
Selected humanization of yeast U1 snRNP leads to global suppression of pre-mRNA splicing and mitochondrial dysfunction in the budding yeast
- Subbaiah Chalivendra1,6,
- Shasha Shi1,6,
- Xueni Li1,
- Zhiling Kuang1,
- Joseph Giovinazzo1,4,
- Lingdi Zhang1,5,
- John Rossi1,
- Jingxin Wang2,
- Anthony J. Saviola1,
- Robb Welty1,
- Shiheng Liu3,
- Katherine F. Vaeth1,
- Z. Hong Zhou3,
- Kirk C. Hansen1,
- J. Matthew Taliaferro1 and
- Rui Zhao1
- 1Department of Biochemistry and Molecular Genetics, University of Colorado Anschutz Medical Campus, Aurora, Colorado 80045, USA
- 2Department of Medicinal Chemistry, University of Kansas, Lawrence, Kansas 66047, USA
- 3Department of Microbiology, Immunology, and Molecular Genetics, University of California, Los Angeles, California 90095, USA
- Corresponding authors: rui.zhao{at}cuanschutz.edu, subbaiah.chalivendra{at}cuanschutz.edu
-
↵6 These authors contributed equally to this work.
Abstract
The recognition of the 5′ splice site (5′ ss) is one of the earliest steps of pre-mRNA splicing. To better understand, the mechanism and regulation of 5′ ss recognition, we selectively humanized components of the yeast U1 (yU1) snRNP to reveal the function of these components in 5′ ss recognition and splicing. We targeted U1C and Luc7, two proteins that interact with and stabilize the yU1 snRNA and the 5′ ss RNA duplex. We replaced the zinc-finger (ZnF) domain of yeast U1C (yU1C) with its human counterpart, which resulted in a cold-sensitive growth phenotype and moderate splicing defects. We next added an auxin-inducible degron to yeast Luc7 (yLuc7) protein (to mimic the lack of Luc7Ls in human U1 snRNP). We found that Luc7-depleted yU1 snRNP resulted in the concomitant loss of Prp40 and Snu71 (two other essential yU1 snRNP proteins), and further biochemical analyses suggest a model of how these three proteins interact with each other in the U1 snRNP. The loss of these proteins resulted in a significant growth retardation accompanied by a global suppression of pre-mRNA splicing. The splicing suppression led to mitochondrial dysfunction as revealed by a release of Fe2+ into the growth medium and an induction of mitochondrial reactive oxygen species. Together, these observations indicate that the human U1C ZnF can substitute that of yeast, Luc7 is essential for the incorporation of the Luc7–Prp40–Snu71 trimer into yU1 snRNP, and splicing plays a major role in the regulation of mitochondrial function in yeast.
Keywords
INTRODUCTION
Gene regulatory mechanisms have played an important role in the evolution of organismal diversity, including the origin of multicellularity (King and Wilson 1975; Carroll 2005; Wittkopp and Kalay 2012; Kianianmomeni 2015). Transcriptional and posttranscriptional processes constitute the early steps of gene expression. In eukaryotes, pre-mRNA splicing is a major gene regulatory mechanism (Girardini et al. 2023). Only 2.8% of the human genome represents coding sequences, whereas 35% corresponds to introns (Hatje et al. 2019). The intronic expansion has resulted in a huge transcriptomic/proteomic diversity, as 90% of human genes are alternatively spliced (Calarco et al. 2007). The spliceosome is comprised of five highly conserved uridylyl-rich small RNA–protein complexes (U snRNPs)—namely, U1, U2, U4, U5, and U6 (Wahl et al. 2009). U1 snRNP is critical for the recognition of introns, specifically, the 5′ splice site (5′ ss) and forming the first commitment complex of the splicing cycle (E complex in humans and CC1/CC2 in yeast). The role of U1 in gene expression is beyond pre-mRNA splicing, including upstream events of gene expression. For example, in mammals, U1 is critical for enhancing transcription initiation (Damgaard et al. 2008; Engreitz et al. 2014; Rose 2019), increasing the rate of transcription elongation (Mimoso and Adelman 2023), determining promoter directionality (Almada et al. 2013), and suppressing premature polyadenylation and transcriptional termination (Kaida et al. 2010; Di et al. 2019). Many human genetic diseases involving splicing defects relate to the failure of 5′ ss recognition by the human U1 (hU1) snRNP (Lorson et al. 1999; Roca et al. 2008; Jüschke et al. 2021). A detailed structural and functional analysis of the hU1 is critical for understanding splicing regulation and implications for human health.
hU1 snRNP often has transient and contextual interaction with alternative splicing factors, many of whose identities and roles are not fully understood. These intricate genetic and molecular interactions may not be easily studied in vitro, cell culture, or other metazoan model systems. Yeast is a facile model to address outstanding questions in splicing regulation, owing to its simplicity and versatility, in addition to the conservation of splicing machinery (Fabrizio et al. 2009; Meyer and Vilardell 2009). As expression of human homologs can complement yeast genes of diverse pathways, yeast has been successfully used to dissect complex biological pathways relevant to human physiology and pathology (Garge et al. 2020; Boonekamp et al. 2022; Hunter et al. 2024) as well as drug discovery (dos Santos and Sá-Correia 2009; Zimmermann et al. 2018). Yeast has fewer (334; ∼5% of the genome) and smaller introns with a highly conserved 5′ ss, and most of them are cotranscriptionally processed (Spingola et al. 1999; Aslanzadeh et al. 2018; Talkish et al. 2019). Being a unicellular eukaryote uncomplicated by tissue type diversity, gene regulation in yeast is tightly linked to the phenotype (Airoldi et al. 2009; Strassburg et al. 2010; Tamari et al. 2014). In turn, gene expression itself is closely aligned to pre-mRNA splicing because of an enrichment of introns in ribosomal protein genes (RPGs) and the high transcription rate of intron-containing genes (both RPGs and non-RPGs) in yeast (Ares et al. 1999; Juneau et al. 2006; Lukačišin et al. 2022). This permits reliable correlation of any introduced alterations in the splicing machinery with changes in molecular, supramolecular, and cellular/organismal phenotypes.
hU1 snRNP contains a 164 nt U1 snRNA that threads through the heptameric ring formed by seven Sm proteins, and three additional proteins U1A, U1C, and U1-70K. Although earlier studies suggested that U1C makes an important contribution to the sequence-specific recognition of 5′ ss (Du and Rosbash 2002), structural analysis of hU1 snRNP revealed that 5′ ss is primarily recognized via RNA–RNA binding to the 5′ end of U1 snRNA (Kondo et al. 2015). U1C uses its zinc-finger (ZnF) domain to bind the sugar-phosphate backbone but not the bases of the 5′ss–U1 snRNA duplex and is required for the duplex stability, particularly when 5′ ss sequences are noncanonical (Kondo et al. 2015). The 5′ end of U1 snRNA that base pairs with 5′ ss sequence is invariant across all eukaryotes, from yeast to human (Kretzner et al. 1987). However, human 5′ ss sequences are highly divergent, unlike the predominantly uniform 5′ ss sequence of yeast introns. It follows that the duplex-stabilizing role of U1C becomes especially relevant in the case of hU1 snRNP.
Yeast U1 (yU1) snRNP contains a much larger snRNA, homologs of all hU1 snRNP proteins, and seven additional stably associated proteins. Cryogenic electron microscopy (cryo-EM) structure of the yU1 snRNP demonstrated that the yU1 snRNP core (composed of U1 snRNA and homologs of the hU1 snRNP proteins) is highly similar to the entire hU1 snRNP, with additional yeast auxiliary proteins stably bound to the core (Li et al. 2017). Of these auxiliary yeast proteins, Luc7 also uses a ZnF domain to bind the 5′ss–U1 snRNA duplex, presumably providing additional stability for the duplex. There are three human homologs (Luc7L, Luc7L2, and Luc7L3) of Luc7 that are known alternative splicing factors (Puig et al. 2007). The human Luc7Ls (hLuc7Ls) bind U1 snRNA as well as an AAGAAG sequence in the exon near weak 5′ ss (Daniels et al. 2021), suggesting that they may also help stabilize the 5′ ss–U1 snRNA duplex similar to yeast Luc7 (yLuc7). In the current study, we have made selected modifications (humanization) of yU1 snRNP components to unravel the mechanistic details of 5′ ss recognition of splicing regulation in eukaryotes. Our studies reveal novel details on the structure–function relationships of two essential U1 snRNP proteins and how these molecular interactions are integrated into organismal responses to external and internal stimuli.
RESULTS
Humanization of yU1C leads to moderate splicing defects
The N-terminal region of U1C containing the ZnF domain is highly conserved between human and yeast, both in sequence (50% identity and 69% similarity) and its role in forming the first commitment or the E complex (Nelissen et al. 1991; Muto et al. 2004; Schwer and Shuman 2014, 2015; Kondo et al. 2015), whereas its C-terminal region is highly divergent (Fig. 1A). We introduced the full-length human U1C into a yeast U1C (yU1C) shuffle strain (Schwer and Shuman 2014) and showed that human U1C cannot substitute yU1C, even though human U1C was well expressed in the shuffle strain (Fig. 1B; Supplemental Fig. S1A). We next replaced the first 36 amino acids of yU1C containing the ZnF domain with that of human (abbreviated as h-yU1C for humanized yeast U1C) using CRISPR in a yeast strain where U1A is TAP tagged with protein A and calmodulin-binding peptide (CBP) (Li et al. 2017). h-yU1C grew similarly as the wild-type (WT) yeast strain at 30°C and 37°C, although it grew slower at low temperature (17°C) (Fig. 1C).
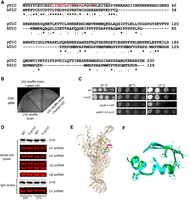
Humanization of yU1C leads to minor cold-sensitive (cs) growth defects. (A) Yeast and human U1C are highly conserved in the ZnF domain as demonstrated by sequence alignment using Clustal Omega (Sievers et al. 2011). (*) identical residues, (:) conserved residues, (.) less conserved residues, (‐‐) gaps. The line above the sequence indicates the yeast ZnF domain that was replaced with the corresponding human sequences. Red font indicates residues interacting with the U1–5′ ss RNA duplex in the cryo-EM structure of the yeast E complex (Li et al. 2019). (B) A yU1C shuffle strain (Schwer and Shuman 2014) alone, transformed with yU1C or transformed with human U1C, was plated on an FOA plate. Only the U1C shuffle strain transformed with yU1C grew. (C) Spot assay (10× serial dilution) of yeast strain with WT background, h-yU1C, prp28-1, and prp28-1 + h-yU1C at different temperatures. (D) U1A (probed by an anti-CBP antibody) and U1 snRNA (probed by solution hybridization) levels in whole-cell lysate and after IgG pull down using the TAP tag on U1A were similar between the WT and h-yU1C strain at both 30°C and 17°C. Other snRNAs serve as internal controls. (E) Overall structure of the U1 snRNPh-yU1C. The ZnF domain of U1C that was humanized is shown in cyan, the 5′ ss in red, the first 10 nt of U1 snRNA in orange, and the N-terminal helix of Luc7 in purple. (F) Superimposition of the ZnF domain of WT (green) and h-yU1C (cyan) backbone. Residues in h-yU1C that interact with the U1–5′ ss RNA duplex are shown in the stick model and labeled.
We pulled down U1 snRNP using the protein A tag on U1A and demonstrated that the h-yU1C strain has similar levels of U1 snRNP as the WT strain at both 30°C and 17°C, by detecting the level of U1 snRNA using solution hybridization (Li and Brow 1993) and the level of U1A through its CBP tag by immunoblotting (Fig. 1D). We purified U1 snRNP from the h-yU1C strain using the TAP tag on U1A and determined its cryo-EM structure in complex with the Act1 pre-mRNA and BBP (branchpoint-binding protein)/Mud2 to 3.5 Å resolution (Fig. 1E; Supplemental Fig. S2A,B), similar to what we have done with the WT yU1 snRNP (Li et al. 2019). The structure demonstrated that h-yU1C bound to the U1 snRNA and 5′ ss duplex similarly to the WT U1 snRNP (Fig. 1E), and the h-yU1C and yU1C structures could be well-superimposed (Fig. 1F). In addition, Luc7 was present in the structure with well-defined density for its N-terminal α-helix that interacted with the Sm ring (Supplemental Fig. S2C), although the rest of the Luc7 density was not as well-defined as in the WT spliceosomal E complex (Li et al. 2019), potentially because of the much smaller particle numbers in the U1 snRNPh-yU1C data set.
To analyze potential splicing changes due to yU1C humanization, we carried out RNA-seq analyses of the h-yU1C and WT strain, from three biological replicates of each. These analyses consistently showed that a number of intron-containing genes in the h-yU1C strain have greater intron retention (IR) than the WT (Fig. 2A; Supplemental Table S1). Of the 190 yeast intron–containing genes with sufficient reads, ∼17% (32 out of 190) showed 20% or higher IR, and ∼43% (83 out of 190) had 10% or higher IR in the h-yU1C strain compared to the WT (Fig. 2A). Yeast strains used for RNA-seq were grown at 30°C but harvested by centrifugation for 7 min at 4°C . We could not rule out the possibility that the splicing changes observed in RNA-seq are caused by the brief exposure of cells to low temperature during harvest. A similar IR was observed in RNAs prepared from cells grown at lower temperature, as analyzed by RT-PCR (Fig. 2B). The IR did not correlate with whether an intron contains the canonical yeast 5′ ss (AG/GUAUGU, where/designates the exon/intron boundary), or whether the intron was perfectly complementary to the 5′ end of U1 snRNA (note that the yeast canonical 5′ ss sequence AG/GUAUGU does not form a perfect complement to the 5′ end of U1 snRNA which has the sequence of ACUUACCU) (Fig. 2C,D; Spingola et al. 1999). On the other hand, IR was greater for genes with shorter intron length or shorter upstream exon (Fig. 2E,F).
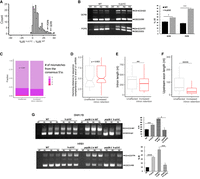
Humanization of yU1C leads to moderate splicing defects. (A) RNA-seq experiments revealed that h-yU1C led to poorer splicing of a moderate number of intron-containing genes as demonstrated by their increased %IR (percentage intron retention). Arrows indicate the two genes selected for RT-PCR validation in B. (B) RT-PCR of POP8 and QCR9 showed that splicing was impaired in the h-yU1C strain at 17°C. POP8 and QCR9 were picked as examples of genes affected by humanizing yU1C in the RNA-seq data as two of the most affected genes. R1–3 represent three biological replicates. (C) There was no significant difference between unaffected and retained introns in how close their 5′ splice sites were to the consensus sequence. P values were calculated using a binomial test considering the fraction of introns with perfect and imperfect matches to the consensus sequence of GTATGT. (D) There was no significant difference between unaffected and retained introns in how close their 5′ splice sites are to the sequence (AGGTAAGTAT) that complements the 10 nt at the 5′ end of U1 snRNA. P values were calculated using a Wilcoxon rank-sum test considering the Hamming distances of introns to the sequence complementary to the 5′ end of U1 snRNA. (E) Increased IR is significantly correlated with shorter intron length. (F) Increased IR significantly correlated with shorter upstream exon. For D, E, and F, introns were identified as having “increased IR” if they had FDR values of <0.05 and an increase in IR value of at least 0.05. Introns that did not meet these criteria were classified as “unaffected.” One hundred and twenty-two introns were classified as having increased IR, and 68 were classified as being unaffected. P values were calculated using a Wilcoxon rank-sum test in D, E, and F. (G) RT-PCR analyses of SNR17B and HRB1 showed that h-yU1C rescued the splicing defect caused by prp28-1. R1–3 represent three biological replicates. (*) P < 0.05, (**) P < 0.01, (****) P < 0.0001.
To understand the mechanism of the splicing changes caused by the h-yU1C, we evaluated the binding affinity between U1 snRNPh-yU1C and the 5′ ss pre-mRNA in comparison to the WT U1 snRNP, using a fluorescence polarization (FP) experiment and a 15 nt RNA (AAGAAAG/GUAAGUAG where/denotes the exon/intron boundary) containing the 5′ ss of Ubc9 whose splicing was impaired (43% increase in IR relative to the WT) in the U1 snRNPh-yU1C strain (Supplemental Table S1). We found that the Kd between U1 snRNPh-yU1C and the 5′ ss RNA is 2.5 nM (2.1–2.9 nM at 95% confidence interval), not significantly different from the Kd between WT U1 snRNP and the same 5′ ss oligo (3.1 nM, 2.6–3.7 nM at 95% confidence interval) (Supplemental Fig. S3). These data indicated that the binding affinities between these two U1 snRNPs and the 5′ ss were clearly not drastically different, although it may be difficult to differentiate small differences in the binding affinities using FP experiments. For example, it could be difficult to ensure the purified U1 snRNPWT or U1 snRNPh-yU1C to be completely identical (which may have different amounts of impurity or active U1 snRNP). Instead, we decided to answer this question using a genetic approach in vivo. We generated a U1 snRNPh-yU1C strain carrying prp28-1, a cs mutant of helicase Prp28. The activity of Prp28 is required for displacing the U1 snRNA from the 5′ ss, thereby allowing the recruitment of U6 snRNA as the splicing cycle proceeds (Staley and Guthrie 1999). We found that the h-yU1C rescues the cs phenotype of prp28-1 (Fig. 1C). Furthermore, RT-PCR analyses of representative genes (SNR17B and HRB1) whose splicing was impaired by prp28-1 confirmed that h-yU1C rescued the splicing defect caused by prp28-1 (Fig. 2G), suggesting that the U1 snRNP and 5′ ss interaction was weakened in the h-yU1C strain.
To further define the key residues contributing to the altered affinity in the h-yU1C strain, we first examined residues in U1C that contact the U1 snRNA and 5′ ss RNA duplex. These residues are mainly concentrated within residues 13–28 in the ZnF (Kondo et al. 2015) and are minimally different in sequence (Fig. 1A, T17 to S and K28 to R from yeast to human) and structure (Fig. 1F) between yeast and human U1C. We generated a T17S/K28R double mutant on chromosomal U1C using CRISPR. Compared to the WT, this strain had no apparent growth defect at 30°C and 18°C (Supplemental Fig. S4A). There was also no significant splicing impairment in the T17S/K28R mutant compared to the WT in the two genes (POP8 and QCR9) we tested using RT-PCR (Supplemental Fig. S4B). These data suggested that residues T17S and K28R were not sufficient to account for the growth and splicing defects observed in the h-yU1C strain. Rather, differences within the entire ZnF domain between the two species likely led to the altered affinity between U1C and the 5′ ss.
Luc7 triple mutant has no significant effect on splicing
Unlike yLuc7, which is an integral component of yU1 snRNP, its three human homologs Luc7L, Luc7L2, and Luc7L3 (Fig. 3A) bind only transiently to hU1 snRNP in a tissue- and development-specific manner (Howell et al. 2007; Daniels et al. 2021). We first attempted to humanize yLuc7 by replacing yLuc7 with its human homologs in a Luc7 shuffle strain. However, none of the shuffle strain transformants with Luc7Ls grew on 5-fluoroorotic acid (FOA) even though the hLuc7Ls were well expressed in the shuffle strain (Fig. 3B; Supplemental Fig. S1B), indicating that yLuc7 could not be replaced by hLuc7Ls.
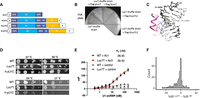
The Luc7 triple mutant has no significant effect on splicing. (A) Domain organization of the yLuc7 and hLuc7 homologs. α represents α-helix. CC represents a coiled-coil region. RE, RS, and R represent Arg/Glu, Arg/Ser, and Arg-rich regions. (B) hLuc7 homologs could not replace the yLuc7, as demonstrated by the lack of growth on a FOA plate after hLuc7Ls were introduced into the yLuc7 shuffle strain. (C) The cryo-EM structure of the yeast E complex (Li et al. 2019) indicated that residues D212, R216, and H220 were in close proximity of the U1–5′ ss RNA duplex. (D) Spot assay with 10× serial dilution showed Luc7T had similar growth as the parental WT strain, with a very mild growth retardation at 17°C that is less severe than h-yU1C. (E) FP experiments demonstrated that U1 snRNPLuc7T and U1 snRNP WT had indistinguishable binding affinity to the 5′ ss oligo. The control oligo is the 5′ end of U1 snRNA with the sequence AUACUUACCU. FP values are baseline (oligo alone) subtracted to better align and compare the two binding curves. (F) Luc7T had no significant effect on splicing. None of the IR differences observed between the Luc7T and WT was significantly larger (FRD < 0.05) than the IR differences observed among the three biological replicates for WT or Luc7T.
The yeast E complex structure we determined (Li et al. 2019) had clear density for the backbone of ZnF2 of yLuc7. However, the side chain density was often missing, making it difficult to unambiguously model the side chain conformation. Residues D212, R216, and H220 are in close proximity of the U1 snRNA and 5′ ss duplex (Fig. 3C), and these residues potentially interact with the RNA duplex. Previous genetic experiments also showed that the R216A mutant affects splicing of the Sus1 gene, which has a weak noncanonical 5′ ss (Agarwal et al. 2016). Because we could not replace yLuc7 with the entire hLuc7Ls, we generated a Luc7 triple mutant (abbreviated as Luc7T) which contained the D212A/R216A/H220C substitutions (we mutated H220 to a Cys instead of Ala to maintain the ZnF) with the objective of weakening the yLuc7 interaction with the 5′ ss.
Yeast strain carrying Luc7T showed no substantial growth defects (in rich media on plates) except for a very subtle growth retardation at 17°C, less severe than h-yU1C (Fig. 3D). We purified U1 snRNPLuc7T from yeast using the TAP tag on U1A and determined its binding affinity to a 15 nt RNA (GAUUCUG/GUAUGUUC where / denotes the exon/intron boundary) containing the 5′ ss of Act1 (Kd = 29.43 nM, 25.95–33.42 nM at 95% confidence interval). This Kd was indistinguishable from that of the U1 snRNPWT and the same oligo (Fig. 3E), suggesting that the three Luc7 residues (D212, R216, and H220) were not critical for binding to the U1—5′ ss RNA duplex.
Consistent with this observation, our RNA-seq analysis of the Luc7T strain revealed no significant splicing differences compared to the WT. Of the 193 yeast intron–containing genes with sufficient reads, none had significant IR difference between Luc7T and WT with FDR < 0.05 (i.e., none of the IR differences observed between the Luc7T and WT was significantly larger than the IR differences observed among the three biological replicates for WT or Luc7T) (Fig. 3F; Supplemental Table S2).
Luc7 depletion leads to the loss of Prp40 and Snu71 in U1 snRNP, and extensive splicing defects
Given that the Luc7T mutant did not significantly interrupt its interaction with the 5′ ss–U1 snRNA duplex, we introduced an auxin-inducible degradation (AID) system (Mendoza-Ochoa et al. 2019) in the h-yU1C strain to deplete Luc7 (an essential protein in yeast). We fused an AID-tag to the C terminus of Luc7 and coexpressed TIR1, an auxin-binding receptor, under the control of a β-estradiol inducible promoter. The addition of IAA (a common auxin) and β-estradiol leads to the expression and activation of TIR1, which in turn targets AID-tagged Luc7 for proteasomal degradation. For simplicity, we designated the resulting strain as Luc7D (for Luc7 degron-tagged), although this strain also had the ZnF domain of yU1C humanized as the h-yU1C strain. After 6 h of β-estradiol and IAA induction, we were able to completely deplete AID-tagged Luc7 (Fig. 4A). The U1 snRNPLuc7D strain showed a slow growth phenotype in the rich YPD medium (Fig. 4B).
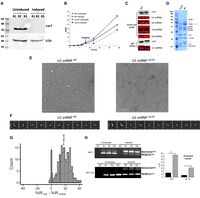
Luc7 depletion leads to the loss of Prp40 and Snu71 in U1 snRNP, and extensive splicing defects. (A) Luc7 was depleted after 6 h of β-estradiol and IAA induction, as shown in western blot of whole-cell lysate using antibodies against the HA tag on Luc7 and CBP tag on U1A. R1–3 represent three biological replicates. (B) The growth of the Luc7D strain was retarded upon induction of Luc7 depletion, relative to the uninduced culture or the WT. (C) U1A (probed by an anti-CBP antibody) and U1 snRNA (probed by solution hybridization) levels in whole-cell lysate and after IgG pull down using the TAP tag on U1A are similar between the WT and Luc7D strains. Other snRNAs serve as internal controls. (D) Coomassie-stained SDS-PAGE gel showed that purified U1 snRNPLuc7D after 6 h induction was depleted of not only Luc7, but also Snu71 and Prp40 (indicated by the purple font), compared to the WT U1 snRNP. (E) Negative stain images showed more smaller (likely broken down) particles in purified U1 snRNPLuc7D as compared to the WT. Red and blue circles highlight representative intact and small particles, respectively. (F) The top 10 class averages (sorted by number of particles in each class) from 2D classification (Punjani et al. 2017) of a small cryo-EM data set showed fewer classes and lower number of intact U1 snRNP particles in purified U1 snRNPLuc7D as compared to the WT. (G) Luc7 depletion after induction led to massive splicing defects (manifested as increased %IR). Of 223 introns with sufficient read coverage to quantify, 197 displayed significantly increased IR (FDR < 0.05, ΔIR ≥ 0.05). Arrows indicate the two genes selected for RT-PCR validation in H. (H) Examples of two genes whose splicing was significantly impaired upon Luc7 depletion as shown by RT-PCR analyses.
Luc7 depletion did not significantly affect the level of U1 snRNP, as revealed by the U1A and U1 snRNA levels in the IgG pull-down fraction (using the TAP tag on U1A) of the cell extract from the Luc7D strain (Fig. 4C). We successfully purified U1 snRNPLuc7D. Gel electrophoresis analysis showed that the complex was depleted in Luc7 as well as Prp40 and Snu71 (Fig. 4D). Negative stain image demonstrates that Luc7D can form U1 snRNP complex, although many particles are significantly smaller than the WT U1 snRNP, likely because of the instability of an incomplete U1 snRNP (Fig. 4E). 2D classification of a small cryo data set also showed fewer classes of intact particles in the U1 snRNPLuc7D sample (Fig. 4F), indicating that U1 snRNPLuc7D was less stable than the WT U1 snRNP.
RNA-seq analysis showed that pre-mRNA splicing was severely impacted in the Luc7D strain after Luc7 depletion compared to the uninduced control (Fig. 4G,H; Supplemental Table S3). Of the 223 intron-containing yeast genes with sufficient read coverage and consistent splicing changes in all triplicates, 197 demonstrated IR (FDR < 0.05, increase in IR of at least 0.05) in the induced sample compared to the uninduced control.
Luc7, Prp40, and Snu71 interact with each other through distinct domains
The loss of Prp40 and Snu71 upon Luc7 depletion prompted us to further investigate the interactions among Prp40, Snu71, and Luc7. Previous biochemical studies demonstrated that Prp40 and Snu71 can form a stable dimer, and Prp40, Snu71, and Luc7 form a trimer (Ester and Uetz 2008; Görnemann et al. 2011; Li et al. 2017). EM structures and biochemical studies also indicated that Luc7 uses its coiled-coil (CC) domain to bind Snu71 (residues 260–312 in the pre-A complex structure) (Li et al. 2019; Zhang et al. 2021). A boomerang-shaped density near U1-70K in the yeast E complex structure was tentatively modeled as FF4-5 domains of Prp40 (Li et al. 2019). A longer stretch of density was observed in similar areas in the yeast pre-A complex structure and was modeled as Prp40 FF1-6 (FF6 contacts U1-70K and FF1 contacts Luc7) based on the shape of the density and cross-linking and mass spectrometry data (Zhang et al. 2021). However, the model is not definitive due to the limited resolution of the density. For example, FF1 and FF2 are modeled right next to each other (close to the CC domain of Luc7) with no room for the long helix connecting the two domains (Zhang et al. 2021). Cross-linking and mass spectrometry of the pre-A complex also indicated that the C-terminal region of Snu71 interacts with FF2 and 3 of Prp40 (Zhang et al. 2021).
We further explored the interaction between Prp40 and Snu71 with an orthogonal approach using pull-down assays and purified GST-fused Prp40 domains and Snu71. We demonstrated that the Prp40 FF4 domain interacts directly and strongly with Snu71 (Fig. 5A). Purified FF2 and FF3 domains were severely degraded and the lack of interaction observed in pull down is inconclusive. Using similar pull-down assays, we showed that the Snu71 region containing residues 382–537 directly interacts with the Prp40 FF4 domain (Fig. 5B). We further demonstrated that the WW domain (residues 1–75) of Prp40 directly interacts with BBP in pull-down assays (Fig. 5C; pull-down results summarized in Fig. 5D). This observation is consistent with previous chemical shift perturbation experiments indicating that the PSPPPVYDA peptide (residues 94–102) in the N-terminal domain of BBP binds directly to the WW domains of Prp40 (Abovich and Rosbash 1997; Wiesner et al. 2002).
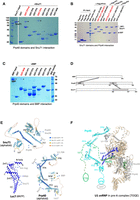
Probing the Prp40, Snu71, and BBP interaction using GST pull-down assays. (A) GST pull-down assays probing the interaction between Prp40 domains and Snu71. Various GST-fused Prp40 domains were expressed and purified from Escherichia coli (WW including both WW1 and 2, FF1, FF2, FF3, FF4, FF5, FF6, FF4-6) and were used to pull-down Snu71. Pull-down samples after extensive wash were visualized by SDS-PAGE and Coomassie staining. “*” denotes bands corresponding to various GST fusion Prp40 domains (GST-FF2 and FF3 have significant degradation). “+” denotes bands corresponding to Snu71. Red labels indicate Prp40 domains that pulled down Snu71. (B) GST pull-down assays probing the interaction between Snu71 domains and Prp40. Various GST-fused Snu71 domains (residues 97–220, 382–537, 538–620, whereas domains covering other regions of Snu71 cannot be expressed in E. coli) were used to pull down FLAG-tagged Prp40 FF4, FF5, FF6, and FF4-6 domains. All proteins were expressed and purified from E. coli. “*” denotes bands corresponding to GST or GST-fused Snu71 domains. “+” denotes bands corresponding to Prp40 FF4-6 or FF4. Red labels indicate the GST-Snu71 domain (382–537) that stably interacts with Prp40 FF4 and FF4-6. (C) GST pull-down assay probing the interaction between Prp40 domains and BBP. Various GST-fused Prp40 domains as in A were used to pull down BBP. Pull-down samples were visualized on SDS-PAGE with Coomassie stain. “*” denotes bands corresponding to various GST-fusion Prp40 domains (GST-FF2 and FF3 have significant degradation). “+” denotes bands corresponding to BBP. Red labels indicate Prp40 domains that pulled down BBP. (D) A summary of interactions among BBP, Prp40, Snu71, and Luc7 (indicated by a solid line connecting regions of two proteins) as observed in A–C. (E) Key interaction regions mapped on the structures of Snu71, Prp40 (both were predicted by AlphaFold2; Jumper et al. 2021), and Luc7 (determined by cryo-EM; Li et al. 2019). (F) A model of how Luc7, Snu71, and Prp40 interact with each other in the framework of U1 snRNP in the pre-A complex (Zhang et al. 2021).
Luc7 depletion leads to mitochondrial dysfunction and galactose toxicity
In a serendipitous observation, we found that the growth medium of the Luc7D strain after Luc7 depletion consistently changed its color to brick red immediately after adding bleach for a safe disposal of the culture (Fig. 6A, top panel). Even the cell pellet turned to rusty red after prolonged induction (e.g., an overnight incubation with β-estradiol and auxin) (Fig. 6A, bottom panel). This indicated an intracellular as well as extracellular release of ferrous iron and its oxidation to red-colored ferric iron by the bleach. We confirmed the release of iron using the iron-specific ferrozine assay (Fig. 6B). Because mitochondria are central for iron homeostasis in all eukaryotes, and many genes that code for mitochondrial proteins have introns, we examined splicing efficiency of these genes in the induced set. Nearly all intron-containing genes encoding proteins critical for mitochondrial biogenesis and function showed increased IR (Fig. 6C,D). Transcript abundance of these and that of the nonintronic genes with mitochondria-related function, as well as almost all genes that regulate iron homeostasis, were also affected to varying levels (Supplemental Table S4). We further evaluated the growth phenotype of the Luc7D strain in the presence of antimycin and oligomycin A, two mitochondrial inhibitors which inhibit electron transport and ATP synthase, respectively. We found that these inhibitors affected the uninduced Luc7D strain much more drastically than the induced (Supplemental Fig. S5), suggesting that Luc7 depletion already induced severe mitochondria dysfunction even without the inhibitors.
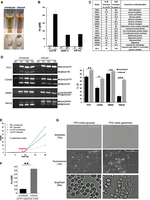
Luc7 depletion leads to mitochondrial dysfunction and galactose toxicity. (A) Luc7 depletion led to Fe2+ release. Both culture supernatants and cell pellets after induction turned rusty red, immediately after the addition of a few drops of 10% commercial bleach. (B) Free iron measurement using ferrozine assay in the Luc7D, prp2-1, and u4-cs1 strains. UI, I, PT, and NPT represent uninduced, induced, permissive temperature, and nonpermissive temperature, respectively. (C) A list of pre-mRNAs with increased IR in RNA-seq analyses of the Luc7D and prp2-1 strain that affected mitochondrial integrity and function. (D) RT-PCR confirmation of RNA-seq results in the Luc7D strain for three genes selected from the list in C and COX5B (which was not in the list of genes affected in the RNA-seq data because of its extremely short exon [1 nt] which cannot be handled by typical sequencing analyses software). (E) Growth curves of auxin and β-estradiol (induced) or ethanol added (uninduced) Luc7D and WT yeast strains in galactose medium (YPG). (F) Significantly more Fe2+ was released into the galactose medium of induced cultures (compared to B) as measured by ferrozine assay. (G) Dihydrorhodamine detection of mitochondrial reactive oxygen species (which manifests as bright fluorescent spots within mitochondria in the fluorescent image) (Laun et al. 2001; Gomez et al. 2014) in the Luc7D strain grown in YPD or YPG upon Luc7 depletion. The intensities of these bright spots were so strong that they were even visible in the bright field image at higher magnification (60×).
Although galactose is equally fermentable as glucose by budding yeast, early steps of galactose utilization require mitochondrial respiration. Galactose elevates heme biosynthesis in yeast, which in turn induces genes required for the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) (Zhang et al. 2017; De-Souza et al. 2020). Because certain mutations leading to mitochondrial dysfunction in mammalian cells are known to cause galactose toxicity (Iannetti et al. 2018), we compared the ability of the Luc7D strain to use galactose under Luc7-sufficient versus Luc7-depleted conditions. As shown in Figure 6E, the strain stopped growing in galactose (a much more dramatic growth inhibition compared to the Luc7D strain in glucose media as shown in Fig. 4B) soon after 6 h induction needed for Luc7 depletion and consequent impairment of mitochondrial function. The strain also released more Fe2+ when induced in the galactose medium than in the glucose medium (Fig. 6F, compare with Fig. 6B), suggesting that growth in galactose exacerbated the damage to mitochondria upon Luc7 depletion. This inference was further tested by staining Luc7D cells induced in glucose (YPD) or galactose (YPG) to detect mitochondria-localized reactive oxygen species (mtROS). As shown in Figure 6G, a severe induction of mtROS was observed in YPG-grown cells when induced for Luc7 depletion.
To understand whether the mitochondria defect is Luc7-specific or a result of a severe splicing defect in general, we performed a similar iron release assay in two other strains carrying splicing factor mutants: prp2-1 (high temperature–sensitive) (Vijayraghavan et al. 1989) and u4-cs1 (cold-sensitive) (Li and Brow 1996), both of which are known to cause widespread splicing defects at nonpermissive temperatures (Li and Brow 1996; Dwyer and Pleiss 2023). We found that both mutant strains had significant iron release at nonpermissive temperatures (Fig. 6B). Evaluation of the genome-wide splicing data published for prp2-1 (Dwyer and Pleiss 2023) showed that essentially all mitochondria-related intron-containing genes affected in the Luc7D strain were also negatively affected at nonpermissive temperature in prp2-1 (Fig. 6C). These data suggest that other splicing mutations that cause severe splicing defects in mitochondrial intron-containing genes also lead to compromised mitochondrial function and iron homeostasis.
DISCUSSION
U1C plays an important role in stabilizing the U1 snRNA and 5′ ss interaction. In structures of both the human and yeast U1 snRNP in complex with the 5′ ss RNA, the ZnF domain located in the N-terminal region of U1C contacts the backbone of the U1 snRNA and 5′ ss RNA duplex (Kondo et al. 2015). The ZnF domain of U1C is highly conserved between the two species, whereas their C-terminal domains are highly divergent (Fig. 1A). The C-terminal domain of yU1C is much longer and forms extensive interactions with Prp42, the latter facilitating the interaction between the other auxiliary proteins and the yU1 snRNP core (Li et al. 2017). Deletion of the CTD of U1C is lethal to yeast (Li et al. 2017). It may not be surprising that the full-length human U1C cannot substitute yU1C (Fig. 1B), potentially because the much shorter and divergent human U1C CTD fails to interact with Prp42.
On the other hand, the ZnF domain of human U1C can functionally substitute that of yU1C, with no effect on yeast growth at 30°C or 37°C, and a slight growth retardation at lower temperatures (17°C). The cryo-EM structure of the U1 snRNPh-yU1C shows that the structure is essentially identical to the WT yU1 snRNP with the exception of different side chains in the U1C ZnF domain. Residues contacting the U1–5′ ss RNA duplex have minimal changes (T17 to S and R28 to K) from yeast to human that are insufficient to account for the growth and splicing defects observed in the h-yU1C strain (Supplemental Fig. S4). However, residues distant from the RNA-binding interface can potentially participate in long-range allosteric signal transmission and affect the RNA-binding affinity, similar to what was observed in the case of U1A (Han et al. 2019). For example, a hydrophobic core composed of F4, L13, and the CH2 groups of R21 exists adjacent to the metal binding pocket in U1C and other canonical ZnF domains and stabilizes the ZnF domain (Muto et al. 2004). Mutations at L13 reduce the interaction between U1 snRNA and the 5′ ss, bypassing the need for RNA helicase Prp28 that disrupts the U1 snRNA and 5′ ss interaction (Chen et al. 2001). In yU1C, F4 is replaced with a Tyr residue, which can potentially affect the interaction between yU1 snRNA and 5′ ss. By engineering a yeast strain carrying both the h-yU1C and prp28-1 (a mutant defective in U1 and 5′ ss unwinding; Staley and Guthrie 1999), we showed that h-yU1C rescues the cs phenotype of prp28-1, indicating that h-yU1C likely had resulted in weaker binding affinity between U1 snRNP and the 5′ ss. It is worth noting that the binding affinity between U1 snRNPh-yU1C and a representative 5′ ss oligo measured by FP experiments is similar to that between U1 snRNPWT and the same oligo (Fig. 3E), potentially because it is difficult to differentiate small differences in these experiments. It is also possible that although the binding affinity between the free U1 snRNP and 5′ ss is similar between U1 snRNPh-yU1C and U1 snRNPWT, this affinity is weaker for U1 snRNPh-yU1C in the context of the pre-B complex (the complex that Prp28 acts on to disrupt the U1 snRNA and 5′ ss interaction), leading to the rescue of the prp28-1 mutant.
Replacing the ZnF domain of yU1C with its human counterpart leads to moderate splicing defects. We have not observed a significant correlation between increased IR and the distance to the yeast consensus 5′ ss or how well it complements the 5′ end of U1 snRNA (Fig. 2C,D), indicating that h-yU1C is not preferentially affecting weaker 5′ ss. Genes with shorter intron or upstream exon seem to be more prone to IR in the h-yU1C strain (Fig. 2E,F), although the mechanism is yet to be understood. It is worth noting that even in WT yeast, genes with long introns (such as the ribosome protein-encoding genes) have been observed to splice more efficiently than those with shorter introns, although the mechanism is also unknown (Pleiss et al. 2007; Xia 2020).
In yU1 snRNP, a second protein Luc7 also uses its ZnF2 domain to help stabilize the interaction between U1 snRNA and the 5′ ss (Plaschka et al. 2018; Li et al. 2019). The human homologs (Luc7L, L2, L3) of Luc7 are alternative splicing factors that only transiently associate with U1 snRNP, and these proteins contain extra RS-rich C-terminal domains that do not exist in yLuc7 (Fig. 3A). The yLuc7 could not be replaced with the full-length hLuc7Ls (Fig. 3B). It is possible that the hLuc7Ls are different enough from yLuc7 such that they can no longer be incorporated into the yU1 snRNP, or the ZnF2 of hLuc7Ls is not sufficient for stabilizing the U1–5′ ss RNA duplex. Alternatively, the C-terminal RS domain of hLuc7Ls may have a dominant negative effect in yeast.
Previous cryo-EM structures of the yeast E complex indicated that residues D212, R216, and H220 of Luc7 are in close proximity to the U1 snRNA and 5′ ss RNA duplex; however, the density for Luc7 is poor and the side chains could not be modeled with high confidence. We generated a Luc7T triple mutant (D212A/R216A/H220C) and showed that U1 snRNPLuc7T has the same binding affinity to the 5′ ss and no significant splicing changes compared to the WT. These observations suggest that these three residues are unlikely to play a major role in the stabilization of the U1 snRNA and 5′ ss interaction.
Using an AID system, we were able to deplete Luc7 in the Luc7D strain (which was combined with the humanized ZnF domain of yU1C). Luc7 is an essential gene, and the fact that the Luc7D strain continued to grow after auxin induction (Fig. 4B) is potentially due to a residual amount of U1 snRNP containing Luc7 remaining in cells, even though the majority of U1 snRNP was depleted of Luc7 (Fig. 4D). U1 snRNP without Luc7 could be assembled, although it also completely lost Snu71 and Prp40 and was less stable than the WT U1 snRNP. This is consistent with previous observations that a Luc7 mutant leads to the reduced association of Snu71 and Prp40 with U1 snRNP (Fortes et al. 1999). By integrating previous cryo-EM structures, cross-linking and mass spectrometry analyses, and the pull-down analyses presented here, we present a model of how these three proteins interact with each other and the rest of the U1 snRNP (Fig. 5E,F). In essence, Luc7 binds to the Sm ring through its N-terminal helix and ZnF1, to the U1–5′ ss RNA duplex through its ZnF2, and to Snu71 through its CC domain. Snu71 binds Prp42 and Snu56 in U1 snRNP through its N-terminal domain (residues 1–50), and to Luc7 through the long helix formed between residues 260 and 312 (Zhang et al. 2021). Prp40 binds to the C-terminal domain of U1-70K through its FF6 domain (Li et al. 2019), to a region within residues 382–537 of Snu71 through its FF4 domain (this paper) and potentially FF2 and 3 domains (Zhang et al. 2021), and to BBP through its N-terminal WW domain. Although both Snu71 (through its N-terminal domain) and Prp40 (through FF6) contact U1 snRNP, the interaction between Luc7 and U1 snRNP seems to be critical for maintaining the trimer within the U1 snRNP, as evident from the loss of Prp40 and Snu71 when Luc7 is depleted (Fig. 4D). Given that Prp40 is critical for bridging the 5′ ss and BPS (Li et al. 2019; Zhang et al. 2021), it is not surprising that Luc7 depletion (and the subsequent loss of Prp40 and Snu71 from U1 snRNP) led to extensive splicing defects in essentially all intron-containing yeast genes with sufficient reads in RNA-seq analysis.
The vast majority of yeast intron-containing genes encode ribosomal proteins, so studies of the functional consequence of widespread splicing defects have usually focused on ribosomal proteins. We showed that another important consequence of substantial splicing defects in yeast is mitochondria dysfunction. Splicing of essentially all intron-containing genes related to mitochondrial function is negatively impacted (Fig. 6C). The resulting mitochondrial dysfunction likely led to impaired iron homeostasis and galactose toxicity. Interestingly, human Luc7L2 KD led to significant downregulation of glycolytic genes (Daniels et al. 2021), and Luc7L2 was found to be a major player in maintaining the balance between OXPHOS in mitochondrial and glycolysis in cytosol, the two major pathways for ATP production (Jourdain et al. 2021). Luc7L2 suppresses OXPHOS through alternative splicing regulation of specific genes (the glycolytic enzyme PFKM and cystine/glutamate antiporter SLC7A11) as well as secondary repression of mitochondrial respiratory complex protein expression. Although the mechanistic details of energy metabolism regulation by yLuc7 (through global suppression of splicing, which in turn affects essentially all intron-containing genes encoding mitochondrial proteins) and human Luc7L2 (through specific alternative splicing regulation of PFKM, whose yeast homology PFK2 is intronless, and SLC7A11, which has no yeast homolog) are different, it highlights the important and conserved role of splicing in the regulation of eukaryotic energy production, which was further confirmed by the mitochondrial dysfunction caused by two other splicing mutations—prp2-1 and u4-cs1 (Fig. 6B,C).
MATERIALS AND METHODS
Yeast strains and growth
All yeast strains used were grown on YPD or appropriate dropout media; 50 µg/mL of ampicillin was routinely incorporated into large-scale liquid cultures to avoid bacterial contamination.
Plasmid shuffling was done by selecting initial transformants (from dropout plates) on 5-FOA. Colonies resistant to 5-FOA were retested on appropriate dropout plates to confirm successful shuffling. FLAG-tagged full-length yeast U1C, yeast Luc7, and human U1C and Luc7Ls (all with native human codons) were cloned into the pRS415 plasmid under a GPD promoter and used for plasmid shuffling experiments in the U1C or Luc7 shuffle strain (Schwer and Shuman 2014; Agarwal et al. 2016).
Humanizing yeast U1C
The first 36 residues of yU1C protein encompassing the ZnF domain were replaced with the corresponding region of the human U1C protein using the CRISPR–Cas9 system in a yLuc7 shuffle strain (Agarwal et al. 2016) containing a C-terminal TAP tag on the endogenous U1A, essentially as described in Laughery and Wyrick (2019). The following three guide RNAs covering the first 108 bp of the coding sequence were cloned into pML107 (Laughery and Wyrick 2019) and used to swap the first 36 amino acid segment with that of the human U1C: 5′-ATGACACGTTGAGCGTT-3′, 5′-GTTCGTAAATCGCACTTGG-3′, 5′-CGTATAACAGCTGACTATT-3′.
The yeast-codon optimized human sequence (lowercase indicates changed bases) with flanking sequences from the yLuc7 gene (italics), as shown below, was cloned into pUC19 by Gibson cloning (NEBuilder, New England Biolabs) to replace the yeast sequence: 5′-tagtgtaggcgatgaaggtgctcaagtaacggagaggaaagagataggcaATGCCaAAaTTTTATTGTGAtTAtTGtGATACtTAttTgACtCATGAtTCTCCATCTGTtAGAAAaACtCAtTGttcTGGAAGaAAACAtAAAGAaAATGTtAAAGAttattatagAaacaaagcaagagacattattaataaacataat-3′.
The same strategy was used to engineer h-yU1C into the prp28-1 strain (Staley and Guthrie 1999).
Generating an inducible Luc7 depletion strain
We introduced an inducible degradation system for yLuc7 in the h-yU1C strain (described above). We engineered an AID + 3×-HA tag at the C terminus of yLuc7 and an auxin receptor (OsTIR1) driven by a β-estradiol inducible promoter (Mendoza-Ochoa et al. 2019) cloned into a single plasmid (pRS415) and transformed it into the h-yU1C strain.
Phenotypic analyses of yeast
OD600 was used to monitor the growth in YPD (glucose as the sugar) or YPG (galactose as the sugar). Spot assay was done on YPD plates with a starting OD600 of 0.5 and 10× serial dilution. Fe2+ was measured using ferrozine, an iron-specific reagent (Riemer et al. 2004). Yeast cells were stained for viability using Evans blue (Gomez et al. 2014). All microscopy work was done using an EVOS FL all-in-one Olympus microscope (Advanced Microscopy Group) as described (Denney et al. 2021).
Total protein isolation and immunoblot analyses
Total cellular protein from mid-log phase grown yeast was extracted using the TCA precipitation protocol (Yaffe and Schatz 1984) as modified by Denney et al. (2021). The protocol preserves the intactness of proteins by suppressing proteolysis and any posttranslational modifications. An equal amount of protein quantified as A280 was resolved by PAGE (10% acrylamide-SDS), blotted onto a PVDF membrane, and probed with α-HA tag (for the detection of AID and HA-tagged Luc7) and α-CBP tag (for the detection of U1A) antibodies. The immune-reactive proteins were detected using HRP-labeled secondary antibodies and chemiluminescence.
Purification of U1 snRNPWT, h-yU1C, Luc7T, Luc7D
Purification of U1 snRNPWT, h-yU1C, Luc7T were carried out essentially as described (Li et al. 2017), taking advantage of the TAP tag on endogenous U1A in these strains. U1 snRNPLuc7D was purified from yeast cells by growing them in 2× YPD to OD600 of 8–10 and inducing them for 6 h by adding 10 µM of estradiol and 750 µM of IAA. The TAP tag on U1A in the WT strain came from the yeast TAP tag strain collection, whereas the TAP tag on U1A in the other three strains was generated by ourselves. The two TAP tags have slightly different lengths, resulting in the U1A protein in WT migrating slightly slower than that of U1A in the Luc7D strain on SDS-PAGE as in Figure 4D.
To analyze U1 snRNP level in cells, 5 mL of U1 snRNPWT, h-yU1C, Luc7T was grown at 30°C or 17°C and purified by one-step IgG purification. Whole-cell and IgG elution were analyzed by western blot with an anti-CBP antibody (GenScript A00635) and solution hybridization with an IRDye-700 labeled oligo complementary to U1 snRNA (CCGTATGTGTGTGTGACC).
Purified U1 snRNPh-yU1C was assembled with ACT1 pre-mRNA and BBP–Mud2 essentially as described (Li et al. 2019). In brief, ACT1 pre-mRNA bound to MBP–MS2 fusion protein was mixed with h-yU1C and IgG Sepharase-6 purified BBP–Mud2 dimer, and calmodulin resin (Agilent). After incubation for 3 h at 4°C, the resin was washed eight times, and then eluted six times with 100 μL eluting buffer. The elutions were pooled for cryo-EM sample preparation.
Fluorescence polarization assay
A serial dilution (1:2) of each U1snRNP was prepared in buffer containing 20 mM HEPES (pH 7.9), 150 mM KCl, 2 mM EDTA, 1 mM MgCl2, 0.01% Triton X-100, and 0.1% PEG3350. Cy5-labeled RNA oligo (GAUUCUGGUAUGUUC, from Integrated DNA Technologies) containing the 5′ ss (underlined) was added into each sample at a final concentration of 3 nM. The mixture was transferred into a 384 well, black, flat-bottom microplates (Greiner Bio-One) and incubated at room temperature for another 15 min (the total time the reaction mixture stayed at room temperature is ∼45 min). The plate was read using an Envision plate reader (PerkinElmer) at excitation/emission = 620/688 nm at 25°C. The readings did not change with an additional 10 min incubation at room temperature, indicating that the binding had reached equilibrium. The experimental data were analyzed using Prism 10 software (GraphPad Software), by fitting the data to a single site binding isotherm equation: Y = start + (end − start) × X/(Kd + X), where Y is the FP value, X is the ligand concentration, and start and end represent the starting and saturating FP values, respectively.
cryo-EM sample preparation and imaging
Three microliters of the freshly purified complex were applied to a plasma-cleaned C-flat holy carbon grid (1.2/1.3, 400 mesh, Electron Microscopy Sciences) with 20 sec glow discharge using air and flash-frozen in liquid ethane with a Vitrobot Mark IV (Thermo Fisher Scientific). The grids were obtained with the chamber at 100% humidity, 2.5 sec blotting time, –6 blotting force, and 15 sec wait time and flash-frozen into liquid ethane with a Vitrobot Mark IV (Thermo Fisher Scientific). The data were collected on a Titan Krios at the Pacific Northwest Cryo-EM Center (PNCC) operated at 300 keV and equipped with a K3 direct detector (Gatan). Movies were acquired at a pixel size of 0.83 A/pix (0.415 A/pix super-res), a defocus range of −0.8 to −2.5 μm, and 50 frames with a total dose of ∼50 e−/Å2.
Image processing, model building, and refinement
Patch motion correction and patch CTF estimation were done in cryoSPARC. A total of ∼1.5 million particles were automatically picked using “Blob Picker” and extracted with a box size of 300 × 300 pixels. After three rounds of reference-free 2D classification, ∼71,607 particles were selected for ab initio reconstruction, and heterogeneous refinement. Then 138,997 good particles are unbinned with a box size of 640 × 640 pixels. After homogenesis refinement and nonuniform refinement, the map reached a resolution of ∼3.49 Å.
The model was built in Coot. To aid subunit assignment and model building, we took advantage of the yeast E complex structure we previously determined (PDB code: 6N7R), which was fitted into the h-yU1C density map. The first 36 amino acids of U1C and the 5′ ss region were adjusted manually to match the density. The RNA components were subsequently adjusted using RCrane (Keating and Pyle 2012). The rest of the subunits fit well and were slightly adjusted for the side chains. The model was refined using PHENIX in real space with secondary structure and geometry restraints (Liebschner et al. 2019). The final model and EM density map were deposited into PDB (8W2O) and EMDB (EMD-43753).
RNA-seq analyses
Yeast cells (WT, h-yU1C, Luc7T, or Luc7D uninduced or induced) were grown to an OD600 of 0.35–0.5. Total RNA was isolated from 10 to 15 × 107 cells. Hot phenol (Green and Sambrook 2021)-extracted RNA was DNase I-treated and purified further using Monarch RNA Cleanup spin-columns (NEB). The library was prepared by poly(A) capture without rRNA depletion, and 150 bp paired-end sequencing was performed by Novogene. For each sample, ∼40 million read pairs were obtained. These reads were trimmed using cutadapt v4.2. AGATCGGAAGAGCACACGTCTGA was trimmed from the 3′ end of read 1, and GATCGGAAGAGCGTCGTGTAGGG was trimmed from the 3′ end of read 2.
Reads were mapped to the yeast reference genome (version R64.3.1), using the STAR program (Dobin et al. 2013). Differential expression analysis was carried out using DE-seq2 (Love et al. 2014). Splicing efficiency values were obtained using JunctionCounts (github ajw2329/junctionCounts) software. This approach uses reads that span splice junctions and reads that cover intronic regions to calculate the fraction of transcripts that contain introns. Differentially included introns were identified by comparing IR values across conditions using a Benjamini–Hochberg corrected t-test. Affected introns were defined as those with an FDR value of <0.05 and an absolute difference in mean IR value across conditions of at least 0.05. Distances of affected 5′ splice sites to the consensus yeast 5′ ss (GTATGT) were calculated using Hamming distances. Values of intron and exon lengths were calculated using genome build R64-1-1 (SacCer3, GCA_000146045.2). The RNA-seq data were deposited in GEO with accession number GSE252348.
RT-PCR analysis
Total RNA, purified as described above, was used in sequential reverse transcription and PCR amplification steps using the Protoscript II first strand cDNA synthesis kit and Q5 Hi-fidelity DNA polymerase (NEB) according to the manufacturer's protocols; 1–2 µg of total RNA was used for reverse transcription and 1–2 µL of the reaction product was amplified by PCR. Primer sets that can amplify both the unspliced and spliced forms of selected mRNA were used in each case. An appropriate number of amplification cycles was chosen for each amplicon, based on the copy number/cell data (available from SGD) and as needed for optimal quantitation of the products from agarose gel images. Signals were quantified using the ImageJ program (https://imagej.net/software/imagej/).
GST pull-down assays using purified protein
To investigate the interaction between Prp40 domains and Snu71 or BBP, Snu71 fused to a C-terminal CBP tag was cloned into the plasmid pRS416/GPD-Snu71-CBP. Snu71 was purified from yeast BCY123 cells harboring the above plasmid using calmodulin resin. BBP was purified from BCY123 cells harboring pRS414/GPD-protA-BBP using IgG Sepharose-6 Fast Flow resin. Various Prp40 domains fused with an N-terminal FLAG tag were cloned into pGEX-6p-1 vector (GE Healthcare) and purified from Escherichia coli as GST fusion proteins. The Prp40 domains constructed are 1–75 (WW); 134–189 (FF1); 198–259 (FF2); 260–335 (FF3); 351–415 (FF4); 425–488 (FF5); 488–552 (FF6); 351–552 (FF4–6). To detect interactions between Prp40 domains with Snu71 or BBP, GST fusion proteins or GST alone were bound on glutathione-Sepharose resin (GE Healthcare) and incubated with 5 μg of purified Snu71 or BBP protein in pull-down buffer (20 mM Tris pH 8.0, 120 mM NaCl, 1 mM Mg2Cl, 1 mM DTT, 0.05% Triton X-100) for 2 h at 4°C. Resins with bound proteins were washed five times with pull-down buffer. Resins were boiled in SDS loading buffer, and proteins were analyzed on SDS-PAGE followed by Coomassie stain.
To detect interactions between Prp40 FF domains with different regions of Snu71, GST fusion of FF4, FF5, FF6, and FF4–6 were purified using glutathione-Sepharose resin and released by PreScission protease. The proteins were further purified by gel filtration using a Superose 6 Increase 10/300 GL column (GE Healthcare). Snu71 fragments with residues 1–96, 97–220, 221–380, 381–537, 537–620 fused with an N-terminal HA tag were cloned into pGEX-6p-1 vector. Snu71 fragments 1–96 and 221–381 failed to be expressed and purified. GST fusion of the other Snu71 fragments or GST alone was coupled to glutathione-Sepharose resin and incubated with 2 µg of each individual purified FF domain in pull-down buffer (20 mM Tris pH 8.0, 120 mM NaCl, 1 mM Mg2Cl, 1 mM DTT, 0.05% Triton X-100) for 2 h at 4°C under rotation. Resins with bound proteins were washed five times with pull-down buffer. Resins were boiled in SDS loading buffer, and proteins were analyzed on SDS-PAGE followed by Coomassie stain and western blot. Western blot was performed using M2 antibody (Sigma-Aldrich).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants R35GM145289 and R01GM126157 (R.Z.); R35GM133385 (J.M.T.); R35GM147498 (J.W.); and R01GM071940 (Z.H.Z.). J.G. was supported by the NIH under the Ruth L. Kirschstein National Research Service Award T32CA174648. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. We thank Dr. Manny Ares for advice on yeast RNA-seq analyses, as well as Drs. Beate Schwer and Stewart Shuman for the U1C and Luc7 shuffle strains. A portion of this research was supported by NIH grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. We acknowledge the staff (in particular Rose Marie Haynes) at the Pacific Northwest Cryo-EM Center for help with data collection as well as the CU Anschutz School of Medicine Cryo-EM and proteomics core facilities (partially supported by the School of Medicine and the University of Colorado Cancer Center Support Grant P30CA046934) for EM and proteomics support. We thank the Drug Discovery and Development Shared Resource (D3SR, especially its Program Director Qiong Zhou) for support of the FP experiments. The D3SR is supported in part by the CU Cancer Center, an NIH NCI Designated Cancer Center (P30CA046934), and the CU AMC Center for Drug Discovery, which was established from a generous gift from the ALSAM Foundation and through CU AMC institutional support. Molecular graphics and analyses were performed with the UCSF Chimera and ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from the National Institute of General Medical Sciences (NIGMS) P41-GM103311 (Chimera, ChimeraX) and NIH R01-GM129325 (ChimeraX).
Footnotes
-
Handling editor: Timothy Nilsen
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079917.123.
- Received December 8, 2023.
- Accepted April 16, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHORS


Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Subbaiah Chalivendra and Shasha Shi are co-first authors of this paper, “Selected humanization of yeast U1 snRNP leads to global suppression of pre-mRNA splicing and mitochondrial dysfunction in the budding yeast.” Subbaiah is a research instructor in Rui Zhao's laboratory at the University of Colorado-Anschutz Medical Campus, Aurora. Shasha is a postdoctoral researcher dedicated to unraveling the fundamental molecular mechanisms underlying splicing through the interdisciplinary approaches of structural biology and molecular biology. Both work in the Department of Biochemistry and Molecular Genetics. Their laboratory is mainly focused on understanding the regulation of pre-mRNA splicing and the impacts of its dysregulation both at molecular and organismal levels.
What are the major results described in your paper and how do they impact this branch of the field?
Although introns were discovered nearly half a century ago, the molecules and mechanisms involved in their processing are not fully understood. In this work, we attempted to unravel the role of U1C and Luc7, two essential and conserved proteins of U1 snRNP (U1, hereafter), across all eukaryotes. As the mammalian splicing machinery is far too complex, we used the budding yeast as a model owing to its simpler biology and amenability for a multidisciplinary approach (particularly genetics). We showed that the zinc-finger domain (ZnF) of yeast U1C (involved in the recognition of 5′ splice site) can be swapped with its human counterpart. In the resultant strain, splicing was significantly affected in >50% of transcripts and growth was cold-sensitive, in spite of ∼70% sequence similarity between the two ZnFs. This indicated that the remaining nonconserved residues of the ZnF (many of them being more hydrophobic in the yeast U1C) play an important role in splicing. To further humanize yeast U1, we selectively depleted Luc7 to mimic the absence of its human homologs in the purified human U1. The loss of Luc7 was accompanied by a loss of two other essential proteins from yeast U1, which also affected splicing in nearly all transcripts and led to growth retardation at all temperatures tested even in rich media. Further, mitochondrial function was severely disrupted, as evidenced by an induction of reactive oxygen species in mitochondria and Fe2+ release into the medium. We further demonstrated that the substitution of three residues in the second ZnF of Luc7 (presumed to be important based on previous work) has no consequence on pre-mRNA splicing or the growth. In summary, our work revealed novel details on structure–function relationships of two essential U1 proteins and how these interactions play into organismal responses to its environment.
What led you to study RNA or this aspect of RNA science?
SC: As a student of biology, I have been interested in exploring the molecular interactions underlying a phenotype. To my understanding, gene regulatory mechanisms have played a predominant role in organismal and phenotypic diversity over the gene content itself (although regulation also involves genes). Transcription and posttranscriptional processing, the first steps of gene expression, involve RNA. It follows that we study RNA. A part of my PhD thesis (lucky to continue this work in my first job) was on chloroplast RNA-binding proteins.
SS: The journey into RNA science is fascinating. It begins with a profound curiosity about the molecular machinery that underpins life itself. The first time I saw the splicing circle alongside the structure of the splicing complexes, I was struck by the precision and beauty of their arrangement. As we all know, unlike DNA, which can form a stable double-stranded structure, RNA can fold into complex shapes because of its single-stranded nature and diverse chemical properties. This remarkable dynamic and flexible nature of RNA enables it to interact with a wide array of molecules, including proteins, other RNAs, and small ligands. You can hardly imagine how those individual proteins assemble at the right time and perfect position, and function coordinately to play vital and precise roles in gene expression regulation.
As I delved deeper into RNA science, I encountered the burgeoning field of RNA therapeutics. The prospect of manipulating RNA molecules to treat diseases at their genetic roots sparked my imagination. From antisense oligonucleotides correcting faulty splicing to messenger RNA vaccines priming the immune system against pathogens, RNA therapeutics is rapidly expanding and has vast potential.
We have all endured challenging times with COVID-19. RNA vaccines prevented illness and death, protected vulnerable populations, brought us hope, and contributed to ending the pandemic. Their widespread use represents a significant milestone in the fight against infectious diseases.
The dynamic nature of RNA presents both challenges and opportunities for understanding fundamental biological mechanisms. So many mysteries are waiting for us to uncover for the marvelous RNA science, and I am happy and proud to be a part of it.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
Based on the high conservation of the ZnF between human and yeast U1C proteins, we expected no phenotype, either molecular or growth. When we got our RNA-seq results, it was a real surprise to see an increased IR in >50% of transcripts because of this modification. That led us to check the growth phenotype and discover its cold sensitivity. Initial efforts to correlate splicing data with in vitro binding data gave some hope in explaining the results. However, subsequent work showed that the assay results do not reflect the splicing defect in the humanized U1C strain. We had to address this (also prompted by helpful reviews) by taking a genetic approach, which allowed us to explain the unexpected results.
Another twist in our research was the serendipitous observation of cultures turning to deep red with the addition of bleach. However, it did not take too long to explain this intriguing color change observed only in the Luc7-depleted strain. We suspected the release of ferrous iron into the medium and promptly confirmed the conjecture using an assay that is specific to iron. This also led us to probe and find mitochondrial dysfunction in more detail.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
SC: A very dedicated and inspiring science teacher in my early college years made us realize how curiosity and concerted investigation can demystify our world. Although it was a very long time ago and my teacher passed away a couple of years ago, the spark still lives on.
If you were able to give one piece of advice to your younger self, what would that be?
SC: Two pieces of advice, if I may. Think fearlessly. Flirt with as many hypotheses as you fancy, but do not be wedded to any of them until you have solid experimental evidence.
SS: When I was a doctoral student, my most common habit was to digest and understand it alone when faced with difficulties or new technologies. Sometimes a problem would bother me for many days. It was not until I could not figure it out on my own that I started asking for outside help.
As I progressed to the postdoctoral stage, I became involved in an increasing number of collaborative projects. I began to realize that actively seeking help and advice from the scientific community is sometimes much more efficient than trying to understand things on my own. A few simple sentences from others may help get to the bottom of the problem or provide direction toward solving it. Of course, I am not denying the importance of self-study; finding a balance is the most important thing. Now, I am a beneficiary of sharing and communicating in science. Keeping an open mind, being willing to share, seeking assistance, and getting advice are effective strategies for making continuous progress in science. Communication makes science more dynamic and interesting.
Looking back, I would encourage my younger self to communicate more and share more.
What are your subsequent near- or long-term career plans?
SS: I am deeply passionate about RNA therapeutics. Following my postdoctoral training, I have accumulated both theoretical knowledge and practical experience in RNA science. I am actively engaged in projects focused on understanding RNA function, especially splicing, using a variety of techniques ranging from cell biology to molecular biology to structural biology.
In the short term, except to continue completing ongoing projects, I will expand my knowledge in the field of RNA therapy and acquire new technologies that can further enhance my understanding and application of RNA-based treatments. I will try to seek out new projects related to RNA therapy.
I am confident that these experiences, together with my enthusiasm, will greatly benefit my future career in the field of RNA therapeutics.
How did you decide to work together as co-first authors?
SC: Shasha and I complement each other in our skills and research outlook. I am a molecular biologist and biochemist with an overarching desire to explain a phenotype. Shasha is a structural biologist with a strong expertise in electron microscopy.
SS: The first is the necessity of the project itself. We were driven by a scientific idea that requires data collection and analysis from different angles. Subbaiah possesses extensive experience in functional analysis, and my expertise lies in structural biology. Our cooperation is complementary based on our respective backgrounds. This enabled us to solve the scientific issues from multiple perspectives. We both like sharing and communicating, and we regularly update each other on developments to push the project forward, which is very important for collaboration. We deeply appreciate each other's contributions. The progression of our project requires extensive collaboration, analysis, and learning new technologies. Throughout this process, we encountered many challenging issues. We helped and supported each other. Working with Subbaiah has been a rewarding experience to me.








