
tRNA pseudouridine synthase D (TruD) from Thermus thermophilus modifies U13 in tRNAAsp, tRNAGlu, and tRNAGln and U35 in tRNATyr
- Department of Applied Chemistry, Graduate School of Science and Engineering, Ehime University, Matsuyama, Ehime 790-8577, Japan
- Corresponding authors: yamagami.ryota.bn{at}ehime-u.ac.jp, hori.hiroyuki{at}outlook.jp
-
Handling editor: Eric Phizicky
Abstract
Pseudouridine is a modified nucleoside found in various RNA species, including tRNA, rRNA, mRNA, and other noncoding RNAs. Pseudouridine is synthesized from uridine by pseudouridine synthases. While the landscape of pseudouridines in RNA has been extensively studied, much less is known about substrate RNA recognition mechanisms of pseudouridine synthases. Herein, we investigate the tRNA pseudouridine synthase D (TruD), which catalyzes the formation of pseudouridine at position 13 in tRNAAsp in Thermus thermophilus, a thermophilic eubacterium. To identify the tRNA substrates of TruD, we compared results of next-generation sequencing experiments combined with bisulfite probing of pseudouridine in tRNAs from both wild-type and a truD gene disruption mutant. Our data reveal that TruD recognizes tRNAAsp, tRNAGlu, and tRNAGln as substrate tRNAs. In addition, we discover that TruD modifies U35 in tRNATyr, which has previously been reported as a substrate of RluF in Escherichia coli. These findings were validated through in vitro assays with recombinant TruD, which further demonstrated that TruD can act on other RNAs, including a CDC8 mRNA fragment, a known substrate of Pus7, the eukaryotic counterpart of TruD. Systematic mutational analysis of CDC8 transcripts reveals that TruD preferentially pseudouridylates the UNUAR sequence in tRNA substrates (N = any nucleotide, R = purine, U = target site). Finally, we identify over 600 mRNA fragments containing this recognition sequence in T. thermophilus ORFs and demonstrate the ability of TruD to act on these potential mRNA substrates. Our findings suggest the possibility that many other RNAs are modified by TruD in vivo.
Keywords
INTRODUCTION
To date, more than 100 types of modified nucleotides have been found in tRNA across the three domains of life (Cappannini et al. 2024). These tRNA modifications are crucial for the stabilization and maturation of tRNA, protein synthesis, and in multicellular organisms are linked to various diseases and biological phenomena such as oncogenesis, viral infection, and immune response (Motorin and Helm 2010; Ohira and Suzuki 2011; Hori 2014; Suzuki 2021; Cui et al. 2022; Anazco-Guenkova et al. 2024; Ohira and Suzuki 2024). Typically, these tRNA modifications are installed by site-specific tRNA modification enzymes, which may exhibit variability in their site and substrate specificity (Awai et al. 2009; Benitez-Paez et al. 2012; Ranaei-Siadat et al. 2013; Hamdane et al. 2014). For example, tRNA (N2, N2-guanine)-dimethyltransferase (Trm1) from Aquifex aeolicus catalyzes the formation of N2, N2-dimethylguanosine (m22G) at two positions (26 and 27) in tRNACys (Awai et al. 2009). The bacterial tRNA (m5U54) methyltransferase (TrmA) has specificity for multiple substrates, acting on both tRNA and transfer-messenger RNA (tmRNA) (Ranaei-Siadat et al. 2013). Similarly, the 2-methyladenosine (m2A) methyltransferase (RlmN) from Escherichia coli catalyzes the formation of both m2A2503 in 23S rRNA as well as m2A37 in tRNA (Benitez-Paez et al. 2012).
Pseudouridine (Ψ) is one of the most abundant RNA modifications identified in various RNA species, including tRNA, rRNA, mRNA, and snRNA (Borchardt et al. 2020). This modification is synthesized by pseudouridine synthases through the C5-ribosyl isomerization of uridine (Rintala-Dempsey and Kothe 2017). There are two types of pseudouridine synthases: guide RNA-dependent H/ACA small nucleolar ribonucleoproteins (snoRNPs), which are only encoded in archaeal and eukaryotic genomes, and guide RNA-independent pseudouridine synthases (Rintala-Dempsey and Kothe 2017). The guide RNA-dependent pseudouridine synthases are composed of box H/ACA RNA, and core proteins Cbf5 (dyskerin in human), Gar1, Nop10, and L7Ae (Nhp2 in eukaryote), and the guide RNAs determine the pseudouridylation sites (Hamma and Ferre-D'Amare 2006; Kiss et al. 2010; Blaby et al. 2011; Majumder et al. 2020). The archaeal Cbf5 can pseudouridylate uridine at position 55 in tRNA in a guide RNA-independent manner, and thus the enzyme also works as a stand-alone pseudouridine synthase (Gurha et al. 2007; Zhou et al. 2011). The guide RNA-independent pseudouridine synthases are further categorized into six families based on their amino acid sequences: TruA, TruB, tRNA pseudouridine synthase D (TruD), RsuA, RluA, and Pus10 (Spenkuch et al. 2014; Rintala-Dempsey and Kothe 2017). The human genome contains genes for at least 13 distinct pseudouridine synthases (Pus1, PusL1, Pus3, TruB1, TruB2, Dkc1, Pus7, Pus7L, RPusD1, RPusD2, RPusD3, RPusD4, and Pus10), which catalyze pseudouridine formation at various positions in RNAs. In contrast, the E. coli genome encodes 11 distinct pseudouridine synthases (TruA, TruB, TruC, TruD, RluA, RluB, RluC, RluD, RluE, RluF, and RsuA), which have been reported to act on tRNA and rRNA (Spenkuch et al. 2014; Borchardt et al. 2020). In Thermus thermophilus, only six genes encoding a pseudouridine synthase (TruA, TruB, TruD, RsuA, RluB, RluD) are found in the genome (Henne et al. 2004).
Several methods have been described for detecting pseudouridine in RNAs. The classical methods utilize nuclease digestion of RNA followed by UV-liquid chromatography (LC) or 2D thin-layer chromatography (TLC) (Keith 1995; Zhang et al. 2022). N-cyclohexyl-N′-(2-morpholinoethyl) carbodiimide (CMC) is a chemical that can probe pseudouridine selectively (Bakin and Ofengand 1993). Upon CMC-adduction, reverse transcription is terminated, enabling the positional identification of pseudouridine through gel-based reverse transcription analysis (Bakin et al. 1994; Motorin et al. 1998). In the last decade, advancements in chemical probing techniques combined with next-generation sequencing (NGS) facilitated genome-wide detection of pseudouridines (Carlile et al. 2014, 2015; Lovejoy et al. 2014; Schwartz et al. 2014; Li et al. 2015; Khoddami et al. 2019; Marchand et al. 2020; Dai et al. 2023; Zhang et al. 2023). These methods have broadened our understanding of the pseudouridine landscape across different RNA species. More recently, methods for the detection of pseudouridines that do not involve NGS have been developed, providing alternative approaches (Lei and Yi 2017; Fang et al. 2024). These studies have revealed that some eukaryotic tRNA pseudouridine synthases exhibit multisubstrate specificities. For example, Pus7, a member of the TruD family, has been shown to act on tRNA, mRNA, U2 snRNA, and 5S rRNA, indicating broad substrate specificity (Behm-Ansmant et al. 2003; Ma et al. 2003; Decatur and Schnare 2008; Carlile et al. 2014). More specifically, Pus7 recognizes the UGUAR sequence (R = purine and U = pseudouridylation site) and the secondary structure around the modification site of the target RNA (Carlile et al. 2014; Purchal et al. 2022). In addition, archaeal Pus7 (aPus7) is reported to pseudouridylate the uridine at position 13 in tRNA in Thermococcus kodakarensis and Sulfolobus islandicus and uridines at positions 13 and 35 in Pyrococcus abyssi (Muller et al. 2009; Hirata et al. 2019; Li et al. 2024). In contrast, whereas TruD is also known to pseudouridylate the uridine at position 13 in tRNA (Kaya and Ofengand 2003), the substrate and sequence specificity of bacterial TruD (also known as bacterial Pus7) remains poorly understood.
The X-ray crystal structures of yeast Pus7 (Purchal et al. 2022) and E. coli TruD (Ericsson et al. 2004; Hoang and Ferre-D'Amare 2004; Kaya et al. 2004) have been determined. These structures provide significant insights into their structural and functional properties (Hoang and Ferre-D'Amare 2004; Kaya et al. 2004; Chan and Huang 2009; Guegueniat et al. 2021; Purchal et al. 2022). Whereas Pus 7 possesses three insertion domains (I, II, and III in Fig. 1A) not present in E. coli TruD, the amino acid residues within the catalytic TruD domain are highly conserved between these enzymes (Fig. 1B; Purchal et al. 2022). In E. coli TruD, K21, F27, D80, N129, and F131 residues (corresponding to K61, F67, D256, N305, and F307 residues in yeast Pus7, respectively) are conserved within the TruD family (Fig. 1B; Purchal et al. 2022). Among these, D80 is also conserved in other pseudouridine synthases and is proposed to act as the catalytic residue that initiates pseudouridylation via a Michael addition-like mechanism (Hoang and Ferre-D'Amare 2004; Spenkuch et al. 2014). Mutational studies in yeast Pus7 suggested that F131 (F307 in Pus7) stabilizes the target uridine through stacking interactions, while K21 (K61 in Pus7) adjusts the position of the substrate RNA (Purchal et al. 2022). Furthermore, kinetic studies of the N305A mutant of Pus7 revealed a significant decrease in pseudouridylation activity, suggesting the importance of N305 (N129 in E. coli TruD) in catalysis (Purchal et al. 2022). In E. coli TruD, mutational analyses proposed that the conserved E31 residue acts as a general base to complete the reaction (Chan and Huang 2009). The conserved K79 and Q87 residues play supportive roles in the pseudouridylation process; although their substitution with other amino acids reduces activity, it does not abolish it entirely. This suggested that these residues are not directly involved in the core catalytic mechanism but assist the reaction (Chan and Huang 2009). Since the key amino acid residues required for catalysis are well conserved between TruD and Pus7, it is believed that both enzymes share the same catalytic mechanism. However, due to the lack of insertion domains in bacterial TruD, the mechanism by which bacterial TruD recognizes its RNA substrates might be different.
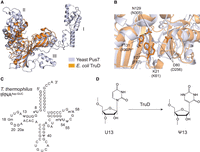
Bacterial TruD pseudouridylates U13 in tRNA. (A) Superposition of structures for yeast Pus7 (white blue) and E. coli TruD (orange) in PyMOL. The three insertion domains in Pus7 are labeled as I, II, and III. (B) Close-up of the catalytic cores in Pus7 (white blue) and E. coli TruD (orange) demonstrating their structural similarity. Conserved amino acid residues for E. coli TruD are compared to those of Pus7 (in brackets). (C) Secondary structure of tRNAAsp-GUC in T. thermophilus with modified positions numbered. (D) TruD pseudouridylates U13 in tRNA.
T. thermophilus is a thermophilic eubacterium and grows at temperatures ranging from 50°C to 80°C (Oshima and Imahori 1974). The tRNAAsp-GUC sequence in T. thermophilus contains three pseudouridines, Ψ13, Ψ40, and Ψ55 (Fig. 1C; Keith et al. 1993). Of these, Ψ13 is synthesized by the TruD protein (Fig. 1D; Hori et al. 2018). In this study, we focus on T. thermophilus TruD with the aim to understand whether specific sequences in the substrate RNAs determine the modification reaction. We found that a selected set of tRNAs are pseudouridylated at U13 and that T. thermophilus TruD is the responsible enzyme for the pseudouridylation of U35 in tRNATyr, which was previously reported to be catalyzed by RluF in E. coli (Addepalli and Limbach 2016).
RESULTS
T. thermophilus TruD can be modeled to a structure similar to that of E. coli TruD
We began this study by obtaining a model for the higher-order structure of the TruD protein of T. thermophilus. We collected primary amino acid sequences of TruD from five different bacteria (T. thermophilus, E. coli, Deinococcus radiodurans, Salmonella Schwarzengrund, and Haemophilus influenzae). We chose these TruD sequences due to the following reasons: for E. coli TruD, the X-ray crystal structure and information of mutation studies of TruD are available, for S. Schwarzengrund and H. influenzae TruD, their amino acid sequences are highly similar to E. coli TruD, and for D. radiodurans TruD, the Deinococcus genus is closely related to the Thermus genus. Multiple sequence alignment analysis showed that catalytically important amino acid residues in E. coli TruD (highlighted in orange in Fig. 1B) are well conserved (Supplemental Fig. S1). For example, the catalytic aspartate residue at position 80 in E. coli TruD is conserved in T. thermophilus TruD (D84; Supplemental Fig. S1). Similarly, other critical residues such as F27, E31, K79, N129, and F131 in E. coli TruD correspond to F31, E35, K83, N132, and F134 in T. thermophilus TruD are also conserved (Supplemental Fig. S1). While K21 in E. coli TruD is replaced with an arginine (R25) in T. thermophilus TruD (Supplemental Fig. S1), the basic side chain of arginine is expected to contribute to RNA binding in the same way as lysine at this position in E. coli TruD. We then modeled the T. thermophilus TruD using AlphaFold3 (Abramson et al. 2024). The resulting model exhibited an overall structural architecture similar to that of E. coli TruD (Supplemental Fig. S2A), where the functionally important amino acid residues within the catalytic domain of the T. thermophilus TruD model closely positioned relative to their counterparts in E. coli TruD (Supplemental Fig. S2B). These observations suggest that the substrate specificity of TruD might be also conserved among the five bacteria.
Disruption of the truD gene does not affect growth of T. thermophilus
To identify pseudouridine sites in tRNAs in T. thermophilus, we constructed a strain in which the truD gene was disrupted by the insertion of the highly thermostable kanamycin-resistance gene (htk gene) into the truD locus (annotated as TTH_RS07750, classically named TTHA1531) of strain HB8 (Fig. 2A). Correct formation of the mutant strain (ΔtruD) was confirmed by PCR analysis using three different sets of PCR primers (Fig. 2B). We observed nonspecific bands in the PCR products amplified with the primer set of P1 and P3 (Fig. 2B, lane labeled P1 and P3), likely due to low primer specificity. However, the other primer set yielded a band corresponding to the theoretical size of the expected PCR fragment (Fig. 2B). Furthermore, dot-hybridization experiments using a truD-specific probe showed the loss of hybridization signal in the ΔtruD genome (Fig. 2C). In addition, we conducted primer extension experiments with and without CMC chemical probing (Fig. 2D,E). In the tRNA fraction from the WT strain, RT termination was observed at position 14 in tRNAAsp upon CMC treatment (Fig. 2E), indicating the presence of pseudouridine at position 13. RT termination at position 14 was absent in the tRNA fraction from the ΔtruD strain, and we confirmed that the ΔtruD strain lost the pseudouridylation activity at position 13 in tRNAAsp. Also, RT termination was observed at position 9 in both WT and ΔtruD samples (Fig. 2E). This termination is caused by s4U at position 8. The apparent band intensities of the RT termination at position 9 were consistent across samples. This result suggests that the s4U8 level is not affected by truD gene disruption. To compare their growth phenotypes, we cultured wild-type T. thermophilus (WT) and the ΔtruD strain in nutrient-rich medium at temperatures of 75°C, 65°C, and 55°C. Our observations indicate that the ΔtruD strain shows a wild-type growth phenotype, suggesting that the disruption of truD gene does not influence cell growth under nutrient-rich conditions (Fig. 2F). It has been reported that the E. coli strain in which the truD gene was disrupted, exhibited no growth phenotype in LB medium (Kaya and Ofengand 2003), which is consistent with our results.
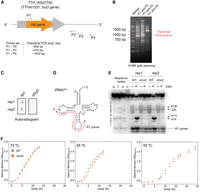
Disruption of the truD gene does not affect growth. (A) The highly thermostable kanamycin-resistance gene htk (orange arrow) was inserted into the truD locus (TTH_RS07750, also known as TTHA1531, gray) by homologous recombination. The gene disruption was tested by PCR with three different primer sets (P1–P2, P1–P3, and P1–P4). The theoretical sizes of each PCR product are provided. (B) The PCR products were analyzed by 2% agarose gel electrophoresis and visualized with SYBR Gold. The circles highlighted in red indicate the theoretical PCR product. (C) The disruption of truD gene in T. thermophilus genome was tested by dot-hybridization, where 32P-labeled truD gene-specific primer was used for a detection probe. (D) Secondary structure of tRNAAsp is shown. The red arrow shows the RT primer-annealing region. (E) tRNA mixture was treated with CMC to probe pseudouridine modifications. The resulting RNAs were used in primer extension reactions. The RT product was analyzed using 15% denaturing PAGE, and RT-terminations were visualized by a phosphor imager. The experiment was independently repeated three times (n = 3). A representative gel image of primer extension experiments is presented. (F) Growth in nutrient-rich medium of WT (gray) and the ΔtruD strain (orange) was monitored at OD600 at 75°C, 65°C, and 55°C. The experiment was independently repeated three times (n = 3). Error bars indicate the standard deviation.
TruD pseudouridylates a subset of tRNAs in T. thermophilus
To identify the substrate tRNAs of TruD in T. thermophilus, we employed PRAISE, a high-throughput pseudouridine detection method (Zhang et al. 2023), on tRNAs extracted from WT and the ΔtruD strain. This approach utilizes a bisulfite/sulfite mixture that leads to sugar-ring opening of pseudouridines resulting in abasic sites in RNAs (Zhang et al. 2023) that leave mutation signals during, reverse transcription. These mutation signals in cDNA can be analyzed through mutational profiling by NGS using ShapeMapper 2 (Busan and Weeks 2018) for variant calling, as done in our previous work (Fig. 3A; Yamagami et al. 2022; Meyer et al. 2023b; Yamagami and Hori 2023a,b). We performed the PRAISE experiments with three biological replicates for the WT strain and two biological replicates for the ΔtruD strains. Each replicate showed a high correlation with the other replicates (Supplemental Fig. S3). We first analyzed tRNAAsp-GUC-1-1 from the WT strain and detected relatively high signals for mutations at positions 13, 40, and 55 (Fig. 3B). These positions are known to be pseudouridylated in cells (Keith et al. 1993). In the ΔtruD strain, we confirmed a loss of the mutation signal at position 13 but observed the signals at positions 40 and 55, demonstrating that U13 is pseudouridylated by TruD (Fig. 3C). We note that mutation signals were detected for nucleotides 5′ adjacent to the pseudouridine sites due to deletion mutations resulting from reverse transcription (Fig. 3B,C; Zhang et al. 2023). Additionally, mutation signals at U8, U20, and U20a were detected in the ΔtruD strain. These Us are modified to s4U8, and dihydrouridines D20 and D20a in T. thermophilus tRNAAsp by ThiI (Sugio et al. 2023) and DusA (Kusuba et al. 2015), respectively, implying bisulfite conversion on these modifications. Consequently, we concluded that TruD pseudouridylates U13 in this tRNA (Fig. 3A,B). We then checked other tRNAs for this modification and found that U13 in tRNAGln-CUG-1-1, tRNAGln-UUG-1-1, tRNAGlu-CUC-1-1, and tRNAGlu-UUC-1-1 showed high mutation rates in the WT strain, while these signals disappeared in the absence of TruD (Fig. 3D; Supplemental Fig. S4). This result indicates that these tRNAs are a substrate of TruD. According to the genomic tRNA database, these four tRNA species are the only tRNAs in T. thermophilus that possess uridine at position 13. Compared to eukaryotic Pus7, which recognizes the UGUAR sequence in target RNAs (Carlile et al. 2014), our data imply that TruD also pseudouridylates UCUAA and ACUAG sequences in vivo (Fig. 3D,E). We note that we observed that the signal intensities of s4U at position 8 and pseudouridine at position 55 were increased in the ΔtruD strain compared to the WT strain (Fig. 3B,C). While the overall mutation signal patterns were similar between WT and ΔtruD strains, some tRNA species showed different signal patterns (Supplemental Fig. S4). For example, increased signal intensities at position 8 were detected in tRNACys, tRNAGly, tRNALeu, and tRNAPro in ΔtruD. These results initially suggested that modification levels at these positions might be changed due to the loss of pseudouridine at position 13. However, since tRNACys, tRNAGly, tRNALeu, and tRNAPro do not possess pseudouridine at position 13, we ruled out this possibility. Instead, we hypothesized that these changes in the modification levels could result from limitations in the quantitativeness of the method. To address this, we performed nucleoside analysis using tRNA fractions from WT and ΔtruD strains (Fig. 4). These analyses detected major tRNA modifications in T. thermophilus, including pseudouridine, m1A, m7G, m5U, Gm, m1G, m2G, and m5s2U (Fig. 4A,B). The apparent peak intensities for these modifications were nearly identical between WT and ΔtruD strains. In addition, the peak corresponding to s4U was detected with UV absorbance at 330 nm, where the s4U levels were also comparable (Fig. 4C,D). These nucleoside analyses demonstrate that overall modification levels are maintained between WT and ΔtruD strains. In summary, the primer extension experiment revealed that the s4U8 level remains unchanged in the ΔtruD strain, and the nucleoside analyses showed that overall modification levels are not changed in the ΔtruD strain. These two results led us to conclude that the changes in s4U and pseudouridine levels observed in certain tRNAs in our PRAISE data are likely due to limitations in the quantification of modified nucleosides.
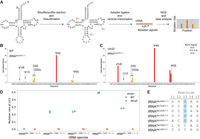
U13 in a subset of tRNAs is pseudouridylated by TruD in T. thermophilus. (A) Overview of the PRAISE experimental procedure. Pseudouridine sites in tRNAs are labeled by bisulfite treatment. After the desulfonation step, the adapter oligonucleotide is attached to the 3′ end of tRNAs, which are converted to cDNA by reverse transcription followed by NGS. Adapter- and quality-trimmed reads were analyzed by ShapeMapper2 (Busan and Weeks 2018). The NGS experiments were independently repeated (n = 3 for WT and n = 2 for ΔtruD). (B and C) Normalized mutation signals at each nucleotide position in tRNAAsp-GUC for WT (B) and the ΔtruD strain (C). (D) Mutation rates at U13 in the substrate tRNAs of T. thermophilus TruD for WT (green dots) and ΔtruD (orange dots). (E) Sequences from positions 11 to 15 in substrate tRNAs (tRNAAsp-GUC-1-1, tRNAGln-CUG-1-1, tRNAGln-UUG-1-1, tRNAGlu-CUC-1-1, tRNAGln-UUC-1-1) and nonsubstrate tRNAs (e.g., tRNAAla-CGC-1-1, tRNAPhe-GAA-1-1, tRNAMet-CAT-1-1) of TruD are aligned with the target uridine highlighted in blue and compared to other tRNAs.
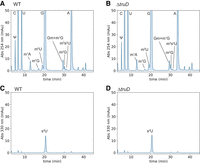
Nucleoside analysis of tRNA fractions from WT and ΔtruD strains revealed that overall modification levels are comparable. (A–D) About 500 pmol tRNA were digested to mononucleoside and analyzed by HPLC. Modified nucleosides were monitored with UV absorbance (A and B) at 254 nm and (C and D) at 330 nm. The peaks were assigned as described in our previous paper (Tomikawa et al. 2010; Ishida et al. 2011; Yamagami et al. 2016; Sugio et al. 2023). The experiments were independently performed three times (n = 3).
T. thermophilus TruD pseudouridylates tRNA transcripts and mRNA fragments in vitro
To assess TruD substrate specificity, we conducted in vitro pseudouridylation assays using purified recombinant T. thermophilus TruD (Fig. 5A). We confirmed that the recombinant TruD pseudouridylates E. coli tRNAGlu-UUC, a known substrate for E. coli TruD (Del Campo et al. 2001; Kaya et al. 2004), and T. thermophilus tRNAAsp-GUC-1-1 (Figs. 4C and 5B). We prepared a tRNAAsp-GUC-1-1 transcript where the target U13 was replaced with C. TruD did not pseudouridylate tRNAAsp U13C (Fig. 5D), confirming that TruD targets the U at position 13 in tRNAAsp. Furthermore, we prepared tRNAGln-CUG-1-1, tRNAGlu-CUC-1-1, and tRNAGlu-UUC-1-1 transcripts and performed pseudouridylation assays. TruD successfully pseudouridylated these transcripts, consistent with our PRAISE data (Supplemental Figs. S5A,B and S6A,B). Notably, tRNAGlu-UUC-1-1 uniquely contains an adenosine at position 11 among the substrate tRNAs we identified (Fig. 3E). We first prepared a U13C mutant transcript of tRNAGlu-UUC-1-1, in which the target U13 was replaced with C (Supplemental Fig. S6A). Pseudouridylation assays confirmed that TruD does not modify the U13C mutant transcript (Supplemental Fig. S6C), indicating that the pseudouridylation site in tRNAGlu-UUC-1-1 is U13. Next, to test whether A11 is critical for TruD substrate recognition, we prepared A11U and A11C mutant transcripts of tRNAGlu-UUC-1-1. The A11U mutant transcript contained a UCUAG sequence at the pseudouridylation site, while the A11C mutant transcript contained a CCUAG sequence (Supplemental Fig. S6A). Pseudouridylation assays revealed that TruD is able to introduce pseudouridine in both mutant transcripts (Supplemental Fig. S6D,E). These results suggest that, at least for tRNAGlu-UUC-1-1, TruD is not likely to recognize the nucleotide at position –2 relative to the pseudouridine site.
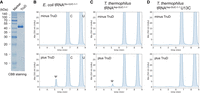
The recombinant TruD pseudouridylates substrate tRNAs. (A) Purified T. thermophilus TruD was analyzed by 15% SDS-PAGE and stained with CBB. (B–D) In vitro pseudouridylation assays with (plus TruD) or without (minus TruD) recombinant TruD using in vitro synthesized (B) E. coli tRNAGlu-UUC, (C) T. thermophilus tRNAAsp-GUC-1-1, and (D) mutant tRNAAsp-GUC-1-1 U13C. Pseudouridine formation was detected by nucleoside analysis using HPLC, where pseudouridine eluted ∼5 min. These experiments were performed independently three times (n = 3) and gave the same results; one representative chromatogram is shown.
Yeast Pus7 is known to modify a CDC8 mRNA fragment (Fig. 6A; Purchal et al. 2022). Therefore, we wondered whether TruD can pseudouridylate mRNA as well. If TruD has a similar substrate specificity to Pus7, TruD should act on this CDC8 mRNA fragment, which we tested in vitro (Fig. 6). As shown in Figure 6B, we found that TruD successfully pseudouridylates the CDC8 mRNA fragment, demonstrating that TruD can modify not only tRNA but also mRNAs. To gain a more detailed insight into the substrate specificity of TruD, we prepared a series of transcripts with a single-point mutation in the CDC8 fragment (Fig. 6C) and examined pseudouridine formation in these transcripts (Fig. 6D–N). We confirmed that the U15C transcript in which the target uridine was replaced with C, was not pseudouridylated by TruD (Fig. 6D). In addition, the U13A transcript was weakly pseudouridylated in vitro (Fig. 6E), consistent with the in vitro and in vivo result for tRNAGln-UUC-1-1 (Fig. 4D; Supplemental Fig. S6). Also, we found that TruD pseudouridylates CDC8 transcripts G14A, G14C, G14U (Fig. 6G–I), and weakly pseudouridylates CDC8 A16G and G17A (Fig. 6K,L). In contrast, TruD does not modify U13C, A16C, and G17C (Fig. 6F,J,M). While the G17A mutation retains a purine nucleotide at position 17, the pseudouridylation activity was detected to be low (Fig. 6L). This low pseudouridylation activity by TruD may result from the disruption of the Watson–Crick base pair between C3 and G17, which could potentially alter the secondary structure of CDC8 and affect its recognition by TruD. Also, we prepared the CDC8 U12C mutant, where U at position 12 (3 nt upstream of the pseudouridylation site) was substituted with cytidine, to examine whether neighboring nucleotides adjacent to the recognition site influence the substrate recognition of TruD. TruD efficiently pseudouridylated the U12C mutant transcript (Fig. 6N), indicating that this position is not critical for substrate recognition. In this research, we introduced 11 distinct single-point mutations in CDC8 transcripts. Among these CDC8 mutants, U15 is the only nucleotide that satisfied the UNUAR sequence. In summary, TruD preferentially pseudouridylates the sequence UNUAR in substrate RNAs (Fig. 6O). While U13A and G17A mutant transcripts were weakly pseudouridylated by TruD, our findings confirm that UNUAR is the primary RNA recognition site for TruD.
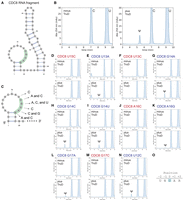
T. thermophilus TruD pseudouridylates CDC8 mRNA fragments in vitro. (A) Secondary structure of the CDC8 mRNA fragment with the recognition sequence of TruD in green. (B) In vitro pseudouridine formation assays using recombinant TruD protein and the CDC8 fragment. (C) Single-point mutations in the CDC8 fragment with neutral (blue) or disruptive (red) effect on pseudouridylation. (D–N) In vitro pseudouridylation assays with (plus TruD) or without (minus TruD) recombinant TruD protein using indicated CDC8 mutants (D) U15C, (E) U13A, (F) U13C, (G) G14A, (H) G14C, (I) G14U, (J) A16G, (K) G17C, (L) G17A, (M) G17C, and (N) U12C. These experiments were performed independently two times (n = 3) and gave the same results; one representative chromatogram was shown. (O) T. thermophilus TruD recognizes the UNUAR sequence in substrate RNAs with the target uridine underlined and highlighted in blue in (N), where N is any nucleotide (A, U, G, C), and R is purine residues (A, G).
To test the recognition sequence of TruD using tRNA substrates, we prepared tRNAAsp-GUC-1-1 mutant transcripts with single-point mutations introduced into the TruD recognition site (Supplemental Fig. S7A). As shown in Figure 5C,D, TruD pseudouridylates tRNAAsp-GUC-1-1 but does not pseudouridylate the U13C mutant (Fig. 5C,D; Supplemental Fig. S7B,C). We further tested tRNAAsp-GUC-1-1 U11A, G12C, A14G, and G15C mutants. Interestingly, TruD pseudouridylates these mutant transcripts (Supplemental Fig. S7D–G). Similar to tRNAGlu-UUC-1-1, which possesses the ACUAG sequence, the U11A mutant transcript contains an AGUAG sequence in its recognition site, and TruD efficiently pseudouridylates this mutant transcript (Supplemental Fig. S7D). Also, TruD pseudouridylates the G12C mutant transcript, indicating that the second nucleotide in the recognition site is not critical for substrate recognition (Supplemental Fig. S7E). The substitution of A14 with G14 decreases the activity to 21%, which was calculated from the peak area of pseudouridine normalized by the peak area of cytidine. This result demonstrates that A14 in the recognition sequence is important for efficient pseudouridylation (Supplemental Fig. S7E). Similarly, the substitution of G15 with C15 decreases the activity to 33%. Overall, these findings highlight the importance of specific nucleotides within the recognition sequence for TruD activity. These results also demonstrate its flexibility in substrate recognition of tRNA. This flexible substrate recognition of tRNA might be caused from structural differences around the target site between tRNA and the CDC8 fragment.
Next, we searched for potential mRNA substrates containing UNUAR sequences within the T. thermophilus open reading frames (ORFs) using RNABOB. This RNA motif search identified over 600 RNA fragments containing the TruD recognition sequence (Fig. 7A; Supplemental File S1). Sequence conservation analysis around the UNUAR motif revealed no conservation of adjacent nucleotides (Fig. 7B). We then predicted the secondary structures of these mRNA fragments (Fig. 7C). As shown in the histogram, their folding energies ranged from −54.3 to −12.4 kcal/mol (Fig. 7C), indicating a diversity of secondary structures. For example, RNA337 and RNA363, which exhibited the lowest folding energies, were predicted to form long helical regions (Fig. 7D,E). In contrast, RNA428 and RNA499, with the highest folding energies, were predicted to form long loops and extended single-stranded regions in their structures (Fig. 7F,G). To further investigate, we prepared these four potential substrates via in vitro transcription and performed pseudouridylation assays (Fig. 7H–O). The assays demonstrated that TruD pseudouridylates all four substrates (Fig. 7H–K). Notably, the substitution of the target uridine at position 43 with cytidine abolished pseudouridylation activity (Fig. 7L–O), confirming that the pseudouridylation site in these substrates is the uridine at position 43.
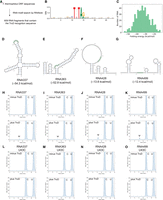
TruD acts on potential substrate mRNA fragments identified from T. thermophilus ORFs. (A) Schematic representation of the identification process for potential TruD substrates. An RNA motif search was conducted using RNABOB to identify sequences containing the recognition sequence. (B) Sequence conservation of the recognition site in the identified potential substrates is analyzed. (C) Minimal folding energies of the potential substrates are plotted on a histogram. Four RNAs were selected for further investigation. (D–G) Secondary structures and folding energies of the selected potential substrates are shown, with the TruD recognition sequence highlighted in green. (H–O) In vitro pseudouridylation assays with (plus TruD) or without (minus TruD) recombinant TruD using in vitro synthesized (H) RNA337, (I) RNA363, (J) RNA428, (K) RNA499, (L) RNA337 U43C, (M) RNA363 U43C, (N) RNA428 U43C, and (O) RNA499 U43C. Pseudouridine formation was detected by nucleoside analysis using HPLC, where pseudouridine eluted ∼5 min. These experiments were performed independently three times (n = 3) and gave the same results; one representative chromatogram is shown.
T. thermophilus TruD acts on U35 in tRNATyr-GUA-1-1
Our in vitro experiments suggested that T. thermophilus TruD recognizes RNAs that possess an UNUAR sequence. We noted that T. thermophilus tRNATyr-GUA-1-1 as well as tRNAThr-CGU-1-1 contain UGUAA at position 33–37 (Fig. 8A) and UGUAG at position 6–10 (Supplemental Fig. S8A), respectively, which fit in the TruD recognition sequence and hypothesized that these tRNAs might be pseudouridylated by TruD in T. thermophilus. To test this, we analyzed our PRAISE data and detected a mutation signal at position U35 in tRNATyr-GUA-1-1 for the wild-type strain (Fig. 8B), which was diminished in the absence of the truD gene (Fig. 8C). In addition, we performed primer extension experiments to confirm the presence of pseudouridine at position 35 in tRNATyr (Fig. 8D,E). Under CMC-treated conditions, we observed RT pauses in primer extension at positions 36 and 38 (Fig. 8E). These RT pauses were caused by CMC-reacted pseudouridine at position 35 and probably by ms2i6A at position 37, respectively. The RT pause at position 36 was completely absent in tRNATyr from the ΔtruD strain (Fig. 8E), confirming that U35 in tRNATyr is pseudouridylated by TruD. Also, to investigate whether that TruD pseudouridylates U35 in tRNATyr-GUA-1-1 under in vitro condition, we performed in vitro pseudouridylation using a tRNATyr-GUA-1-1 transcript and a mutant version with an altered modification site. Pseudouridylation assays with and without purified TruD (Fig. 8F–I) showed, as expected, a pseudouridine peak upon treatment with TruD with the tRNATyr-GUA-1-1 transcript (compare Fig. 8F,G). In contrast, the pseudouridine peak was completely absent when we used the mutant transcript tRNATyr-GUA-1-1 U35C (Fig. 8H,I). These in vitro results demonstrate that T. thermophilus TruD recognizes U35 in tRNATyr-GUA-1-1 as a target uridine. As for tRNAThr-CGU-1-1, our PRAISE data showed no differences in tRNAThr-CGU-1-1 between the WT and ΔtruD strains (Supplemental Fig. S8B,C). A strong signal at position 8 was observed in both samples, which is likely derived from s4U8, since the signal did not disappear upon truD gene disruption. Also, we tested the in vitro pseudouridylation using a tRNAThr-CGU-1-1 transcript. The results demonstrated that TruD does not pseudouridylate tRNAThr-CGU-1-1. Thus, TruD does not recognize tRNAThr-CGU-1-1 as a substrate. Overall, we conclude that UNUAR is the consensus recognition site for T. thermophilus TruD, which is not necessarily at a particular position in the tRNA and might be modified by this enzyme in vivo when it is present in mRNA.
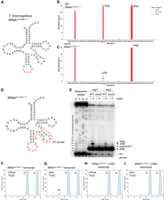
tRNATyr-GUA-1-1 is pseudouridylated by TruD in vivo and in vitro. (A) Cloverleaf structure of T. thermophilus tRNATyr-GUA-1-1 transcript with the sequence in red containing the TruD recognition sequence. (B and C) Normalized mutation signals at each nucleotide position in tRNATyr-GUA for (B) WT and (C) the ΔtruD strain. (D) Secondary structure of tRNATyr is shown. The red arrow shows the RT primer-annealing region. (E) tRNA mixture was treated with CMC to probe pseudouridine modifications. The resulting RNAs were used in primer extension reactions. The RT product was analyzed using 15% denaturing PAGE, and RT-terminations were visualized by a phosphor imager. The experiment was independently repeated three times (n = 3). A representative gel image of primer extension experiments is presented. (F–I) In vitro pseudouridylation assays with (plus TruD) or without (minus TruD) recombinant TruD using (F, G) tRNATyr-GUA or (H, I) mutant tRNATyr-GUA U35C transcripts. These experiments were independently repeated three times (n = 3) and gave the same results. Of the three trials, one representative chromatogram was shown.
DISCUSSION
Recent advancements in pseudouridine detection have enabled genome-wide analysis of pseudouridine sites in RNAs. Many pseudouridines have been identified in tRNA, rRNA, and mRNA in eukaryotic cells by high-throughput techniques. These pseudouridines are known to increase RNA stability and regulate RNA splicing and protein synthesis in eukaryotes. In contrast, the pseudouridine landscape and sequence specificities of pseudouridine synthases in bacteria have not been fully unveiled.
In this study, we conducted both in vivo and in vitro experiments to determine the sequence specificity of T. thermophilus TruD for its substrate RNAs. We found that TruD preferentially recognizes the UNUAR sequence as well as ACUAG and, to a lesser extent, AGUAG in RNAs and has the potential to act on mRNAs containing a recognition sequence. Our findings suggest that TruD replaces Us with pseudouridine in bacterial mRNA given the calculated 1/128 probability of the UNUAR sequence occurring randomly in RNA. Indeed, our RNA motif search identified over 600 potential substrate mRNA fragments within T. thermophilus ORFs (Fig. 7A). Additionally, four representative potential substrate RNAs were pseudouridylated by TruD in vitro (Fig. 7H–O), further supporting the hypothesis that TruD acts on mRNAs in vivo. Moreover, TruD might have a broader substrate specificity compared to Pus7, a eukaryotic TruD-ortholog, which recognizes UGUAR and the local RNA structure around the target uridine (Purchal et al. 2022). Similar to Pus7, local RNA structure might also modulate the substrate specificity of TruD. In our pseudouridylation assays, secondary structure surrounding the UNUAR sequence appeared to affect TruD activity. For instance, the apparent peak areas of pseudouridine in RNA337 and RNA363, which have the lowest folding energies among the substrates, were smaller than those in RNA428 and RNA499 (Fig. 7H–O). These observations suggest that secondary structure plays a critical role in regulating TruD activity. To investigate this further, genome-wide detection of pseudouridine in total RNAs, coupled with their structural analysis, will be necessary.
The structural model of T. thermophilus TruD we obtained with AF3 was highly similar to the overall architecture of E. coli TruD (Supplemental Fig. S2). Furthermore, catalytically important amino acid residues were highly conserved between T. thermophilus and E. coli TruD proteins (Supplemental Figs. S1 and S2). Therefore, our findings regarding the sequence specificity of T. thermophilus TruD might reflect a general property of bacterial TruD proteins.
In yeast, the pseudouridine synthase Pus7 is responsible for the pseudouridylation of U35 in tRNATyr, and this pseudouridylation is reported to be intron-dependent (Johnson and Abelson 1983; Behm-Ansmant et al. 2003). In archaea, aPus7 as well as the Cbf5-associated box H/ACA RNA–protein complex are responsible for this pseudouridylation (Muller et al. 2009; Li et al. 2024). In contrast, in bacteria, while we found that U35 in tRNATyr in T. thermophilus is pseudouridylated by TruD (Fig. 8), in E. coli the same target is modified by RluF, which was originally identified as being responsible for pseudouridylation of uridine at position 2604 in 23S rRNA (Del Campo et al. 2001; Addepalli and Limbach 2016). A different enzyme from TruD, RluF belongs to the RsuA family (Del Campo et al. 2001). The crystal structure of RluF complexed with an rRNA fragment revealed that RluF mainly interacts with the sugar-phosphate backbone and strictly recognizes the target uridine in the sequence AGUUC (Alian et al. 2009). Given the different structural requirements that determine the recognition of the target sequence, RluF is likely to have different enzymatic properties compared to TruD. Interestingly, although the rluF gene is not encoded in the T. thermophilus genome, the pseudouridine at position 2616 (at position 2605 in E. coli) is conserved (Mengel-Jorgensen et al. 2006). Thus, TruD and other pseudouridine synthases compensate for the lack of the rluF gene and pseudouridylate the uridine at position 35 in tRNATyr and at position 2616 in 23S rRNA, respectively, in T. thermophilus.
In this study, we investigated the TruD activity using various tRNA transcripts. T. thermophilus tRNA contains TruD recognition sequences at three distinct positions: at position 6–10 in tRNAThr, at position 11–15 in tRNAAsp, Glu, Gln, and at position 33–37 in tRNATyr (Fig. 9A–C). Using the X-ray crystal structure of yeast tRNAPhe (Shi and Moore 2000), we analyzed the 3D structures of these positions. In tRNATyr, the nucleotide at the target position 35 is thought to be exposed to the solvent side and does not form hydrogen bonds (Fig. 9C). In contrast, the target uridine at positions 8 and 13 interacts with adjacent nucleotides through hydrogen bonding. For instance, U13 forms a Watson–Crick base pair with A22, where N3 and O4 atoms of U13 interact with N1 and N6 atoms of A22, respectively (Fig. 9D). Similarly, U8 forms a non-Watson–Crick base pair with A14, where O2 and N3 atoms of U8 interact with N6 and N7 atoms of A14, respectively (Fig. 9E). Notably, the O2 atom of U13 and the O4 atom of U8 remain unpaired, potentially making them available for enzyme interactions. The X-ray crystal structure of pseudouridine synthase RluB in complex with substrate RNA proposed a catalytic mechanism for pseudouridylation (Czudnochowski et al. 2014). The reaction begins with a conserved aspartate by attacking C6 of the target uridine, forming a covalent intermediate and breaking the N-glycosidic bond. The uracil then rotates 180°, forming a new carbon-carbon bond between C5 and C1′. Finally, the aspartate is released. In the RluB–RNA complex structure, the O4 atom of the target uridine interacts with the backbone amide of the catalytic Asp110, while the O2 atom interacts with the main chain of Val196 and water molecules. These two O atoms play crucial roles in accepting and donating protons during pseudouridine formation (Czudnochowski et al. 2014). Given that pseudouridine synthases share similar catalytic mechanisms, our observations suggest that the O2 status of target uridine in substrate RNA could be critical for the target base recognition and/or pseudouridine formation in the TruD reaction, and this could explain why TruD recognizes U13 but not U8 in tRNA. However, further structural studies are required to elucidate the detailed substrate-binding mechanisms.
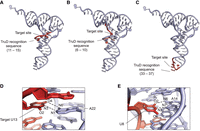
A hypothetical mechanism illustrating how TruD distinguishes the recognition sequence in substrate RNAs from nonsubstrate RNAs. (A–C) The three distinct sites of the TruD recognition sequence found in T. thermophilus tRNAs are mapped onto the crystal structure of yeast tRNAPhe (PDB id: 1EHZ). The TruD recognition sites are highlighted in red. (D) A close-up view of the Watson–Crick interaction between U13 and A22. The dashed lines show hydrogen bonding interaction. Note that the original C13–G22 base in yeast tRNAPhe was replaced with U13–A22 using PyMOL. (E) A close-up view of the non-Watson–Crick interaction between U8 and A14. The dashed lines show hydrogen bonding interaction.
We examined the growth phenotype of the ΔtruD strain and found that its growth rate is similar to that of WT (Fig. 2F). Thus, the role of the pseudouridylation by TruD in T. thermophilus remains unclear. While the disruption of the truD gene does not appear to affect the growth rate, it is possible that mRNA/protein expression levels and translation efficiencies might be changed. Alternatively, thymidylate synthesis is expected to be a rate-limiting factor of DNA synthesis (Escartin et al. 2008; Yamagami et al. 2018), and thus, DNA synthesis might be rate-limiting for growth in the case of WT as well as the ΔtruD strain. To address these aspects, genome-wide mRNA analysis, proteome analysis, and analysis of DNA synthesis would be required.
In sum, our study highlights significant advances in understanding the sequence specificity of T. thermophilus TruD for its substrate RNAs. Our chemical probing experiments and biochemical experiments revealed that T. thermophilus TruD preferentially pseudouridylates substrate RNAs containing the sequence UNUAR. In agreement with this, we observed that T. thermophilus TruD pseudouridylates U35 in tRNATyr. Future research involving comprehensive pseudouridine mapping and structural analysis of RNA would provide deeper insights into the regulatory roles of these pseudouridine modifications in prokaryotic systems.
MATERIALS AND METHODS
Structure modeling of T. thermophilus TruD
The structure of T. thermophilus TruD was modeled with AF3 (https://alphafoldserver.com/about) and visualized with PyMOL (The PyMOL Molecular Graphics System, Version 3.0, Schrödinger, LLC).
Construction of T. thermophilus truD gene deletion (ΔtruD) strain
The T. thermophilus HB8 strain used as wild type was a kind gift from Dr. Tairo Oshima (Kyowa-kako, Japan) (Oshima and Imahori 1974). The T. thermophilus truD gene (TTH_RS07750 or classical name; TTHA1531) was disrupted by replacement with the highly thermostable kanamycin nucleotidyltransferase (HTK) gene (Hoseki et al. 1999) using plasmid TDs08B04 (RIKEN BioResource Research Center) (Yokoyama et al. 2000). Homologous recombination was performed as described by Hashimoto et al. (2001). The recombination region was assessed by polymerase chain reactions with primers P1, P2, P3, and P4 (Fig. 2B) and 2% agarose gel electrophoresis, while a PCR product spanning the region, amplified with primers P1 and P2, was sequenced. The disruption of the truD gene in the genome of the mutant was further confirmed by dot hybridization using a 32P-labeled truD gene-specific probe (5′-AAGGCCCACTTGTCCAGGTGC-3′).
Growth curves of wild-type T. thermophilus and the ΔtruD strain
Cells were cultured in 30 mL medium consisting of 0.8% hipolypeptone, 0.4% yeast extract, and 0.2% NaCl, adjusted to pH 7.5 with NaOH and supplemented with 0.35 mM CaCl2 and 0.17 mM MgCl2 after autoclaving. The cultures, inoculated with 1 mL of precultured wild-type or ΔtruD cells, were incubated at 55°C, 65°C, and 75°C. The optical densities at 600 nm were monitored using a U-5100 Spectrophotometer (Hitachi). All experiments were independently repeated three times and standard deviations were determined. For further experiments, we used T. thermophilus cells grown at 65°C.
Preparation of tRNA from T. thermophilus
Total RNAs were extracted by the acid guanidinium thiocyanate-phenol-chloroform method as described previously (Yamagami et al. 2016; Yamagami and Hori 2023a). The tRNA fraction was purified by 10% PAGE containing 7 M urea, eluted from the gel in a buffer containing 10 mM Tris-HCl pH 7.6, 1 mM EDTA, 250 mM NaCl, and 0.001% SDS at 4°C overnight, and recovered by ethanol precipitation.
Primer extension
The tRNA fraction was denatured in a 45.8 µL solution containing ∼600 pmol tRNA and 5 mM EDTA at 90°C for 3 min. Then, 200 µL BEU buffer (pH 9.0) containing 7 M urea, 4 mM EDTA, 50 mM Bicin, and 0.5 M cyclohexyl-3-(2-morpholinoethyl) carbodiimide (CMC) was added to the mixture and incubated at 45°C for 40 min. The RNA was recovered by ethanol precipitation. The RNA pellet was dissolved in 40 µL sodium carbonate buffer (pH 10.4) containing 50 mM sodium carbonate and 2 mM EDTA. The RNA solution was incubated at 50°C for 2 h. The intact RNA was purified by 10% denaturing PAGE (7 M urea). The RNA was recovered from the gels in TEN250 buffer containing 10 mM Tris-HCl (pH 7.6), 1 mM EDTA, 250 mM NaCl, and 0.001% SDS at 4°C for 16 h, followed by ethanol precipitation. The RNA was dissolved in 5 µL water. For negative control experiments, the reaction without CMC was also performed with the same procedure. Reverse transcription was performed in a 5 µL reaction containing 50 mM Tris-HCl (pH 8.2), 150 mM KCl, 10 mM DTT, 6 mM MgCl2, 1 mM each dNTP, 32P-labeled RT primer (>5000 cpm), 1 µL tRNA, and 50 U SuperScript III (Thermo Fisher Scientific) at 55°C for 1 h. Sequence ladders for tRNAAsp and tRNATyr were prepared by a 5 µL reverse transcription reaction containing 50 mM Tris-HCl (pH 8.2), 150 mM KCl, 10 mM DTT, 6 mM MgCl2, 1 mM each dNTP, 1 mM ddATP, ddTTP, ddCTP, or ddGTP, 32P-labeled RT primer (>5000 cpm), 50 ng tRNAAsp or tRNATyr transcript, and 50 U SuperScript III performed at 55°C for 1 h. After the RT reaction, 0.2 µL 2 M NaOH was added, and the mixture was heated at 95°C for 5 min to degrade the tRNA. The reaction products were separated with 15% denaturing PAGE (7 M urea), and the primer extension was visualized by a phosphor imager. The nucleotide sequences of RT primers used in the primer extension experiments are listed in Supplemental Table S1.
Detection of pseudouridine in tRNA by PRAISE
To determine pseudouridine sites in T. thermophilus tRNAs, the PRAISE protocol (Zhang et al. 2023) was followed with minor changes. In brief, 990 µL 85% sulfite/15% bisulfite solution and 10 µL 100 mM hydroquinone were mixed. One microgram of the tRNA fraction in 50 µL of the bisulfite mixture was incubated at 70°C for 5 h. For desulfonation, 950 µL water was added to the reaction mixture and the tRNA fraction was purified by Q-Sepharose column chromatography, where buffer A (20 mM Tris-HCl pH 7.6), buffer B (20 mM Tris-HCl pH 7.6, 400 mM NaCl), and buffer C (20 mM Tris-HCl pH 7.6, 800 mM NaCl) were used for column equilibration, wash, and RNA elution, respectively. The eluted RNAs were recovered by ethanol precipitation and dissolved in 100 µL water. The RNA solution was mixed with 100 µL 1 M Tris-HCl pH 9.0 and incubated at 75°C for 30 min. The RNA was recovered by ethanol precipitation and used for library preparation (involving adapter ligation and reverse transcription) and NGS (Fig. 3A) as described previously (Yamagami et al. 2022; Meyer et al. 2023a; Yamagami and Hori 2023a,b). The NGS experiments were independently repeated (n ≥ 2) and, since both replicates showed a good correlation to each other (Supplemental Fig. S3), their fastq files were merged and utilized for the data analysis.
Data analysis
Analysis of NGS reads was performed as described previously (Yamagami et al. 2022). Reference sequences of tRNA genes encoded in the T. thermophilus genome were retrieved from GtRNAdb (https://gtrnadb.ucsc.edu/) (Chan and Lowe 2016) and used for reference sequences and names of tRNA. The 3′ adapter sequence and low-quality reads were removed by Cutadapt (Martin 2011). ShapeMapper 2 was used for variant calling and the calculation of normalized mutation rates with default parameters (Busan and Weeks 2018). For the normalization of mutation signals, the mutation rate at each nucleotide position was divided in ShapeMapper2 by the average mutation rate of nucleotides belonging to the top 10% with highest mutation rates (see https://github.com/Weeks-UNC/shapemapper2). The mutation rates on uridines were visualized with a custom Python script.
Nucleoside analysis
Nucleoside analysis was performed after complete digestion of tRNA with nuclease P1, RNase A, and bacterial alkaline phosphatase as described previously (Tomikawa et al. 2010; Ishida et al. 2011; Yamagami et al. 2016; Sugio et al. 2023).
Expression and purification of T. thermophilus TruD
The T. thermophilus TruD-pET11b expression vector was purchased from RIKEN BioResource Center and transformed into E. coli BL21 (DE3) Rosetta 2 (Novagen). TruD was expressed according to the manufacturer's manual for which wet cells (2.5 g) were suspended in 12.5 mL buffer A (50 mM Tris-HCl pH 7.6, 50 mM KCl, 5 mM MgCl2, and 6 mM 2-mercaptoethanol), including one tablet of protease inhibitor (Roche), and disrupted at 4°C with an ultrasonic disruptor (model VCX-500, Sonics and Materials, Inc.). Cell debris was removed by centrifugation at 6000g at 4°C for 20 min, and the supernatant was loaded onto a Q-Sepharose Fast Flow column with a volume of 5 mL (Cytiva). TruD was eluted by a linear gradient from 50 to 1000 mM KCl in buffer A. The eluates were assessed by 15% SDS-PAGE and TruD-containing fractions were combined. After adjustment of the KCl concentration to ∼50 mM by dilution with buffer A without KCl, the sample was loaded onto a HiTrap Heparin HP column (5 mL, Cytiva) from which TruD was eluted by a linear gradient from 50 to 1000 mM KCl in buffer A. The eluted fractions with TruD, as assessed by 15% SDS-PAGE, were combined, and heated at 70°C for 20 min. After removal of denatured protein by centrifugation at 9000g at 4°C for 20 min, the supernatant was concentrated using a Vivaspin Turbo 15 filter device (Sartorius) and loaded onto a HiLoad Superdex 75 preparation grade gel-filtration column (with a volume of 120 mL, Cytiva), which had been equilibrated in buffer B (50 mM Tris-HCl pH 7.6, 5 mM MgCl2, 200 mM KCl, and 6 mM 2-mercaptoethanol). TruD-containing fractions (as assessed by 15% SDS-PAGE) were combined and loaded onto a HiTrap Heparin HP column from which the protein was eluted by a linear gradient from 50 to 1000 mM KCl in buffer A. Fractions with purified TruD were combined and concentrated using a Vivaspin Turbo 15 filter device, glycerol was added to a final concentration of 50% v/v, and aliquots were stored at −30°C. Recombinant TruD (2 µg) was analyzed by 15% SDS-PAGE and stained with CBB.
Preparation of in vitro transcribed RNAs
The tRNA transcripts and CDC8 mRNA fragments were prepared as reported previously (Matsuda et al. 2024). The transcripts were PAGE-purified using 10% polyacrylamide gel (7 M urea). The oligonucleotides used for preparing the transcripts are listed in Supplemental Table S1. For the wild-type and mutated templates in the case of CDC8 or tRNAAsp-GUC, one reverse primer was used.
In vitro pseudouridylation activity of T. thermophilus TruD
In vitro pseudouridylation experiments were conducted in a 100 µL reaction containing 50 mM Tris-HCl pH 7.6, 50 mM KCl, 5 mM MgCl2, 6 mM 2-mercaptoethanol, 4.25 µM RNA, and 0.5 µM TruD at 55°C for 1 h. After the reaction, the RNA was purified by phenol extraction and recovered by ethanol precipitation. The recovered tRNA was digested to nucleosides. The formation of pseudouridine was analyzed by an HPLC system, as described previously (Ishida et al. 2011). In our LC system, retention time of pseudouridine was determined to be ∼5–5.5 min. The in vitro experiments were independently repeated three times (n = 3).
Identification of RNA motif containing UNUAR sequence
ORFs in T. thermophilus genome were retrieved from NCBI. The RNA sequences containing the UNUAR motif were identified within these ORFs using RNABOB, an RNA motif search tool (available at http://eddylab.org/software.html). Based on the stem–loop structure of the D-arm in tRNA, we designed the following structure descriptor: h1, s1, h1; h1′ 0:3 NUN:NNN; s1 0 UAR[10], where h1 is helix 1, s1 is a single-stranded region, and h1′ is the complementary element of helix 1. In the descriptor, helix 1 was specified to form three base pairs, similar to the canonical D-stem in tRNA secondary structures. However, these nucleotides and their counterparts in helix 1 were not required for forming base-pairing, as described by 0:3 NUN:NNN. Additionally, the UAR sequence was specified to be in a single-stranded region, as described by s1 0 UAR[10], where [10] allows for any nucleotide up to 10 nt at that position. Thus, this structure descriptor enables the identification of UNUAR sequences located in both the single-stranded region and portions of helical structures. The resulting RNA fragments containing UNUAR sequences are summarized in Supplemental File S1. The sequences were further analyzed by WebLogo3 (Crooks et al. 2004), where we analyzed sequence conservation around the UNUAR sequences. Then, secondary structures of the RNA fragments were predicted using RNAstructure software (Reuter and Mathews 2010) to calculate minimal folding energy.
DATA DEPOSITION
All sequencing data used in this study are available at the DNA Data Bank of Japan (DDBJ) Sequence Read Archive (SRA), under accession numbers DRR624973–DRR624974 for WT data and DRR598188–DRR598189 for ΔtruD data. All data used in this study are available from the corresponding authors upon request.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
The authors thank Dr. Naohito Tokunaga at the Advanced Research Support Center (ADRES at Ehime University) for assistance with the next-generation sequencing experiments. We also thank Mone Nobeoka and Yusuke Kuwana for assistance with the library preparation of PRAISE samples and purification of TruD, respectively. We are grateful to Teppei Matsuda and Dr. Chie Tomikawa (Ehime University) for preparing T7 RNA polymerase. This work was supported by a Grant-in-Aid for Scientific Research from the JAPAN Society for the Promotion of Science (JSPS) (22K15035 to R.Y.; 18K19302, 20H03211, and 24K09381 to H.H.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080405.125.
- Received January 24, 2025.
- Accepted March 20, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








