
Deciphering the influence of the [4Fe–4S] cluster of tRNA thiolation enzymes on tRNA binding
- Sylvain Gervason1,3,
- Sambuddha Sen1,3,
- Jean-Luc Ravanat2,
- Sylvain Caillat2,
- Djemel Hamdane1,4 and
- Béatrice Golinelli-Pimpaneau1
- 1Laboratoire de Chimie des Processus Biologiques, Collège de France, CNRS UMR 8829, Sorbonne Université, Paris cedex 05, France
- 2University of Grenoble Alpes, CEA, CNRS, Grenoble INP, IRIG, SyMMES, F-38000 Grenoble, France
- Corresponding authors: djemel.hamdane{at}sorbonne-universite.fr, beatrice.golinelli{at}college-de-france.fr
-
↵3 These authors contributed equally to this work.
Abstract
Iron–sulfur clusters [Fe–S] play crucial roles in diverse biological reactions, often serving as prosthetic groups for enzymes. Specifically, certain tRNA-modifying enzymes utilize these clusters to catalyze the thiolation of specific nucleosides. While the participation of [4Fe–4S] clusters in such catalytic processes is known, their potential influence on tRNA binding remains unexplored. In this study, we examine the impact of the cluster on the affinity for tRNA of TtuI from the archeon Methanococcus maripaludis, an enzyme responsible for the formation of 4-thiouridine at position 8 in tRNAs of archaea and bacteria, as well as Escherichia coli TtcA that catalyzes the biosynthesis of 2-thiocytidine at position 32 in bacterial tRNAs. For this purpose, we compare the change of fluorescence properties of judiciously located tryptophans upon tRNA binding between the apo-enzyme (lacking the cluster) and the holo-enzyme (incorporating a reconstituted cluster). Our results indicate that the presence of the [4Fe–4S] cluster does not alter the affinity of the thiolases for tRNA, thus ruling out any direct involvement of the cluster in tRNA binding and emphasizing the purely catalytic role of the [4Fe–4S] cluster in tRNA thiolation.
Keywords
INTRODUCTION
[Fe–S] clusters stand as the oldest metallocofactors on earth (Camprubi et al. 2017), playing key roles in a diverse array of cellular processes. They are essential participants in electron transfer reactions (Yakovlev et al. 2007), contribute to redox and non-redox catalysis (Beinert et al. 1996), and to regulatory mechanisms by acting as sensors (Mettert and Kiley 2015; Crack and Le Brun 2018). While the [2Fe–2S] and [4Fe–4S] states represent the most common forms of [Fe–S] clusters (Beinert 2000), more complex arrangements, with additional heavy metal ions, are present in enzymes such as carbon monoxide dehydrogenases and nitrogenases (Dinis et al. 2016; Jeoung and Dobbek 2018). The irons within the cluster are most often coordinated by the thiolate of cysteine residues of the protein or nitrogen atoms of histidine/arginine residues but occasionally by other ligands, such as the oxygen atoms of aspartate residues (Bak and Elliott 2014; Zhou et al. 2021). In some enzymes, the [4Fe–4S] cluster is coordinated by three protein residues, whereas the fourth iron atom is bound to an external ligand, such as the S-adenosylmethionine (SAM) cofactor in SAM-radical enzymes (Broderick et al. 2023), or a water molecule in aconitase (Castro et al. 2019). This diversity in coordination underscores the adaptability of [Fe–S] clusters, which allows them to ensure a wide range of chemical functions.
A variety of enzymes involved in the posttranscriptional modification of tRNAs, which is crucial for the efficiency and fidelity of genetic translation, rely on [4Fe–4S] clusters (Kimura and Suzuki 2015). They catalyze a large diversity of reactions, encompassing methylations (m2A37 by RlmN), acetylations (cm5U34 by Elp3), the formation of an imidazoline ring (imG14 by Tyw1), methylthiolations (ms2i6A37 by MiaB), and thiolations of tRNA nucleosides (s2U34 by MnmA and NcsA, s4U8 by TtuI, s2C32 by TtcA, and s2U54 by TtuA) (Bouvier et al. 2014; Arragain et al. 2017; Chen et al. 2017; Zhou et al. 2021; He et al. 2022; Bimai et al. 2023). While several of these enzymatic systems are radical SAM-dependent, with clusters involved in the formation of a 5′Ado• radical, it has been shown that the [4Fe–4S] clusters of tRNA thiolases that catalyze the simple substitution of an oxygen atom by a sulfur atom exhibit non-redox activity and are not destroyed to serve as a sulfur donor for the thiolation reaction (Gervason et al. 2024). All known tRNA thiolation enzymes that catalyze non-redox reactions use ATP to specifically activate the target nucleotide by forming an adenylated intermediate (Fellner et al. 2018).
In non-redox tRNA thiolation reactions, the [4Fe–4S] cluster, coordinated by three residues only, likely acts as a Lewis acid to bind a sulfur atom, independently of its oxidation state (Gervason et al. 2024). However, the possible influence of the cluster on the enzyme affinity for its tRNA substrate has not been explored yet. This is an interesting question since, if the [4Fe–4S] cluster contributes to tRNA recognition or to the flipping of the target base, this would influence the overall enzyme activity. The lack of information of this potential function of the cluster is primarily due to the extreme sensitivity of these clusters to oxygen, making it challenging to anaerobically employ classical techniques, such as electrophoretic mobility shift assays, to determine the affinity of the modification enzymes for their tRNA substrates. In this study, we address this gap by successfully determining, under anaerobic conditions, the dissociation constants (Kd) for tRNA of two [4Fe–4S]-dependent tRNA thiolases, which target different positions within tRNA (within the tRNA core or in the anticodon loop) by taking advantage of the fluorescence of suitably located tryptophan residues in these enzymes.
RESULTS AND DISCUSSION
U8-tRNA thiolase MmTtuI
The biosynthesis of s4U8-tRNA was attributed to enzymes initially labeled as ThiI due to their association with thiamin biosynthesis in Escherichia coli (Mueller et al. 1998). However, it has been established that the rhodanese-like domain (RHD) of ThiI, crucial for thiamine synthesis, is not conserved in the U8-tRNA thiolase family (Martinez-Gomez et al. 2011). Consequently, it is now acknowledged that U8-tRNA sulfurases should not be designated as ThiI (Bender 2011), and we have renamed them as TtuI for tRNA thiolation uridine I (He et al. 2022). Intriguingly, the synthesis of s4U8-tRNA in various archaea involves a [4Fe–4S]-dependent mechanism (He et al. 2022), unlike E. coli TtuI (EcTtuI), where a persulfide-transfer mechanism is initiated by the formation of a persulfide on a crucial cysteine within the RHD (Mueller et al. 2001). Moreover, the catalytic mechanism of TtuI from species lacking the RHD, such as Bacillus anthracis and Thermotoga maritima, remains enigmatic due to the absence of conserved cysteines that could coordinate a [Fe–S] cluster. Biochemical and spectroscopic studies have shown that U8-tRNA thiolase from Methanococcus maripaludis (MmTtuI) can assemble a [4Fe–4S] cluster to yield a holo-protein capable of thiolating a tRNA transcript in vitro using inorganic sulfide as a sulfur source (He et al. 2022). The catalytic activity of MmTtuI required the presence of the cluster along with ATP and Mg2+ (He et al. 2022). Based on these findings, a mechanism akin to that of other [4Fe–4S]-dependent tRNA thiolation enzymes (Gervason et al. 2024) has been proposed, wherein the [4Fe–4S] cluster can bind the sulfur atom of the sulfur donor as a thiol, which serves as a sulfur donor (He et al. 2022).
MmTtuI is as an ideal system to investigate the role of a [4Fe–4S] cluster in tRNA binding through fluorescence analysis. Indeed, this enzyme features a sole tryptophan residue (W282), which is exposed to the surface, as evidenced by our structural model of the holo-enzyme derived from the 3D dimer of the apo-protein predicted by AlphaFold (Fig. 1A; Jumper et al. 2021). Furthermore, W282 resides in close proximity to several positive patches on the surface of MmTtuI, likely delineating the interaction zone with tRNA (Fig. 1B). Fluorescence measurements of apo-MmTtuI, following excitation at 295 nm, corresponding to tryptophan residues, revealed a spectrum with a peak at 357 nm (Fig. 1C), consistent with a highly polar environment for W282 (Vivian and Callis 2001). Notably, denaturation of apo-MmTtuI with 8 M urea did not induce any shift in the fluorescence maximum (Fig. 1C), indicating that W282 is fully solvent-exposed. We subsequently explored whether the fluorescence of this tryptophan was responsive to the addition of tRNA substrate. As depicted in Figure 2A, adding four molar equivalents of M. maripaludis tRNALys transcript (MmtRNALys, Supplemental Fig. S1A, left), previously identified as a substrate for holo-MmTtuI (He et al. 2022), to apo-MmTtuI resulted in a threefold decrease in fluorescence intensity compared to the apo-protein alone, accompanied by a slight shift of the peak toward 355 nm (Supplemental Fig. S2A). Thus, the presence of tRNA likely alters the environment of W282, making its fluorescence a potential probe for monitoring substrate binding to the enzyme. The titration of apo-MmTtuI with MmtRNALys yielded a hyperbolic decrease in fluorescence intensity (Supplemental Fig. S2B), allowing us to determine a Kd of 0.63 ± 0.1 µM for apo-MmTtuI and MmtRNALys (Fig. 2B; Table 1). This Kd value is comparable to the reported range of 0.25–1.9 µM for binding of tRNAPhe to EcTtuI (Lauhon et al. 2004; Tanaka et al. 2009).
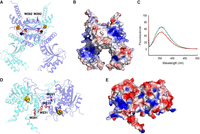
Environment of tryptophan residues in MmTtul and EcTtcA. (A) AlphaFold2 model of the holo-MmTtuI dimer in complex with ATP and Mg2+. One monomer is colored in cyan and the other in blue. Residues W282 and ATP are depicted as sticks (in magenta and red, respectively), while the [4Fe–4S] cluster and Mg2+ ions are shown in sphere representation with sulfur atoms colored in yellow, iron atoms in orange, and Mg atoms in black. (B) Electrostatic surface of the holo-MmTtuI dimer (negative charge in red, positive charge in blue). (C) Fluorescence spectra obtained at λex = 295 nm of native apo-MmTtuI (in green) and after denaturation with 8 M urea (in red). (D) AlphaFold2 model of the holo-EcTtcA dimer. One monomer is colored in cyan and the other in blue. Residues W231 and W291 are depicted as red sticks, while the [4Fe–4S] cluster is shown in sphere representation. (E) Electrostatic surface of the holo-EcTtcA dimer.
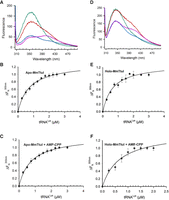
Monitoring of MmtRNALys binding to apo- and holo-MmTtuI by fluorescence. (A) Fluorescence spectra obtained at λex = 295 nm of apo-MmTtuI (0.5 µM) alone (in green), apo-MmTtuI + 3 mM AMP-CPP (in red), apo-MmTtuI + 2 µM tRNALys (in blue), and apo-MmTtuI + 3 mM AMP-CPP + 2 µM tRNALys (in magenta). (B) and (C) Saturation curves of W282 fluorescence at 355 nm normalized to tRNALys concentration for apo-MmTtuI in the absence (B) and presence of 3 mM AMP-CPP (C). (D) Fluorescence spectra obtained at λex = 295 nm of holo-MmTtuI (0.5 µM) alone (in green), holo-MmTtuI + 3 mM AMP-CPP (in red), holo-MmTtuI + 2 µM tRNALys (in blue), holo-MmTtuI + 3 mM AMP-CPP + 2 µM tRNALys (in magenta). (E) and (F) Normalized saturation curves of fluorescence at 355 nm, for holo-MmTtuI in the absence (E) and presence of 3 mM AMP-CPP (F).
Kd values of the protein/RNA complexes studied
We also investigated whether the binding of the nonhydrolyzable ATP analog (α-β-methylene-adenosine 5′-triphosphate [AMP-CPP]) had any effect on the stability of the apo-MmTtuI/MmtRNALys complex. As shown for apo-MmTtuI alone, the fluorescence of apo-MmTtuI was partially quenched upon MmtRNALys binding in the presence of AMP-CPP (Fig. 2A; Supplemental Fig. S2C,D). However, AMP-CPP did not alter the Kd of apo-MmTtuI for MmtRNALys (Table 1), as shown by the titration curve in Figure 2C.
To further substantiate our methodology, we examined the capacity of apo-MmTtuI to bind a mini-RNA, which was designed based on the mini-RNAs that had been shown to be substrates of EcTtuI (Lauhon et al. 2004; Tanaka et al. 2009). Indeed, the 39 nt mini-RNA of E. coli tRNAPhe corresponding to the acceptor-stem and the T-arm, called TPHE39A, was shown to bind to EcTtuI with only a two- to fourfold increase in Kd (depending on the methodology used) compared to the full-length tRNAPhe (Lauhon et al. 2004; Tanaka et al. 2009), whereas it was a substrate for EcTtuI with an activity of one-third that of full-length tRNAPhe (Lauhon et al. 2004). In addition, TtuI from T. maritima (TmTtuI) was identified as another enzyme capable of effectively catalyzing the formation of s4U8 on TPHE39A but the affinity of the enzyme/TPHE39A complex was not reported (Neumann et al. 2014). Intriguingly, the crystal structure of TPHE39A revealed significant changes in the secondary structure of the T-stem compared to that of the full-length tRNAPhe (Tanaka et al. 2009), suggesting that such conformational change in the T-stem does not disturb RNA binding. Similarly, we tested whether MmTtuI could bind the 41 nt mini-RNA called TLYS41A, which includes the target uridine U8, the acceptor-stem, a bulged loop, and the T-stem loop of MmtRNALys (Supplemental Fig. S1A, right). Indeed, fluorescence analysis indicated that apo-MmTtuI can interact with TLYS41A, albeit with a threefold lower affinity compared to full-length MmtRNALys (Table 1; Supplemental Fig. S3).
Previous studies have highlighted the importance of magnesium ions, in addition to the [4Fe–4S] cluster and ATP, for MmTtuI to be catalytically active (He et al. 2022). According to our model of the complex of MmTtuI with ATP and Mg2+ (Fig. 1A,B), the Mg2+ binding site is predicted to be located near the tRNA-binding site. Consequently, we investigated the impact of Mg2+ ions on the binding of both TLYS41A and the full-length MmtRNALys to apo-MmTtuI by fluorescence (Supplemental Fig. S3). Our results showed a 3.8-fold and 3.5-fold decrease in Kd, for TLYS41A and MmtRNALys, respectively, in the presence of Mg2+ (Table 1).
Next, cluster reconstitution was performed with apo-MmTtuI to generate holo-MmTtuI and investigate the potential role of the cluster in tRNA binding. The UV/visible absorption spectrum of the protein after [Fe–S] cluster reconstitution revealed a characteristic peak ∼410 nm consistent with the presence of a [4Fe–4S] cluster bound within holo-MmTtuI (Supplemental Fig. S4A). The iron quantification (3.2 Fe atoms per monomer) together with the 410/280 nm absorbance ratio of 0.26 indicated that the protein underwent successful cluster reconstitution in the [4Fe–4S] cluster state, as reported previously (He et al. 2022). As shown in Supplemental Figure S5A, holo-MmTtuI catalyzed the formation of s4U in the tRNALys transcript. Interestingly, we found that holo-MmTtuI also modifies TLYS41A (Supplemental Fig. S5A), but with an activity approximately six times lower than that observed with the full-length tRNA. Nevertheless, this indicates that a mini-RNA is a substrate for MmTtuI, as previously reported for EcTtuI and TmTtuI (Lauhon et al. 2004; Tanaka et al. 2009).
We then applied our fluorescence approach to investigate the binding of tRNA to holo-MmTtuI. The fluorescence spectrum of the holo-protein displayed a peak maximum at 344 nm, indicating a significant 9 nm blue shift compared to the apo-protein (Fig. 2D). This finding suggests that the presence of the [Fe–S] cluster creates a less polar environment for W282. As with apo-MmTtuI, the addition of MmtRNALys reduced the fluorescence of holo-MmTtuI, regardless of the presence of AMP-CPP (Fig. 2D). Moreover, the analysis of the isotherms for tRNA binding to holo-MmTtuI, depicted by the normalized fluorescence change at 355 nm as a function of tRNALys concentrations, indicated that, similar to apo-MmTtuI, AMP-CPP did not significantly alter the Kd for tRNALys, which remained ∼0.5–0.7 µM (Fig. 2E,F; Table 1). Altogether, apo- and holo-MmTtuI have a comparable affinity for MmtRNALys (Table 1), in the presence or absence of AMP-CPP, suggesting that the [4Fe–4S] cluster has no discernible effect on the overall binding of tRNA.
We subsequently questioned whether the [Fe–S] cluster could have a local effect on the recognition of U8 by TtuI enzymes. Indeed, analysis of the X-ray structures of unbound tRNAs (Fernández-Millán et al. 2016) indicates that the U8 position is not readily accessible to solvent, suggesting the necessity of a conformational change of the U8-containing region and/or the flipping-out of the target base by TtuI to facilitate its thiolation. To investigate this possibility, we replaced adenine 9 with a 2-aminopurine (2AP) in MmtRNALys (2AP9-MmtRNALys) (Fig. 3A) and first examined whether this modified tRNA could serve as a substrate for MmTtuI. Interestingly, as shown in Supplemental Figure S5A, MmTtuI not only modifies 2AP9-MmtRNALys but the activity is enhanced when 2AP is present at position 9 instead of adenine. 2AP is expected to undergo fluorescence quenching upon base stacking, and its unstacking to induce a significant increase in fluorescence. Indeed, substituting adenine with 2AP was shown to facilitate the study of the flipping mechanism of the A57, A58-tRNA methyltransferase TrmI from Pyrococcus abyssi, with the flipping of adenine 57 by TrmI resulting in a 2.5-fold enhancement in 2AP fluorescence (Hamdane et al. 2014). Thus, we monitored the effect of the addition of MmTtuI to 2AP-tRNALys on the fluorescence of this fluorophore when excited at 320 nm. As depicted in Figure 3B, the addition of a fourfold molar excess of apo-MmTtuI to 2AP9-MmtRNALys resulted in a 30% increase in 2AP fluorescence. Adding the same quantity of holo-MmTtuI to 2AP9-MmtRNALys produced a comparable 25% enhancement in 2AP fluorescence. The observation of a 9 nm red shift in the maximum fluorescence in the presence of the [Fe–S] cluster suggests a more polar environment for 2AP9 when bound to holo-MmTtuI rather than to apo-MmTtuI. Due to its positive charge, the cluster likely generates a local electric field that alters the electronic environment of 2AP. In an effort to rationalize the relatively modest increase in fluorescence of 2AP9-MmtRNALys upon binding MmTtuI, we generated an AlphaFold3 model of MmtRNALys and analyzed the environment of U8. As shown in Figure 3C, the U8 and A9 bases are separated by the U13 base so that the change of fluorescence of 2AP9 upon binding of MmTtuI cannot be directly related to the flipping of U8 but rather to a conformational change of the U8–U13 loop. Interestingly, given that the binding of 2AP9-MmtRNALys to apo-MmTtuI and holo-MmTtuI is similar (Fig. 3B), the presence of the [4Fe–4S] cluster does not significantly alter the conformational changes around A9 occurring upon binding MmTtuI. Since 2AP9-MmtRNALys is a good substrate of MmTtuI (Supplemental Fig. S5A), flipping of U8 likely occurs but the experimental structure of the holo-MmTtuI/2AP9-MmtRNALys complex is needed to validate this hypothesis.
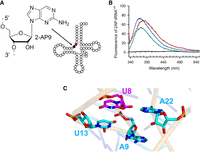
Sensitivity of the [4Fe–4S] cluster of MmTtuI to the environment of the U8 target base in MmtRNALys. (A) Chemical structure of 2-aminopurine (2AP) and its position (in black) in 2AP9-MmtRNALys, adjacent to the target base U8 (in red). (B) Fluorescence spectra obtained at λex = 320 nm of 2AP9-MmtRNALys (0.5 µM). 2AP9-MmtRNALys alone (in green), in the presence of apo-MmTtuI (2 µM) + 3 mM AMP-CPP (in blue) or in the presence of holo-MmTtuI (2 µM) + 3 mM AMP-CPP (in red). Apo-MmTtuI in the absence of 2AP9-MmtRNALys (in black). (C) Region surrounding U8 in the AlphaFold3 model of MmtRNALys (pTM score of 0.44). The target base U8 is shown in magenta.
C32-tRNA thiolase EcTtcA
We used TtcA from E. coli (EcTtcA), a [4Fe–4S]-dependent C32-tRNA thiolase (Bouvier et al. 2014) to expand our investigation on the role of the iron–sulfur cluster in tRNA thiolation enzymes. TtcA enzymes catalyze the insertion of sulfur within C32, next to the anticodon loop, in tRNAs (Supplemental Fig. S1B), in the presence of ATP. Unlike MmTtuI, EcTtcA possesses two tryptophan residues, W231 and W291 (Fig. 1D) that are located near the putative tRNA-binding site, according to their location next to a positive region of the electrostatic interface (Fig. 1E). The s2C32 modification is present in four E. coli tRNAs (Cappannini et al. 2024), including tRNAArg (ICG) (EctRNAArg) (Supplemental Fig. S1B, left). It was previously shown that the [4Fe–4S] cluster and ATP were essential for s2C biosynthesis by EcTtcA within a bulk tRNA isolated from an E. coli ΔttcA strain, using cysteine desulfurase IscS and cysteine, in the presence of dithiothreitol, as the sulfur source (Bouvier et al. 2014). First, we could efficiently reconstitute a [4Fe–4S] cluster in EcTtcA, as shown by the UV-visible spectrum of holo-EcTtcA (Supplemental Fig. S4B), in agreement with the quantification of Fe (3.6 atoms per monomer). The holo-enzyme was active on an EctRNAArg transcript but also on a 17 nt anticodon loop called mini-RNAArg (Supplemental Fig. S1B, right), in the presence of ATP and inorganic sulfide as the sulfur source (Supplemental Fig. S5B).
We determined the Kd of the apo- and holo-forms of EcTtcA for the EctRNAArg transcript by monitoring protein fluorescence, as detailed previously for MmTtuI. Fluorescence analysis revealed that the addition of EctRNAArg transcript to either apo- or holo-EcTtcA induced fluorescence quenching in a concentration-dependent manner (Fig. 4). The resulting titration isotherms yielded similar Kd values for the apo- and holo-forms (3.5 and 4 µM, respectively, Table 1), suggesting that, as with MmTtuI, the [4Fe–4S] cluster did not significantly affect tRNA binding by EcTtcA.
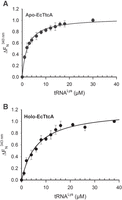
Monitoring of EctRNAArg binding to apo- and holo-EcTtcA by fluorescence. (A) and (B) Normalized saturation curves of fluorescence at 343 nm for apo-EcTtcA (A) and holo-EcTtcA (B).
In conclusion, we report here the first measurement of the affinity of two [4Fe–4S]-dependent tRNA-modifying enzymes for their tRNA substrates, in the apo, and holo states under anaerobic conditions. Our results show that the [4Fe–4S] clusters of MmTtuI and EcTtcA play no obvious role in tRNA binding. Additionally, it is likely that the cluster of MmTtuI is not involved in flipping the target U8 base for modification, a process that appears to be driven by the enzyme itself. These findings suggest an exclusively chemical role of the [4Fe–4S] cluster in the thiolation of tRNA. Finally, our results regarding two tRNA thiolation enzymes possessing tryptophan residues located near the tRNA-binding site have unveiled a robust fluorescence method for determining Kd values of these enzymes for their RNA substrates under various conditions, which could be extended both to other [Fe–S]-dependent or [Fe–S]-independent RNA modification enzymes to determine their affinity constants for their RNA substrate.
MATERIALS AND METHODS
The nonhydrolyzable ATP analog AMP-CPP (α-β-methylene-adenosine-5′triphosphate) was bought from Jena Bioscience and the RNA transcripts from Horizon Discovery Biosciences.
Heterologous gene overexpression and aerobic purification of MmTtuI and EcTtcA
Apo-MmTtuI was produced and purified as previously described (He et al. 2022). The gene coding for EcTtcA was synthesized by GenScript and cloned between the NdeI and BamHI sites in the pET15b plasmid, in order to add an N-terminal cleavable histidine tag to the protein. Apo-EcTtcA was produced and purified after cleavage of the histidine tag by the H3C protease according to He et al. (2022).
In vitro [Fe–S] cluster reconstitution and anaerobic purification of holo-MmTtuI and holo-EcTtcA
The reconstitution of the [4Fe–4S] cluster and purification of holo-MmTtuI and holo-EcTtcA were performed in a glove box (MBraun) containing <0.5 ppm O2. After incubation of as-purified proteins (100 µM) with 15 mM dithiothreitol for 15 min, a fivefold molar excess of ferrous ammonium sulfate and sodium sulfide was added. Cluster reconstitution was followed by the appearance of a band at around 410 nm in the UV-visible spectrum, which was recorded with an XL-100 UVICON spectrophotometer equipped with optical fibers, and incubation was stopped after 3 h. After centrifugation for 10 min at 20,000g, holo-MmTtuI and holo-EcTtcA were loaded onto a Superdex 200 Increase 10/300 GL gel filtration column (Cytiva) equilibrated in 25 mM HEPES pH 7.5, 150 mM NaCl. The Fe content in the holo-proteins after cluster reconstitution was quantified using the Fish method (Fish 1988).
Activity tests
The RNA transcripts were refolded by heating for 2 min at 85°C and then cooling on ice for 4 min. Holo-MmTtuI and holo-EcTtcA (5 µM) were incubated with their full-length or mini-RNA transcripts (15 µM) for 1 h at 37°C in 50 µL 20 mM HEPES pH 7.5, 150 mM NaCl in the presence of 0.25 mM ATP, 2.5 mM MgCl2, and 1 mM Na2S in an MBraun glove box. The reaction was stopped by adding 1 µL of 3 M formic acid, and the pH adjusted to around 6.5 by adding 3 µL of 1 M Tris pH 8.5. After the reaction, tRNA was digested and s4U or s2C was separated by HPLC-MS/MS and quantified using synthetic s4U or s2C standard, as described previously (Bimai et al. 2023).
Fluorescence studies
The emission fluorescence spectra were recorded in a quartz cuvette at 37°C on a Cary Eclipse fluorescence spectrophotometer (Varian) in an MBraun glove box. After exciting at the Trp wavelength of 295 nm or at 320 nm for experiments with 2AP-containing RNA, the fluorescence emission was recorded every nm from 335 to 550 nm. The excitation and emission slits were set to 5 and 10 nm, respectively. In all experiments, enzyme (1 µM) was incubated with RNA transcript (from 0 to 3.2 µM) in 700 µL of 25 mM HEPES pH 7.5, 150 mM NaCl, 3 mM MgCl2 in the presence or absence of AMP-CPP (3 mM). MgCl2 was omitted only when the effect of MgCl2 on tRNA binding was studied (Supplemental Fig. S3). Before recording the spectra, enzyme and refolded RNA were incubated for 5 min to reach the equilibrium. The resulting spectra were corrected for the contribution of the buffer. ΔFN (normalized value of fluorescence) = (ΔF at a given RNA concentration)/(ΔF_max).
Structural modeling
The AlphaFold models of MmTtuI and EcTtcA were calculated with the Google Colab platform and AlphaFold2_advanced option (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/beta/AlphaFold2_advanced.ipynb#scrollTo=ITcPnLkLuDDE) that does not use templates (homologous structures), and refined using the Amber-relax option to enhance the accuracy of the side chains geometry. The default mode of sampling options was used: num_models=5, ptm option, num_ensemble=1, max_cycles=3, tol=0, num_samples=1, 1:1 value as the homo-oligomer assembly option. The models were ranked according to their pLDDT confidence values (between 0 and 100, from low to high confidence).
The models of holo-MmTtuI and holo-EcTtcA were built by taking the position of the cluster of holo-MmNcsA (PDB code 6SCY) (Bimai et al. 2023) after superposition of its ATPase region with that of the MmTtuI and EcTtcA AlphaFold models, respectively. Similarly, the ATP and Mg2+ positions of TmTtuI in complex with ATP and Mg2+ (PDB code 4KR7), after superposition of TmTtuI with the AlphaFold model of MmTtuI, were used to build the complex of MmTtuI with ATP and Mg2+.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the Centre National de la Recherche Scientifique, Sorbonne Université, as well as the “Initiative d'Excellence” program from the French State (grant “DYNAMO,” ANR-11-LABX-0011-01) and the French National Research Agency (grant sulfo-tRNA, ANR-22-CE44-0012). We thank Nisha He for preparing apo-MmTtuI.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080292.124.
- Received October 25, 2024.
- Accepted March 4, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








