complementarity is critical for the formation of long-range RNA–RNA kissing-loop interactions that regulate viral replication and translation (Friebe et al. 2005).

Phylogenetic analysis and sequence conservation of the 5BSL3.2 element of the HCV CRE domain. (A) Phylogenetic tree of representative HCV genotypes (1a, 1b, 2a, 2b, 3a, 3b, 4, 5, 6a, 7, and 8) based on the nucleotide sequences of the 5BSL3.2 region. The corresponding aligned sequences are shown adjacent to the tree. (B) Sequence logo representation of the 5BSL3.2 RNA element illustrating nucleotide conservation across different HCV genotypes. The height of each nucleotide indicates its relative frequency at that position. (C) Predicted secondary structure of the 5BSL3.2 element, highlighting the conserved apical loop and stem regions critical for long-range RNA–RNA interactions within the HCV genome.
Additionally, highly conserved G-C-rich stretches within both the lower and upper stems further support the structural and functional importance of these regions. These features are consistently preserved across the diverse HCV isolates examined, suggesting strong evolutionary pressure to maintain these elements. The consensus secondary structure derived from our comprehensive data set is consistent with earlier phylogenetic analyses conducted on a smaller subset of HCV sequences (You et al. 2004).
The 5BSL3.2 element spans 48 nt and is located within the coding region for the NS5B RNA-dependent RNA polymerase, specifically encompassing amino acids 555–571. This element forms part of a larger cruciform RNA structure that encodes NS5B residues 539–591, which include the polymerase's C-terminal domain, a linker region, and the membrane-associating segment (Schmidt-Mende et al. 2001). The absence of variation in both the size and sequence composition of the apical loop and bulge across the analyzed HCV isolates (Supplemental Fig. S2) points to intense evolutionary constraint acting to preserve the structural integrity and functional capacity of the 5BSL3.2 element, highlighting its indispensable role in the viral life cycle.
UP1 domain of hnRNPA1 is sufficient for 5BSL3.2 RNA binding
Although previous studies have demonstrated the involvement of hnRNPA1 in recognizing the cis-acting replication element (CRE, nucleotides 9280–9330) within the Hepatitis C Virus (HCV) genome, a detailed understanding of the specific domains of hnRNPA1 required for this interaction has remained incomplete. To delineate the domain architecture necessary for 5BSL3.2 RNA element (nucleotides 9265–9308) binding, we generated and systematically evaluated a series of hnRNPA1 deletion constructs against the 5BSL3.2 RNA element. The following deletion mutants of hnRNPA1 were designed: (i) hnRNPA1 (1–249), comprising both RNA recognition motif (RRM) domains and the C-terminal RGG-rich region; (ii) hnRNPA1 (1–196), corresponding to the UP1 fragment that includes both RRM1 and RRM2 connected via the flexible linker; (iii) hnRNPA1 (11–88), encompassing only RRM1; and (iv) hnRNPA1 (89–179), containing the linker region and RRM2 domain (Fig. 1A).
Electrophoretic mobility shift assays (EMSAs) demonstrated that hnRNPA1 (1–249) binds to the 5BSL3.2 RNA, producing a distinct and well-resolved RNA–protein complex band, indicative of stable and specific interaction. Interestingly, truncation of the C-terminal RGG domain, as seen in the hnRNPA1 (1–196) construct, did not adversely affect the binding affinity or complex formation, suggesting that the RGG region is dispensable for 5BSL3.2 recognition under these conditions (Fig. 3A–D). In contrast, constructs expressing individual domains either RRM1 (11–88) or the linker-RRM2 region (89–179) exhibited significantly diminished RNA-binding activity (Fig. 3E–H), highlighting the necessity of both RRMs for efficient 5BSL3.2 RNA recognition. These results imply that cooperative action between RRM1 and RRM2 is required for stable and specific CRE (5BSL3.29265–9308) binding, and that neither domain alone is sufficient for productive interaction.
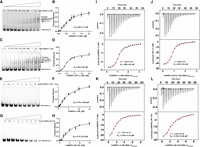
Binding analysis of hnRNPA1 constructs to the HCV 5BSL3.2 RNA element. (A,C,E,G) Electrophoretic mobility shift assays (EMSAs) showing complex formation between 5BSL3.2 RNA and increasing concentrations of (A) hnRNPA1 (1–249), (C) UP1 fragment (1–196), (E) RRM1 domain (1–88), and (G) RRM2 domain (89–179). The arrows indicate RNA–protein complexes. Protein concentrations (μM) are noted above each lane. (B,D,F,H) Quantification of EMSA data. The fraction of RNA bound was plotted as a function of protein concentration and fitted to a one-site binding model to determine apparent dissociation constants (KD). (I–L) Isothermal titration calorimetry (ITC) analysis of 5BSL3.2 RNA binding to (I) hnRNPA1 (1–249), (J) UP1 (1–196), (K) hnRNPA1 (1–88), and (L) hnRNPA1 (89–179). (Upper panels) Raw thermograms; (lower panels) integrated binding isotherms fitted to a single-site binding model. Derived KD values and binding stoichiometries (n) are indicated.
To quantitatively define the thermodynamic parameters of these interactions, we performed isothermal titration calorimetry (ITC) using the same set of hnRNPA1 deletion mutants. The ITC profiles for hnRNPA1 (1–249) binding to the 5BSL3.2 RNA displayed classic sigmoidal binding isotherms, which were best fitted to a single-site binding model, consistent with a 1:1 protein:RNA stoichiometry (Fig. 3I; Table 1). The derived thermodynamic parameters at 25°C revealed that the interaction is primarily driven by favorable enthalpic contributions, accompanied by an unfavorable entropic term, indicative of the formation of a highly ordered and specific CRE (5BSL3.29265–9308) –hnRNPA1 complex. Notably, the binding behavior of hnRNPA1 (1–196) closely mirrored that of the hnRNPA1 (1–249), exhibiting similar affinity and thermodynamic signatures (Fig. 3J; Table 1). This further reinforces the EMSA-based conclusion that the UP1 fragment, comprising both RRM1 and RRM2, represents the minimal functional unit necessary and sufficient for CRE (5BSL3.29265–9308) binding. Conversely, the individual RRM-containing constructs hnRNPA1 (11–88) and hnRNPA1 (89–179) exhibited weak binding in ITC, consistent with their reduced affinity observed in EMSA assays, suggesting that single RRMs interact poorly with the 5BSL3.2 RNA (Fig. 3K,L; Table 1). Moreover, the thermodynamic parameters obtained for the individual constructs indicate (ΔG value) significantly weaker binding, suggesting that the absence of interdomain cooperativity between RRM1 and RRM2 compromises the stability and efficiency of RNA recognition.
Thermodynamic parameters determined from ITC measurements
Collectively, these findings delineate the critical requirement of both RRM domains acting cooperatively for high-affinity, specific recognition of the CRE (5BSL3.29265–9308) element of HCV. This establishes UP1 fragment as a minimal yet fully competent binding module for CRE (5BSL3.29265–9308) engagement.
Spectroscopic analysis of CRE 5BSL3.2 reveals two stem regions
To investigate the structural features of the CRE 5BSL3.2 element critical for its interaction with hnRNPA1, we first validated its predicted secondary structure using a combination of UV thermal melting, circular dichroism (CD) spectroscopy, and nuclear magnetic resonance (NMR) spectroscopy. The secondary structure of the 5BSL3.2 element (nucleotides 9265–9308) was initially predicted by RNAfold, which suggested the formation of two stem regions separated by an internal bulge and capped by an apical loop.
UV thermal melting experiments were performed to experimentally assess the stability of these predicted structural elements. The thermal denaturation profile of the full-length 5BSL3.29265–9308 RNA was monitored at 260/280 nm as the temperature increased from 20°C to 95°C. The 5BSL3.2 RNA exhibited a stronger absorbance maximum at 280 nm than at 260 nm, consistent with its guanine-rich stem region; therefore, the measurements were reported at 280 nm. The resulting melting curve displayed two distinct transitions with melting temperatures (Tm) of 72.8 ± 0.9°C and 90.3 ± 0.3°C, respectively (Supplemental Fig. S3B,C). This biphasic melting behavior suggests the presence of two thermodynamically independent base-paired regions within the RNA. To assign these transitions to specific structural domains, we performed additional melting experiments on a truncated construct containing only the apical stem–loop region of 5BSL3.2apical stem-loop. This construct exhibited a single melting transition at 67.9 ± 0.8°C, clearly indicating that the first transition observed in the full-length RNA corresponds to the unfolding of the upper stem region (Supplemental Fig. S3E,F). Consequently, the higher-temperature transition (∼90°C) is attributable to the unfolding of the lower stem region. Together, these UV melting data experimentally confirm that the 5BSL3.2 element indeed forms two discrete helical regions separated by a central bulge, consistent with the in silico secondary structure predictions (Supplemental Fig. S3A).
To further validate the structural conformation, CD spectroscopy was performed on the full-length 5BSL3.29265–9308 RNA at 20°C. The recorded CD spectrum exhibited the characteristic features of an A-form RNA duplex, including a prominent positive ellipticity peak near 260 nm and a negative peak around 210 nm (Supplemental Fig. S3G), as reported for canonical A-form RNA helices (Gratzer and Richards 1971; Wells and Yang 1974). These spectral features confirm that the 5BSL3.2 RNA adopts an A-form geometry under the experimental conditions used.
In addition, proton (1H) NMR spectroscopy was utilized to obtain more detailed information on the base-pairing status and stability of the secondary structure. The 1D 1H NMR spectrum of the full-length 5BSL3.2 RNA revealed 13–14 distinct imino proton resonances in the region typically associated with Watson–Crick base-paired protons (Supplemental Fig. S3H), supporting the presence of well-formed and stable helices as suggested by the UV and CD data. To optimize conditions for two-dimensional NMR analysis, 1H NMR spectra were acquired at various temperatures, revealing significantly sharper and more resolved peaks at 303 K, which was subsequently used for recording 2D NOESY spectra. However, NOE cross-peaks corresponding to AU and some GC base pairs were either absent or poorly resolved, likely due to signal broadening in AU-rich regions and dynamic fraying at specific sites (data not shown), as indicated by the 1D imino proton spectra at 303 K. Overall, the NMR data align well with previous findings (Friebe et al. 2005).
Overall, the combined results from UV melting, CD spectroscopy, and NMR analysis confirm the predicted secondary structure of 5BSL3.29265–9308. These experiments strongly support the proposed secondary structure model of 5BSL3.29265–9308, featuring two distinct and stable stem regions separated by a bulge.
Structural insights into the 5BSL3.2 apical stem–loop by NMR spectroscopy
To determine the three-dimensional structure of the 5BSL3.2 element (nucleotides 9265–9308), we in vitro transcribed a 48 nt RNA construct, incorporating a nonnative GC base pair to enhance transcription efficiency. To validate its secondary structure, 1H–1H NOESY spectra were collected. However, several expected NOE cross-peaks corresponding to Watson–Crick base pairs in the upper helix were weak or absent, most likely due to rapid solvent exchange during the mixing time. Additionally, the overall spectral quality of the 1H–1H NOESY (acquired in 100% D2O) was of low quality, potentially due to local dynamics introduced by the bulge region in the RNA. We first acquired one-dimensional (1D) 1H NMR spectra of a shorter, 26 nt RNA construct representing the apical stem–loop of 5BSL3.2, which contained a nonnative GC pair introduced to enhance transcription yield. The 1D spectra revealed signals corresponding to the expected number of base pairs in the upper helix, except the terminal GC and AU pairs, for which no imino proton resonances were detected, likely due to rapid exchange with solvent (Fig. 4B). Subsequently, two-dimensional (2D) 1H–1H NOESY spectra showed characteristic NOE cross-peaks and chemical shifts consistent with GC and AU base-pairing within the stem region, supporting the presence of a stable helical structure despite the absence of terminal base pair signals (Fig. 4B).
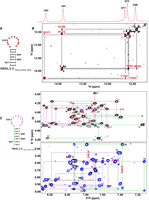
NMR analysis of the 5BSL3.2 apical stem–loop. (A) Predicted secondary structure of the 5BSL3.2 apical stem–loop, highlighting residues for which imino proton resonances were assigned. (B) Imino proton region of the NOESY spectrum (mixing time: 250 msec) recorded in H2O at 303 K. The corresponding one-dimensional spectrum is displayed above. Sequential imino–imino cross-peaks are labeled with residue numbers. (C) Predicted secondary structure of the 5BSL3.2 apical stem–loop, color-coded based on NMR assignments: left stem region in green, right stem region in red, and apical loop in purple. (D) NOESY spectra recorded in D2O at 303 K (sample was prepared using rNTPs in which the ribose H3′–H5′′ positions and the H5 position of pyrimidines were selectively deuterated). The upper panel shows intranucleotide H2′–H6/H8 and internucleotide H2′–H6/H8 cross-peaks; the lower panel presents intranucleotide H1′–H6/H8 and internucleotide H1′–H6/H8/H2 correlations. Assigned cross-peaks are annotated with residue numbers; adenosine H2 protons are marked in red.
Chemical shift assignments for the nonexchangeable protons of the 5BSL3.2 apical stem–loop were achieved using a nucleotide-selective deuteration strategy. The accompanying figure displays a representative 1H–1H NOESY spectrum recorded in 100% D2O at 303 K and pH 6.5 (Supplemental Fig. S4B). The spectrum exhibits sharp and well-resolved NOE cross-peaks, allowing for identification of sequential H8/H6 (i) to H1′ (i−1) NOEs across the stem region spanning nucleotides 9270–9275 and 9289–9294. Additionally, the presence of adenines (A9272, A9274, and A276) involved in AU base pairs is confirmed by characteristic A-form of dsRNA NOE cross-peaks, specifically between the H2 proton of adenine and the H1′ proton of the nucleotide at position i + 1 on both the same and opposite strands. The data also showed strong H6–H5 correlations in the (1H,1H) TOCSY spectrum, which helped distinguish pyrimidines from purines (Supplemental Fig. S4C). However, since some H8/H6 (i) to H1′ (i−1) NOE cross-peaks were masked by strong TOCSY cross-peaks, the D2O NOESY results were further validated using a selectively deuterated RNA sample, in which the ribose H3′–H5″ positions and the H5 position of pyrimidines were deuterated.
In the NOESY spectra of the 5BSL3.2 apical stem–loop, the sequential NOE walk across the loop region shows either weak or absent NOEs between H8/H6 (i) and H1′ (i + 1), whereas the corresponding H8/H6 (i) to H2′ (i + 1) interactions are observed (Fig. 4D). This reduction in NOE intensity is likely due to C2′-endo conformation, the geometry of the ribose positions H2′ (i + 1) closer to H8/H6 (i), while H1′ (i + 1) is further away, weakening H8/H6 (i) to H1′ (i + 1) NOEs. The line broadening caused by local dynamics occurring on the millisecond timescale in the flexible loop region affects the weaker NOEs between H8/H6 (i) and H1′ (i + 1) but do not affect the spatial NOEs between H8/H6 (i) and H2′ (i + 1), due to the closer proximity between H8/H6 (i) and H2′ (i + 1) (Fig. 5D). Supporting this, NOESY spectra acquired in 100% D2O, with and without selective labeling [H8/H6 (i), while H1′ (i + 1)] revealed weak or no NOEs between residues A9276–U9277, U9277–A9278, A9278–U9279, U9279–A9280, A9280–U9281, A9283–C9284, C9284–A9285, A9285–G9286, and C9288–U9289. Notably, NOESY spectra acquired in 100% D2O revealed strong NOE interactions between U9281–C9282 and C9282–A9283, indicative of continuous stacking in this segment of the apical loop. Strong NOEs were also observed between G9286–C9287 and C9287–C9288.

Superimposition of the 20 lowest-energy structures of the RNA element showing the apical loop region in two views rotated by 180°. The loop residues (U9277–C9288) are highlighted in red, while the remaining stem residues are shown in gray. The labeled nucleotides indicate residues within the loop region. The conformational ensemble illustrates the structural variability and orientation of the loop nucleotides relative to the stable helical stem.
In addition, sequential connectivity was analyzed in the ribose region, revealing intra-residue H1′–H2′ correlations along with inter-residue back-sequential interactions. The sugar puckering assignment is further supported by Ribose TOCSY spectra, where the observed homonuclear ³JH1′−H2′ coupling constant of ∼8 Hz results in clear cross-peaks for residues adopting the C2′-endo conformation, in contrast to the weaker ∼2 Hz coupling in C3′-endo sugars, which typically do not yield cross-peaks (Zhang et al. 2012). Strong H1′–H2′ TOCSY cross-peaks were observed for residues A9278, A9280, U9281, C9282, C9285, A9286, and C9288, while U9279 and Cytosine* exhibited a medium–weak intensity cross-peak, suggesting a mixed C2′/C3′-endo sugar pucker for these two residues (Supplemental Fig. S4A). Collectively, our NMR data indicate that the 5BSL3.2 apical stem–loop adopts a well-defined A-form helical stem, whereas the loop region is characterized by conformational flexibility with evidence of sequential base stacking.
The final ensemble of 20 lowest-energy structures was selected from a larger pool of accepted simulated annealing structures generated using Xplor-NIH (Fig. 5). These structures showed no significant NOE violations (>0.2 Å) or torsional angle violations (>5°) and satisfied all experimental restraints, including NOE-derived distance constraints and dihedral angles Table 2. Superimposition of the 20 lowest-energy conformers was performed over the well-defined helical regions. The resulting ensemble revealed a well-converged global fold, which exhibited a stable A-form helical geometry flanking a flexible loop. The apical loop displayed conformational heterogeneity, consistent with the presence of dynamic ribose sugar puckers and unpaired nucleotides. Root mean square deviation (RMSD) values calculated over the stem region for heavy atoms indicated high structural precision, whereas the loop region showed increased variability, reflective of its intrinsic flexibility Table 2.
NMR distance restraints and statistical analysis
Mapping of RNA elements identifies the apical loop within 5BSL3.2 as the recognition site for UP1
To delineate the RNA elements within the 5BSL3.2 (nucleotides 9265–9308) responsible for UP1 recognition, a series of RNA variants were engineered, and their binding affinities to UP1 were assessed using isothermal titration calorimetry (ITC). The constructs included the 5BSL3.2 apical stem–loop, 5BSL3.2 Mut1, 5BSL3.2 Mut2, 5BSL3.2 Mut3, 5BSL3.2 Mut4, and a single-stranded apical loop sequence. The correct folding of all RNA variants was confirmed by circular dichroism (CD) spectroscopy (Supplemental Fig. S5), ensuring structural integrity for reliable binding studies.
To identify the specific RNA region involved in UP1 interaction, an apical stem–loop construct was generated by removing the bulge and lower stem regions from the full-length 5BSL3.2 RNA. This truncated construct displayed a binding affinity comparable to that of the wild-type RNA, suggesting that the apical loop alone mediates UP1 recognition (Fig. 6A). To probe the importance of specific sequence motifs, the AU-rich stretch on the 5′ face of the apical loop was substituted with cytosines (5BSL3.2 Mut1), given the established preference of UP1 for AU-rich sequences. This mutation resulted in a ∼2.8-fold decrease in binding affinity compared to the wild-type RNA, indicating that this AU-rich region contributes to UP1 recognition (Supplemental Fig. S6A; Supplemental Table S1). Interestingly, the apical loop from the JFH1 HCV isolate (genotype 2a), which contains a U-stretch instead of the AUAU sequence, exhibited a binding affinity similar to 5BSL3.2 Mut1, suggesting that adenines are particularly important for UP1 binding (Supplemental Fig. S6E; Supplemental Table S1).
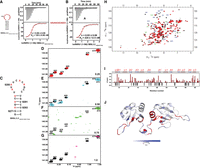
Interaction of hnRNPA1 with the apical stem–loop of the 5BSL3.2 element from the HCV CRE domain. (A,B) ITC profiles showing the binding of hnRNPA1 (1–196) to (A) the wild-type 5BSL3.2 apical stem–loop and (B) a mutant 5BSL3.2 loop containing the UAUUACACACAC sequence. The top panels show the raw heat changes upon injection of RNA into the protein solution, and the bottom panels show the integrated binding isotherms fitted to a one-site binding model. The derived thermodynamic parameters (n and KD) are indicated. (C) Predicted secondary structure of the 5BSL3.2 apical stem–loop used for NMR analysis. (D–G) Overlay of 2D 1H–13C HSQC spectra of selectively labeled adenosine residues in the apical stem–loop RNA titrated with increasing molar ratios (0.25 [red], 0.5 [cyan], 0.75 [green], and 1.0 [magenta]) of hnRNPA1 (1–196). Chemical shift perturbations upon protein binding are indicated. (H) Superposition of 2D 1H–15N HSQC spectra of hnRNPA1 (1–196) in the free state (black) and in complex with 5BSL3.2 RNA (red). Assigned backbone amide resonances are labeled. (I) Histogram of combined 1H–15N chemical shift perturbations (Δδ) plotted as a function of hnRNPA1 residue number upon RNA binding. The secondary structure elements of hnRNPA1 are indicated above the plot. (J) Mapping of chemical shift perturbations onto the 3D structure of hnRNPA1. The color gradient reflects the magnitude of the combined chemical shift changes (Δδ) according to the scale bar shown (white to blue). Residues showing amide signal broadening due to exchange upon RNA binding are shown in red.
Considering that hnRNPA1 (the precursor of UP1) shows a preference for purines and AG motifs, the AG sequence on the 3′ face of the apical loop was replaced with cytosines in 5BSL3.2 Mut2, leading to a ∼2.7-fold reduction in binding affinity (Supplemental Fig. S6B; Supplemental Table S2). This confirms the necessity of purine residues in this region for optimal recognition. Similarly, replacement of an adenine on the upper face of the loop with cytosine (5BSL3.2 Mut3) also decreased binding affinity, reinforcing the importance of adenines in UP1–RNA interaction (Supplemental Fig. S6C; Supplemental Table S2). To further evaluate the role of the apical loop sequence, a construct (5BSL3.2 Mut4) was designed in which the entire 12 nt loop was substituted with cytosines (Supplemental Fig. S6D; Supplemental Table S2). This modification resulted in an approximately fivefold reduction in UP1 binding, underscoring the critical role of purines, particularly adenines, in mediating this interaction. Consistently, an apical loop CCC construct, in which only the apical loop of the apical stem–loop RNA was replaced with cytosines, exhibited a similarly reduced affinity, confirming that the apical loop is the principal recognition element (Supplemental Fig. S6G; Supplemental Table S2).
To test the significance of loop length and sequence specificity, the 12 nt apical loop was replaced with a UCCU tetraloop (5BSL3.2 apical loop UCCU). This substitution completely abolished UP1 binding (Supplemental Fig. S6F; Supplemental Table S2), demonstrating that both the length and sequence of the native apical loop are essential for UP1 recognition. To determine whether UP1 recognition is governed by sequence or structural context, UP1 was titrated into a 12 nt single-stranded apical loop RNA, devoid of stem constraints (Fig. 6B; Table 1). Remarkably, this construct exhibited a binding affinity comparable to the full-length 5BSL3.2 RNA (Fig. 3J; Table 1), strongly suggesting that sequence identity, rather than secondary structure, dictates UP1 recognition. While the sequence appears to drive UP1 binding, the stem likely contributes to maintaining the loop conformation and overall RNA architecture required for specific and physiologically relevant recognition.
Analytical size exclusion chromatography (SEC) was employed to monitor complex formation between UP1 and the 5BSL3.2 constructs in solution. Increasing UP1 concentration while maintaining RNA at 5 µM resulted in a progressive shift in elution volume, with a distinct complex peak appearing at a 0.25 molar ratio. Complete RNA binding was achieved at a 1:1 ratio for the wild-type RNA (Supplemental Fig. S7A). In contrast, mutant RNAs displayed incomplete complex formation even at a 1.5:1 UP1:RNA ratio, reflecting their weaker binding affinities (Supplemental Fig. S7B–F).
Collectively, these results demonstrate that adenine-rich sequences within the 12 nt apical loop are critical for UP1 binding, and cytosine substitutions markedly reduce this affinity. The data establish that apical loop sequence, rather than structure, predominantly governs UP1 recognition, with purine (particularly adenine) residues playing a central role in the specificity of this 5BSL3.2–hnRNPA1 protein interaction.
UP1 preferentially recognizes the adenines of the 5BSL3.2 apical loop
To gain residue-specific insights into the molecular interaction between UP1 and the 5BSL3.2 apical stem–loop RNA (Fig. 6C), we performed NMR-based 1H–13C HMQC titration experiments. For these measurements, the 5BSL3.2 apical stem–loop (comprising 26 nt) was selectively labeled with [13C]-adenine to monitor the four adenine residues (A9278, A9280, A9283, and A9285) positioned within the apical loop region. Stepwise titration was carried out by the gradual addition of unlabeled UP1 protein to the labeled RNA. The overlaid C8–H8 spectral region of the 1H–13C HMQC spectra showed distinct signals corresponding to both the free and protein-bound forms of the adenines at a UP1:RNA molar ratio of 0.25:1 (Fig. 6D), suggesting an initial encounter complex formation without complete saturation of the RNA binding sites. As the molar ratio was increased to 0.5:1 (Fig. 6E), progressive line broadening and notable chemical shift perturbation (CSP) were detected, particularly for adenines in the apical loop, implying their direct involvement with UP1. Interestingly, the adenines A9272 and A9274, located within the double-stranded helical region forming stable Watson–Crick base pairs, displayed negligible CSPs and line broadening, consistent with their inaccessibility to protein binding due to their base-paired conformation. At a 1:1 molar ratio, the resonances corresponding to all apical loop adenines (A9278, A9280, A9283, A9285) became fully broadened beyond detection, indicative of their engagement in dynamic or intermediate exchange binding with UP1 on the NMR timescale (Fig. 6G). This broadening pattern supports the notion that UP1 preferentially interacts with the exposed adenines within the apical loop, while leaving the stem region undisturbed, thereby maintaining the global secondary structure integrity of the RNA.
Overall, these titration experiments reveal that UP1 specifically targets the apical loop adenines of 5BSL3.2, highlighting a loop-directed mode of recognition without disrupting the RNA stem structure, an important feature for its potential functional role in regulating RNA–protein interactions in the biological context.
Recognition of the 5BSL3.2 apical stem–loop by UP1
NMR analysis
NMR titration experiments were performed to elucidate the binding interface between the UP1 protein and a 12 nt linear RNA fragment derived from the 5BSL3.2 element, which includes the UP1 recognition motif. Stepwise titration of the RNA into 15N-labeled UP1 revealed a slow exchange regime on the NMR timescale, further supporting the formation of a tight complex. Notably, residues in RRM1, such as G20, G56, and G58, and in RRM2, including G110, G111, and M137, exhibited marked CSPs even at a substoichiometric ratio (0.25:1 RNA:UP1) (Supplemental Fig. S8A). As RNA concentration increased, perturbations became more pronounced, extending to additional residues including K15, S54, F59, and V90 (Supplemental Fig. S8B,C). Importantly, residues in the C-terminal region of RRM2 V163, I164, Y167, I175, and R178 also displayed significant CSPs consistent with previous studies (Fig. 6H; Barraud and Allain 2013). This region is known to be flexible in the unbound UP1 structure, but upon RNA binding, it adopts a well-defined α-helix (α3), a structural element not present in the free form of the protein. This RNA-induced folding of α3 highlights the conformational plasticity of UP1 upon RNA interaction (Kooshapur et al. 2018).
At equimolar concentrations, CSPs predominantly localized to the canonical RNA-binding β-sheet surfaces of both RRMs, indicating that both domains are essential for high-affinity binding, consistent with the nanomolar binding affinity determined by ITC. Intriguingly, residues in the interdomain linker between RRM1 and RRM2 underwent severe line broadening upon RNA binding, indicative of conformational dynamics occurring on the microsecond-to-millisecond timescale (Fig. 6I,J). The negative ΔΔ(ppm) values observed for several residues correspond to those that are completely broadened in the complex, reflecting intermediate exchange and strong interaction with RNA. These residues are positioned in the RNPs of RRMs, and the linker region likely represents key contact points or regions that undergo substantial conformational rearrangements upon RNA binding.
We further performed mutational analyses of hnRNPA1, focusing on the RRM2 region, to assess the direct impact of these mutations on binding affinity. Specifically, amino acids in RNP2 (I107A, G110A, G111A), RNP1 (F150A), the C-terminal region (V163A, I164A, Y167A) of RRM2, and the linker region (R75D, R88D) were mutated. ITC data indicated that these residues contribute to the interaction with the apical loop of 5BSL3.2 RNA, as alanine substitutions reduced binding affinity by approximately three- to fivefold compared to wild-type hnRNPA1 (residues 1–196) (Supplemental Fig. S9; Table 2). In conclusion, the data clearly show that both RRM domains play a direct role in RNA recognition. Additionally, the observed chemical shift changes in residues within the interdomain linker suggest that RNA binding induces conformational dynamics and structural adaptation at this interface.
Crystal structure of UP1–5BSL3.2Apical stem loop (5′-AG-3′) RNA interactions
The crystal structure of UP1 in complex with the 5BSL3.2Apical stem loop (5′-AG-3′) RNA element was determined using X-ray diffraction to a resolution of 1.75 Å (Supplemental Fig. S10; Supplemental Table S4). Crystals belonged to the monoclinic space group P21, comprising only of dinucleotide sequence 5′-AG-3′ as the rest of the RNA was hydrolyzed during the crystallization process. The final electron density maps allowed unambiguous modeling of both the protein and RNA components with a final R and Rfree of 0.168 and 0.2. The RNA ligand occupies a conserved binding groove within UP1, consistent with previous structural observations (RCSB PDB: 4YOE), particularly the UP1–ESS3 complex. This binding pocket is formed by the canonical RNA Recognition Motif 1 (RRM1) and the interdomain linker connecting RRM1 to RRM2, which together adopt a clamp-like conformation that embraces the RNA (Supplemental Fig. S10A).
The RNA nucleotides adenine (A) and guanine (G) are well-resolved, both adopting a syn conformation that facilitates a network of stabilizing interactions with residues in the RRM1 domain and the linker region. Interestingly, both A and G are in a syn conformation as observed in the previously reported UP1–ESS3 complex (Morgan et al. 2015). Adenine and guanine are stabilized in the pockets by multiple interactions. Notably, Phe17, His101, and Phe59 participate in π–π stacking interactions with the planar RNA bases, anchoring the nucleotides securely within the hydrophobic cleft (Supplemental Fig. S10B,D). Adenine forms hydrogen bonds with backbone atoms of Arg88 and Val90, while guanine forms hydrogen bonds with Ser95, Gln12, and Lys15 side chains. These interactions contribute to the overall selectivity of these purine bases in the RNA-binding cleft. Ribose moieties of the RNA are stabilized by hydrogen bonds made by their 2′-OH atoms with side chains of His101, Ser 95, and Arg 92 residues. Phosphate groups are stabilized by interactions with Arg55. Met46, through hydrophobic interactions, assists in shaping the binding groove and supporting nearby polar interactions (Supplemental Fig. S10D). Structural superimposition of this UP1–5BSL3.2Apical stem loop (5′-AG-3′) complex with that of UP1–ESS3 complex structure (PDB ID: 4YOE) reveals a highly conserved RNA-binding mode, highlighting the functional importance of this clamp-like conformation for RNA recognition (Supplemental Fig. S11).
Structural modeling for UP1 and 5BSL3.2 RNA complex
HADDOCK-based modeling of the UP1–RNA complex revealed that the interaction extends beyond RRM1 and the interdomain linker region. The orientation of UP1 on the 5BSL3.2 terminal loop was guided by the crystal structure, which defined the binding interface between the RNP residues of RRM1 and the linker with the 5′-AG-3′ nucleotides. Notably, the orientation and involvement of RNP residues from RRM1 and the linker in the HADDOCK model closely resemble those observed in the crystal structure (Morgan et al. 2015), confirming that this region provides the primary RNA recognition surface. In contrast, the distinct feature of our HADDOCK-derived model lies in the inclusion of RRM2, whose positioning was defined using NMR-derived ambiguous interaction restraints (AIRs) rather than crystallographic data.
The docked ensemble indicates that adenine residues within the RNA engage with key aromatic and basic residues on the surface of RRM1, consistent with previous NMR chemical shift perturbation and crystallographic observations. Importantly, the spatial arrangement in the top-ranked HADDOCK models demonstrates that RRM2 lies in close proximity to the RNA-binding site, with a distance of ∼7 Å between the RNA and residues within RRM2 (Fig. 7). This is in contrast to the model proposed by Tolbert et al., where RRM2 is positioned ∼20 Å away from the apical loop (Morgan et al. 2015). The closer positioning of RRM2 in our model suggests that it may directly contribute to RNA recognition, supporting a cooperative binding mechanism in which both RRM1 and RRM2 act together to engage the 12 nt purine-rich apical loop of the 5BSL3.2 element.
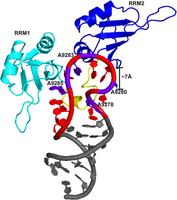
Model for 5BSL3.2Apical loop stem and UP1. The loop adenine residues are highlighted in purple, while the remaining loop residues are shown in red. The interactions of UP1 with the adenines of the loop region involve not only RRM1 and the linker region (cyan), but also suggest the proximity of RRM2 (blue), as indicated by 5BSL3.2 apical loop and RRM2 distance of ∼7 Å.
DISCUSSION
HCV replication is critically dependent on conserved long-range RNA–RNA interactions that govern genome architecture and function. A particularly important interaction is the kissing-loop pairing between the apical loop of the 5BSL3.2 cis-acting element and the 3′X tail (SL2), which facilitates genome circularization and promotes replication (Tabata et al. 2020). Given the structural sensitivity of this interaction, host RNA-binding proteins (RBPs) have the potential to modulate viral replication by remodeling these regulatory RNA elements. In this study, we addressed the central biological question of whether hnRNPA1, a host RBP known to influence RNA structure and metabolism, binds to the 5BSL3.2 element and interferes with this essential viral RNA–RNA contact. Our findings reveal a mechanism by which hnRNPA1 negatively regulates HCV replication by directly binding to the terminal loop of 5BSL3.2 RNA and potentially affecting its ability to form a kissing-loop interaction with the 3′X tail (SL2). This was supported by MST binding assays, which showed that increasing concentrations of hnRNPA1 progressively weaken the 5BSL3.2–3′X tail (SL2) RNA complex. These results suggest that hnRNPA1 competes with the 3′X tail (SL2) for binding to the apical loop, sterically or conformationally blocking this critical RNA–RNA interaction.
Structural and biophysical studies further delineate the molecular basis of this recognition. hnRNPA1 engages the adenosine-rich apical loop of 5BSL3.2 cooperatively through both its RNA recognition motifs (RRMs). Notably, a single 5′-AG-3′ motif within the loop, together with flanking adenosines, provides an optimal binding platform. While earlier work demonstrated that hnRNPA1's RRM1 alone is sufficient to recognize short UAG motifs (Morgan et al. 2015; Kooshapur et al. 2018), our NMR and ITC data indicate that both RRM1 and RRM2 contribute significantly to the high-affinity binding of the extended 5BSL3.2 loop. This mode of dual RRM engagement resembles that observed in the context of pri-miR-18a, where two UAG motifs are cooperatively recognized by hnRNPA1 (Kooshapur et al. 2018). Furthermore, our studies indicate that the C-terminal RGG domain is dispensable for interaction with the CRE element. Previous structural studies have revealed that hnRNPA1 maintains a similar overall domain arrangement and interface in both its free and RNA-bound forms, with some conformational adjustments and fine-tuning upon RNA engagement (Kooshapur et al. 2018). Our data further support this model, highlighting specific adaptations in domain positioning to facilitate high-affinity and selective recognition of the 5BSL3.2 RNA. While our data align with previous findings on 12-mer RNA, where both RRMs of hnRNPA1 engage cooperatively to recognize two UAG motifs, as reflected by comparable line broadening of amide resonances in both RRM1 and RRM2, we also observe distinct differences that reflect adaptation to the adenosine-rich sequence in 5BSL3.2. These differences highlight the structural versatility of hnRNPA1, revealing its ability to undergo subtle conformational rearrangements to accommodate diverse RNA targets. This inherent plasticity likely underlies hnRNPA1's capacity to interact with a wide range of cognate RNAs and regulate multiple biological processes with specificity.
Our study underscores the critical role of the apical loop within the 5BSL3.2 stem–loop of the CRE domain in the HCV genome, which has been previously implicated in viral replication through various mutational analyses (Friebe et al. 2005). This loop engages in long-range base-pairing with the 3′ X-tail (SL2), an interaction essential for efficient viral replication (Friebe et al. 2005). Phylogenetic analyses in our study reveal a remarkable conservation of the 5BSL3.2 secondary structure across different HCV genotypes, indicating an additional layer of evolutionary constraint and emphasizing the value of integrating phylogenetic methods with RNA structural probing to uncover functionally relevant features. Furthermore, our mutational and NMR studies demonstrate that hnRNPA1 specifically binds to the apical loop of 5BSL3.2 via its tandem RRMs, without any detectable involvement of the bulge region. This direct interaction with the terminal loop appears to interfere with the formation of the kissing-loop complex, a critical RNA–RNA interaction required for replication. Notably, NMR chemical shift perturbation (CSP) data highlight that adenosine residues within the apical loop are key determinants of hnRNPA1 recognition. Mutation of these adenosines to cytosines abolishes binding, as evidenced by NMR, ITC, and analytical size exclusion chromatography, confirming the specificity of this interaction.
Interestingly, our data reveal that the residues 5′AG3′ (A9285 and G9286) within the 5BSL3.2 RNA element undergo a conformational rearrangement upon binding to hnRNPA1. In the free RNA, NMR analysis, including 13C-HMQC experiments, clearly shows that both adenine (A9285) and guanine (G9286) adopt an anticonformation, as indicated by the absence of the characteristic downfield shift at the C8 position that typically signifies a syn conformation (Supplemental Fig. S12). This observation contrasts with the conformation captured in the hnRNPA1–RNA complex crystal structure, where both A9285 and G9286 are switched to a syn conformation (Supplemental Fig. S10B), suggesting that binding to hnRNPA1 induces this specific structural change in the RNA. Interestingly, in an earlier NMR study of the free ESS3 viral RNA, the 5′G3′ was suggested to adopt a syn conformation (Levengood et al. 2012), whereas our data do not support such a shift for either residue under free conditions. These findings suggest that the 5′AG3′ segment of 5BSL3.2 RNA undergoes a protein-induced conformational switch to accommodate hnRNPA1 binding, enabling specific recognition by the protein. This conformational adaptability of the RNA likely plays an important role in the molecular mechanism by which hnRNPA1 discriminates its cognate RNA targets, emphasizing the dynamic nature of RNA structure in mediating protein interactions.
Our combined crystal and NMR data give a clearer understanding of how hnRNPA1 recognizes 5BSL3.2 RNA and also help explain earlier structural findings. The crystal structure of our complex, which features a short RNA fragment from the apical loop region, closely resembles the first crystal structure of the UP1 domain bound to RNA reported by Tolbert and colleagues (Morgan et al. 2015). In that study, a 7 nt loop containing a single UAG motif was shown to interact primarily with RRM1 and the inter-RRM linker, with little to no contribution from RRM2. Consistently, our structure with improved resolution reveals a similar binding pattern, limited to RRM1 and the linker. However, it is worth noting that in both cases, the RNA was partially hydrolyzed, and only 2 nt are resolved in the density, restricting our ability to infer interactions across the full-length loop.
In contrast, our solution NMR studies using a 12 nt apical loop containing a single AG motif, along with additional adenines spaced throughout the loop, show substantial chemical shift perturbations in both RRM1 and RRM2. These results align with the findings of Kooshapur et al., who demonstrated through NMR and crystallographic analyses that both RRMs of hnRNPA1 are involved in recognizing the 12 nt terminal loop of pri-miRNA-18a (Kooshapur et al. 2018). Notably, RRM2 engagement appears to be especially important when multiple purines are present and properly spaced within the RNA loop. Our data are consistent with the quantitative affinity profiling by Jain et al. (2017), which demonstrated that hnRNPA1 binds more strongly to RNAs containing multiple, well-spaced AG motifs. This supports a cooperative binding mechanism, suggesting that both RRMs function together, rather than a single RRM and the linker, to enhance higher binding affinity and specificity. Our structural and biophysical data indicate that, in addition to RRM1 and the linker, RRM2 plays a key role in multivalent RNA interactions, particularly when purines are distributed across extended loop regions. This modular and context-dependent mode of RNA recognition by hnRNP A1 likely represents a versatile strategy that enables the protein to selectively bind a diverse array of RNA targets across various cellular environments.
Finally, our data support a mechanistic model in which the kissing-loop interaction between the apical loop of 5BSL3.2 and the 3′X tail is progressively attenuated in a concentration-dependent manner by hnRNPA1. Increasing levels of hnRNPA1 reduce the strength of this specific RNA–RNA interaction, likely by competitively binding to the viral RNA 5BSL3.2 terminal loop (Fig. 8). This highlights the role of hnRNPA1 as a regulatory host factor that modulates the formation or stability of critical long-range RNA–RNA contacts within the HCV genome. Such modulation may influence essential steps in the viral replication cycle, underscoring the dynamic interplay between viral RNA structures and host proteins that fine-tune HCV replication efficiency. We further hypothesize that this negative regulation of viral replication may act in parallel with hnRNPA1, competing with NS5B for CRE binding and thereby reducing the efficiency of the viral polymerase in RNA replication, as proposed in previous studies (Ríos-Marco et al. 2016). Although our studies indicate that hnRNPA1 binds specifically to the apical loop, with no involvement of the bulge region, NS5B has been shown to recognize the bulge region of the 5BSL3.2 RNA (Zhang et al. 2005; Kanamori et al. 2010). Future studies are needed to more thoroughly investigate the potential competitive interaction of these two proteins at this CRE domain.
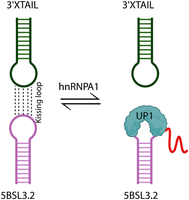
Model illustrating the disruption of kissing-loop interactions. The interactions between 5BSL3.2 and the 3′X tail are disrupted with increasing concentrations of hnRNPA1 (UP1) protein.
A limitation of the present work is that it focuses exclusively on structural and biochemical analyses of the hnRNPA1–5BSL3.2 interaction and does not include replication studies. While our NMR, MST, EMSA, ITC, biophysical experiments, and functional assay clearly demonstrate specific binding of hnRNPA1(UP1) to the conserved apical loop of 5BSL3.2, we were not able to assess the direct impact of this interaction on viral RNA replication or infectivity, as this would require access to HCV replicon or infectious systems that were beyond the scope of the current study. Future studies using subgenomic replicons will be essential to determine how hnRNPA1 binding influences HCV replication dynamics and to validate the mechanistic model proposed here.
In conclusion, this study provides mechanistic insights into how hnRNPA1 selectively binds the apical loop of the 5BSL3.2 element of the HCV genome, thereby it may interfere with the critical long-range RNA–RNA interaction essential for replication. The cooperative engagement of both RRMs, the RNA-induced conformational plasticity, and the evolutionary conservation of the apical loop underscore the biological relevance and structural complexity of this interaction. These findings offer a broader perspective on how host RBPs dynamically interact with viral RNAs to regulate pathogenesis and may inform future antiviral strategies targeting host-viral RNA interfaces.
MATERIALS AND METHODS
Phylogenetic analysis of 5BSL3.2 elements
The complete genome sequences of HCV containing the 5BSL3.2 elements were retrieved from the NCBI database. These data included up to 10 sequences of each genotype, which were then used for phylogenetic analysis of the 5BSL3.2 elements. Sequences corresponding to specific genotypes/isolates were subjected to alignment using the ClustalW multiple-sequence alignment algorithm in Geneious (version 2023.2.1). Geneious interface was then used to build consensus sequences and logos for each isolate, relying on the predominant nucleotide at each site. Conserved regions were established for every isolate by utilizing a threshold frequency (indicated by logo height) within the consensus logo derived from aligned sequences of that isolate. Additionally, a universal consensus sequence and logo were generated by aligning the consensus sequences of each isolate using Geneious as described above.
The MEGA-X software created a phylogenetic tree using the consensus sequences from all genotypes/isolates. Before constructing the tree, the consensus sequences were aligned to assess the extent of phylogenetic relatedness among all genotypes/isolates. Afterward, the aligned data were employed to interpret a phylogenetic tree using the maximum likelihood (ML) method. To enhance the robustness of the tree, 1000 bootstrap replicates were conducted. Additionally, a partition analysis was carried out, and the average genetic distance was computed using MEGA-X software. The resulting tree was then visualized using the iTOL server. One sequence from each genotype was then folded using the RNAstructure web server.
5BSL3.2 and the mutant RNA preparation
The RNA constructs used in this study, including 5BSL3.2 and its mutant variants (Table 3), were produced via in vitro transcription. Synthetic DNA templates corresponding to the 5BSL3.2 and mutant RNA constructs were purchased from IDT. In vitro transcription was performed, and purified RNA samples were collected as previously described (Kumar et al. 2023). The NMR sample was prepared using an equimolar mix: ATP, GTP (ribose-3′,4′,5′,5′-D4,98%); CTP, UTP (5-D1, ribose-3′,4′,5′,5′-D4,98%) (Cambridge Isotope Laboratories, Inc.) along with uniformly 13C/15N-labeled rNTPs (Sigma-Aldrich). The purified RNA samples were stored at −20°C until further use. The 5BSL3.2 RNA, the terminal base pair 5′C-G was switched to 5′G-C, and 5′A-U was replaced with 5′GC for efficient transcription.
All the RNA constructs used in this study
Affinity-based RNA pull-down
HepG2 cells were maintained in Dulbecco's modified Eagle medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS; Life Technologies) under standard culture conditions. For protein extraction, cells were washed with 1× phosphate-buffered saline (PBS; Gibco) and lysed using RIPA buffer on ice for 30 min. The resulting lysate was clarified by centrifugation at 13,000 rpm for 10 min, and the supernatant was collected. Protein concentrations were quantified using the BCA Protein Assay Kit (Pierce).
In vitro transcribed RNAs were labeled at the 3′ end with biotin using the Pierce RNA 3′ End Biotinylation Kit, following the manufacturer's protocol. For the RNA pull-down experiment, 100 pmol of each biotinylated RNA was incubated with ∼500 µg of HepG2 lysate (preincubated with RNase inhibitor and yeast tRNA) on a rotator at 4°C for 5 h. RNA–protein complexes were captured using magnetic streptavidin beads (Dynabeads MyOne Streptavidin; Invitrogen) with lysate for 90 min at 4°C. Beads were washed five times with 1× RIPA buffer supplemented with a protease inhibitor cocktail (PIC), and bound proteins were eluted in 25 µL of 2× SDS loading buffer. The eluates were analyzed by western blot (n = 3) using anti-hnRNPA1 antibody (Cell Signaling Technology 5380).
Protein purification
The hnRNPA1 constructs, encompassing residues hnRNPA1 1–249, 1–196 (UP1), and residues 11–88, 89–179 were subcloned from the pET9d-hnRNPA1 vector into the pET28a (+) vector. UP1 mutant constructs were generated using site-directed mutagenesis using Kapa HiFi HotStart ReadyMix (Roche). For the MST experiments, the UP1 protein was tagged with GFP. These constructs were subsequently expressed in Escherichia coli (E. coli) Rosetta cells, producing fusion proteins tagged with an N-terminal (His)6 tag. The transformed bacterial cultures were cultured in LB media supplemented with kanamycin (50.0 µg/mL) and chloramphenicol (25 µg/mL) at a temperature of 37.0°C until reaching an OD600 of 0.4. Subsequently, induction was done by 0.2 mM IPTG at a temperature of 30°C at an OD600 of 0.6–0.8, with continuous shaking at 200 rpm overnight. The cells were harvested and lysed, and (His)6-tagged proteins were purified using the same buffer conditions as previously described (Kumar et al. 2023). Followed by purification using Superdex 75–200 pg gel filtration columns (Cytiva) and eluted into the binding buffer (10 mM K2HPO4, 120 mM KCl, 10 mM NaCl, 0.5 mM EDTA, 1 mM TCEP/DTT) under conditions reported previously (Kumar et al. 2023), the protein samples were stored at 4°C prior to use. UP1 protein for MST experiments was eluted from a gel filtration column in 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 0.05% Triton-X, and 1 mM DTT. Protein purity was evaluated using SDS-PAGE (Supplemental Fig. S1). U-[15N]-labeled proteins were expressed in M9 media supplemented with 1 g/L of 15N-labeled NH4Cl (98%) as the sole nitrogen source to produce U-[15N] UP1. Protein expression and purification were performed according to the standard procedure described above; additionally, the His-tag was cleaved from UP1 by the addition of TEV protease overnight at 4°C, followed by further purification. For all NMR experiments, the final volume in the NMR tube included 10% D2O. The ProtParam program was used to extract the theoretical extinction coefficient, and the protein concentration was calculated using the Thermo Scientific NanoDrop 1000.
NMR data acquisition, processing, and analysis
Two-dimensional NMR experiments were carried out using a Bruker Neo spectrometer operating at 600 MHz proton frequency and equipped with a triple-resonance cryogenically cooled probe. To assess RNA conformational homogeneity and purity, one-dimensional 1H NMR experiments were performed. The acquired NMR data were processed using NMRPipe/NMRDraw (Delaglio et al. 1995) and analyzed with NMRViewJ software (Johnson and Blevins 1994). Exchangeable 1H spectra were recorded at 303 K using the Watergate NOESY pulse sequence with a mixing time (τm) of 250 msec. Nonexchangeable protons were assigned following well-established protocols. 1H–1H NOESY (τm = 250 msec), 1H–1H TOCSY (τm = 75 msec) and for Riboses 1H–1H TOCSY (τm = 120 msec) spectra were collected in 100% D2O at 303 K. Samples were labeled with deuterated equimolar rNTPs, rRTPs (3′,4′,5′,5″), and rYTPs (5,3′,4′,5′,5″). To confirm NOE assignments, 1H–13C heteronuclear multiple quantum coherence (HMQC) spectra were measured for various 5BSL3.2Apical stem loop constructs. Chemical shift assignments were determined for all aromatic protons and ribose protons (H1′–H2′). All the experiments carried out are listed in Supplemental Table S1.
Structure calculation and refinement
Distance restraints for NMR structure calculations of the 26 nt 5BSL3.2 apical stem–loop RNA were obtained from 2D 1H–1H NOESY spectra (mixing time: 250 msec), using RNA samples synthesized with either fully protonated or partially deuterated ribonucleotides. NOE cross-peaks were grouped based on their intensities into four categories: strong (1.8–3.0 Å), medium (2.5–4.5 Å), weak (3.5–6.0 Å), and very weak (3.5–7.5 Å). For residues within the apical loop, only very weak NOEs were considered for restraint generation. Sugar pucker conformations were assigned using TOCSY spectra, based on the intensity of H1′–H2′ cross-peaks. Nine out of 12 loop residues displayed medium to strong cross-peak intensities, suggesting a C2′-endo pucker; the remaining loop nucleotides were modeled in the C3′-endo conformation. Additional A-form-specific distance restraints, involving H8/H6–H3′/H4′ proton correlations, were incorporated, consistent with the sugar pucker data and canonical helical geometry. Backbone dihedral angle restraints (±20°) consistent with A-form RNA were applied to all residues except those in the loop region (positions 9276–9288). Glycosidic torsion angles were restricted to the anticonformation (180° ± 90°) for all nucleotides.
Hydrogen-bonding and base pair planarity restraints (force constant: 20 kcal·mol−1·Å−2 in Xplor-NIH) were applied to all Watson–Crick base pairs, as identified from 1D proton chemical shifts and NOESY spectra recorded in both H2O and D2O. Additional loose hydrogen bond restraints were applied to the terminal G-C base pair of the stem and the A-U base pair in the apical loop. In total, 2000 structural (200 structures produced in each of 10 independent runs, initialized with distinct random seeds) models of the 26 nt apical stem–loop (5BSL3.2) RNA were generated using a multistage simulated annealing protocol within the Xplor-NIH v2.25 software package (Schwieters et al. 2003), utilizing the standard nucleic-2.0.par topology and parameter files. The simulations were conducted within the NMRBox platform (Maciejewski et al. 2017).
A rigorous two-stage calculation strategy was employed to ensure both comprehensive conformational sampling and high-resolution refinement (Maciejewski et al. 2017). The initial “global folding” stage began with high-temperature torsion angle dynamics (TAD) at 8000 K for 75 psec, starting from a randomized, extended RNA conformation. This was followed by a slow-cooling TAD phase where the system was annealed from 8000 to 1000 K over 300 psec, during which the van der Waals repulsive term was gradually increased. The protocol then transitioned to full Cartesian space dynamics for refinement of local geometry, with cooling from 1000 to 300 K over 60 psec. Finally, each structure underwent Powell energy minimization using high force constants for all experimental restraints. To achieve robust convergence, the global folding protocol was carried out in 10 independent runs, each initiated with a distinct random seed and producing 200 structures, resulting in an exhaustive total of 2000 conformers. From this extensive pool, the 20 lowest-energy structures that exhibited no NOE violations >0.2 Å and no dihedral violations >5° were selected to initiate the second “gentle folding” refinement stage. These selected conformers were subjected to a second, complete simulated annealing protocol that used milder thermal conditions to refine the already well-folded structures. This refinement included a high-temperature TAD step at 1000 K, followed by cooling in both torsion angle (1000–300 K) and Cartesian space (300–0 K), and concluded with a final energy minimization.
Following refinement, the final structures were pooled, and an ensemble of the 20 conformers with the lowest overall energy and no restraint violations was selected to represent the solution structure of HCV 5BSL3.2Apical stem loop. The stereochemical quality of the final ensemble was rigorously assessed using the MolProbity tool (Chen et al. 2010). The 20 lowest-energy structures were analyzed using X3DNA (Lu and Olson 2008) and visualized in PyMOL (DeLano 2002) Molecular Graphics System.
UV melting method for calculating Tm
Thermal melting assays were conducted on a UV-Vis spectrophotometer (Agilent Cary 3500). The 5BSL3.2 and 5BSL3.2Apical stem loop RNA constructs were prepared in the same binding buffer as reported previously (Kumar et al. 2023). The RNA samples were refolded by heating at 95°C for 2–3 min, then immediately incubated on ice. The samples were added to a cuvette with a 10 mm path length. At a ramp rate of 0.5°C/min, melting measurements at 260/280 nm were collected spanning the temperature range of 20°C–95°C. The first derivatives of melting curves were used to determine Tm. The melting data for all constructs were collected in triplicate.
Gel mobility shift assay
The gel mobility shift assay was carried out by incubating 200–500 nM of 5BSL3.2 RNA with increasing concentrations of recombinant
different hnRNPA1 protein constructs at 21°C for 2 h in the same binding buffer conditions as previously described (Kumar et al. 2023). The reaction products were separated by electrophoresis on a 6% polyacrylamide gel in 1× TBE at 100 V for 1.2 h at a gel
temperature of 4°C. Band shifts were visualized after staining with SYBR Gold using the GelDoc XR+ (Bio-Rad), and band widths
were quantified using Image Lab software. For the RNA–protein complex, the fraction bound was used for binding affinity calculation,
while for single RRMs, where the complex was not detected, however, the amount of free RNA was decreased with increasing RRM
concentration; thus, the binding affinity was calculated using the unbound fraction. All gel mobility assays were carried out at least three times. Subsequently, the data were analyzed using a nonlinear regression
method, and the KD was determined by the Hill slope method in GraphPad Prism 8.
All gel mobility assays were carried out at least three times. Subsequently, the data were analyzed using a nonlinear regression
method, and the KD was determined by the Hill slope method in GraphPad Prism 8.
Analytical gel filtration assay
Gel filtration assay was conducted using a Superdex 200 10/300 GL column (GE Healthcare). A gel filtration assay was used to examine the mobility of the 5BSL3.2 and different mutant RNA complexes with the UP1 protein. The experiments were carried out with aliquots of 100 µL of free 5BSL3.2 RNA and RNA–UP1 complexes with UP1 protein at ratios ranging from 0 to 1.5 (0, 0.25, 0.5, 0.75, 1, and 1.5) injected into the column at a flow rate of 0.5 mL/min. Similarly, various mutant RNA and mutant RNA–UP1 complexes were injected. Prior to injecting, the complexes were incubated at 21°C for 2 h in the same binding buffer as used in the gel mobility shift assay. The approximate molecular weight of complexes was determined using a low molecular weight calibration kit (Cytiva).
Circular dichroism (CD) spectroscopy
CD spectra were recorded using a J-815 spectropolarimeter (JASCO Inc.) to assess the secondary structure of the RNA constructs. Measurements were performed in a quartz cuvette with a path length of 0.1 cm, using RNA samples at a final concentration of 5 µM. The spectra were collected over a wavelength range of 190–320 nm under constant nitrogen flow to minimize atmospheric interference. All samples were prepared in binding buffer to maintain the native folding environment and to ensure consistent conditions across measurements. Data acquisition was carried out at a scanning speed of 100 nm/min, with each spectrum representing the average of five consecutive scans to improve the signal-to-noise ratio. The temperature during all measurements was maintained at 20°C using a Peltier temperature controller to avoid thermal fluctuations that could impact RNA conformation. Baseline correction was applied by subtracting the buffer spectrum recorded under identical conditions.
Isothermal calorimetry titration of UP1
Titrations were performed at a temperature of 25°C utilizing a MicroCal PEAQ-ITC instrument from MicroCal, LLC. The protein and RNA constructs were exchanged in the same binding buffer as reported in the gel shift assay. The RNA constructs were annealed by heating at 95°C for 3 min, followed by rapid cooling on ice. (His)6 UP1 and UP1 (1–249) constructs at 60–80 µM were titrated into 300 µL of 2.5–5 µM of 5BSL3.2 and mutant RNA constructs throughout 19 injections of 2 µL each. All titrations were conducted in triplicate. Titrations of (His)6-RRM1 and (His)6-RRM2 at 150–200 µM into 5BSL3.2 (3 µM) were conducted. All titrations were conducted in triplicate. A control experiment involving protein titration into buffer verified that any heat generated due to titrant dilution was negligible. Utilizing the fitted offset option, the control heat was automatically deducted from the titration, and integrated heat data from the titration were fitted using the MicroCal PEAQ-ITC analysis program. A one-set site binding model was applied to fit the isotherms.
NMR titrations of UP1–5BSL3.2Apical stem loop
Two-dimensional 1H–13C HMQC titration experiments were carried out to investigate the interaction between selectively labeled RNA and the UP1 protein. Specifically, 13C-selectively labeled 5BSL3.2 apical stem–loop RNA constructs were titrated with increasing concentrations of unlabeled UP1 protein. The titration points included four different protein-to-RNA molar ratios: 0.25, 0.50, 0.75, and 1.0, ensuring a progressive evaluation of binding interactions. All titrations were performed in 5 mM K2HPO4 buffer at pH 6.5, with the temperature maintained at 303 K to ensure structural stability of the RNA.
For the complementary protein titration, 2D 1H–15N HSQC spectra were acquired using uniformly 15N-labeled UP1 (U-15N-UP1) to monitor backbone amide chemical shift perturbations upon RNA binding. These spectra were recorded on a Bruker Avance
II 600 MHz NMR spectrometer equipped with a triple-resonance cryogenically cooled probe at a temperature of 298 K. The protein
samples were prepared in the same binding buffer with the addition of 10% D2O for the spectrometer lock signal. All NMR data were processed using TopSpin 4.1.1 (Bruker), and spectral analysis was conducted
with NMRFAM-SPARKY (Lee et al. 2015). Chemical shift assignments for the free UP1 protein were referenced from previously published 1H–15N HSQC data (Barraud and Allain 2013). For the titration, the unlabeled 12 nt linear apical loop derived from 5BSL3.2 was gradually added to the 15N-labeled UP1 protein sample to achieve the desired molar ratios. After each addition, the mixture was incubated for 10–20
min to allow equilibration before acquiring the NMR spectra, ensuring complete binding and stable complex formation prior
to data collection. Chemical shift perturbations (CSPs) were calculated by observing 1H chemical shift changes of UP1 upon
binding to a 12 nt linear apical loop compared with the free UP1 at a 1:1 molar ratio.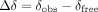
Crystallization, X-ray data collection, processing, model building, and refinement
Purified UP1 (hRNPA1; 1–196 aa) was mixed with 5BSL3.2Apical stem loop in a 1:1.2 molar ratio in SEC buffer (10 mM K2HPO4, 120 mM KCl, 10 mM NaCl, 0.5 mM EDTA, 1 mM TCEP), and the complexed protein was separated from unbound protein using a Superdex 200 10/300 GL. Fractions containing complexed protein were pooled and concentrated to 15 mg/mL. Crystallization trials of the UP1–5BSL3.2Apical stem loop were set up in sitting drop plates using the Mosquito robot (TTP LabTech). Plates were incubated at 20°C in a vibration-free incubator (RUMED). Initial screening was done with commercially available screens (Crystal Screen I and II, JCSG+, PACT Premier). Diffraction quality crystals appeared in conditions containing 0.2 M potassium formate and 20% w/v PEG3350, pH 7.3. Crystals were cryo-cooled in the reservoir condition containing 20% glycerol. X-ray diffraction data for UP1–5BSL3.2Apical stem loop (5′-AG-3′) complex crystals were collected at the XRD2 beamline (Lausi et al. 2015), Elettra Sincrotrone Trieste, using Dectris PILATUS 6M detector at a wavelength of 0.99 Å. The collected data were processed using the autoPROC (Vonrhein et al. 2011). Phases were determined using the molecular replacement method using Phaser-MR from the Phenix suite (Adams et al. 2010; Liebschner et al. 2019), with RNA recognition motifs (RRMs) serving as the search model. Automated model building was performed with the AutoBuild module, followed by iterative manual adjustments using Coot (Emsley et al. 2010) and refinement with Phenix refine (Bricogne et al. 2011). Crystallographic refinement statistics are provided in Supplemental Table S3. Structural figures were prepared using UCSF Chimera (Pettersen et al. 2004).
Docking calculations
The structural model of the UP1-HCV_5BSL3.2 protein–RNA complex was generated using the HADDOCK 2.5 software suite (Dominguez et al. 2003; Honorato et al. 2024) with the CNS 1.31 engine (Dominguez et al. 2003; HADDOCK3 2022). Initial coordinates for UP1 protein (Barraud and Allain 2013) and HCV_5BSL3.2 RNA were sourced from Xplor structure calculations. Histidine protonation states in UP1 were manually set, and nonpolar hydrogens were subsequently removed. The docking was guided by experimentally derived ambiguous interaction restraints (AIRs) obtained from NMR titration experiments. Specifically, AIRs were generated from 13C adenine-selective HMQC titration data of the 5BSL3.2 apical stem–loop with unlabeled UP1, and from 1H–15N HSQC titration data of 15N-labeled UP1 with the unlabeled linearized apical loop RNA. To ensure robustness, 50% of the restraints were randomly excluded in each docking trial as part of a cross-validation procedure. To maintain RNA fold integrity, standard nucleic acid planarity and hydrogen-bonding restraints were active throughout all stages. The OPLS nonbonded force field was employed, with a distance-dependent dielectric constant to model electrostatics. The HADDOCK protocol began with a rigid-body minimization stage that generated 10,000 initial models, incorporating a 180° rotational sampling step to reduce orientational bias. The 200 best-scoring structures from this stage proceeded to semiflexible simulated annealing, where residues at the interface, defined automatically from the AIRs, were allowed conformational freedom during a multistep cooling protocol. Finally, these 200 models were refined in an explicit water shell through a short molecular dynamics simulation to optimize interface electrostatics and hydrogen bonding. The resulting structures were clustered using a fraction of common contacts (FCC) metric with a 0.60 cutoff, requiring a minimum of four members per cluster. The final model was selected from the top-ranked cluster based on HADDOCK scores, while also meeting the NMR restraints defined by complete line broadening and chemical shift perturbation (CSP) patterns used to identify active and passive residues as well as crystal structure restraints. Further, a weighted combination of van der Waals, electrostatic, desolvation, and restraint violation energies, and their structural quality were validated using PROCHECK (Laskowski et al. 1993, 1996). All structural visualizations were rendered using PyMOL.
Microscale thermophoresis (MST) assay
MST assays were carried out at least three times using a Monolith NT.115 system (NanoTemper Technologies). The Cy5-labeled 5BSL3.2 RNA and 3′X tail (SL2) RNA solutions were prepared in 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 10 mM MgCl2, 0.05% Triton-X, and 1 mM DTT. The Cy5-labeled RNA and 3′X tail RNA were annealed by heating at 95°C for 3 min, followed by snap cooling on ice.
Control experiments
A series of dilutions of the unlabeled 3′X tail (SL2) was made in Cy5-labeled 5BSL3.2 (100 nM), with a final concentration of 3′X tail (SL2) ranging from 10 µM to 0.09 nM. The complexes were kept at RT for at least 10–15 min and then added to Monolith premium capillaries to carry out MST analysis. Similarly, a series of dilutions of unlabeled UP1 protein (ranging from 10 µM to 0.09 nM) in Cy5-labeled 5BSL3.2 RNA (100 nM) was prepared and kept at RT for 2 h. Temperature jump (TJump) analysis was used to examine the data, and the resulting values were normalized and displayed against the concentrations of 3′X tail (SL2) RNA and UP1 protein, respectively. A single-site model was then used to fit the curve and get the dissociation constant.
Cy5-labeled 5BSL3.2-UP1 and 3′X tail (SL2) (competition)
A sample solution of Cy5-labeled 5BSL3.2 RNA (100 nM) and UP1 protein at a molar ratio of 1:1, 1:2, and 1:5 was prepared in MST buffer and kept at 21°C for 2 h. Subsequently, a twofold dilution series of 3′X tail (SL2) RNA ranging from 10 µM to 0.09 nM was prepared, and the complex was incubated at 21°C for 10–15 min. Temperature jump (TJump) analysis was used to analyze the data, and the resulting values were then normalized and plotted against the concentration of the 3′X tail (SL2). A single-site model was then used to fit the curve and calculate the dissociation constant.
DATA DEPOSITION
All data are available within the manuscript and supplemental figures/tables. The crystal data underlying this article can be accessed with data set ID: D_1300061575 and PDB ID: 9VVA. The NMR solution data underlying this article can be accessed at PDB ID 9W1L, Extended PDB ID pdb_00009W1L BMRB entry 53244, 5BSL3.2APICAL_stem_loop.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the Department of Biotechnology, Government of India–Ramalingaswami fellowship grant (BT/RLF/Re-entry/HRD/35/2019), Department of Biotechnology (DBT) to N.J. and CSIR OLP2303. A.K. is supported by a Senior Research Fellowship (SRF) from the Department of Biotechnology (DBT), India. We also thank Dr. Akila Mayeda (Fujita Health University) for providing the constructs for hnRNPA1. We thank the beamline staff at the Elettra XRD2 beamline, particularly Dr. Raghurama P. Hegde, for beamline support. Access to the XRD2 beamline at the Elettra Sincrotrone Trieste was made possible through a grant-in-aid from the Department of Science and Technology (DST), India, vide grant number DSTO-1668. We also thank Dr. Blanton Tolbert for critically reading this paper. This study made use of NMRbox: National Center for Biomolecular NMR Data Processing and Analysis, a Biomedical Technology Research Resource (BTRR), which is supported by National Institutes of Health (NIH) grant P41GM111135 (National Institute of General Medical Sciences [NIGMS]). Graphical abstract and Figure 8 images were created using BioRender software (https://BioRender.com).
Author contributions: N.J. conceived the study, designed the experiments, and secured funding. A.K. carried out the experiments, with V.S. assisting in cell culture, P.K. and S.T. with the SDM studies, and P.D. with the analytical SEC experiment. A.K. and N.J. performed data analysis and interpreted the results. M.V.D. set up NMR experiments, collected the data, and performed preliminary analysis with N.J., while A.K. and N.J. analyzed all NMR data, and S.P. conducted the simulations. L.P. performed crystallization screening, and both L.P. and A.A. analyzed the crystal structure. N.J. wrote the manuscript, with S.M., M.V.D., and A.A. contributing to its revision. All authors have reviewed and approved the final version of the manuscript for submission.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080770.125.
-
Freely available online through the RNA Open Access option.
- Received September 15, 2025.
- Accepted November 13, 2025.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Ajit Kumar is the first author of this paper, “Loop of fate: structural and mechanistic insights into hnRNPA1 binding to the hepatitis C virus RNA.” Ajit is a graduate student at the CSIR-Institute of Genomics and Integrative Biology, New Delhi, India, in the lab of Dr. Niyati Jain and Dr. Souvik Maiti. The focus of his research is to understand the molecular mechanisms of RNA–protein interactions and their role in disease progression.
What are the major results described in your paper and how do they impact this branch of the field?
Through this project, we identified that the hnRNPA1 protein shows a strong preference for binding to the 5BSL3.2 apical stem–loop, a key structural element required for long-range RNA–RNA interactions. Our detailed analysis of the hnRNPA1 UP1 domain revealed its high-affinity specific interaction with the 5BSL3.2 apical loop, which may interfere with the long-range pairing between 5BSL3.2 and the 3′ X-tail RNA, ultimately impacting viral replication. This study offers a new approach for antiviral intervention.
What led you to study RNA or this aspect of RNA science?
Viruses rely on host machinery to survive and replicate, yet few host proteins are known to negatively regulate viral fitness. hnRNPA1 is one such protein, widely recognized for its ability to suppress viral replication. Understanding the precise molecular mechanism by which hnRNPA1 modulates HCV replication could provide valuable insights and ultimately enable the development of RNA-based therapeutic strategies against HCV infection.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
Several key moments have shaped my interest in science and my development as a researcher. My introduction to RNA structural biology, the excitement of hands-on experimentation, and the realization that RNA–protein interactions can profoundly influence cellular processes and be targeted for RNA-based therapies deepened my curiosity. Discovering new mechanisms during my PhD, particularly in RNA structure, gene expression regulation, and viral replication, further strengthened my passion and solidified my commitment to the field of science.
What are your subsequent near- or long-term career plans?
I am joining as a postdoctoral researcher at SUNY Downstate Health Sciences University, Brooklyn, New York, to deepen my expertise in RNA biology. In the long term, I aim to establish myself as an independent scientist and eventually lead my own research group, focusing on understanding RNA-driven regulatory mechanisms and developing RNA-based therapeutic strategies.








