
Functional analysis of the zinc finger modules of the Saccharomyces cerevisiae splicing factor Luc7
- Tucker J. Carrocci1,
- Samuel DeMario2,
- Kevin He2,
- Natalie J. Zeps1,
- Cade T. Harkner4,
- Guillaume F. Chanfreau2,3 and
- Aaron A. Hoskins1,4
- 1Department of Biochemistry, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA
- 2Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, California 90095, USA
- 3Molecular Biology Institute, University of California, Los Angeles, Los Angeles, California 90095, USA
- 4Department of Chemistry, University of Wisconsin–Madison, Madison, Wisconsin 53706, USA
- Corresponding author: ahoskins{at}wisc.edu
Abstract
Identification of splice sites is a critical step in pre-messenger RNA (pre-mRNA) splicing because the definition of the exon/intron boundaries controls what nucleotides are incorporated into mature mRNAs. The intron boundary with the upstream exon is initially identified through interactions with the U1 small nuclear ribonucleoprotein (snRNP). This involves both base-pairing between the U1 snRNA and the pre-mRNA as well as snRNP proteins interacting with the 5′ splice site (5′ss)/snRNA duplex. In yeast, this duplex is buttressed by two conserved protein factors, Yhc1 and Luc7. Luc7 has three human paralogs (LUC7L, LUC7L2, and LUC7L3), which play roles in alternative splicing. What domains of these paralogs promote splicing at particular sites is not yet clear. Here, we humanized the zinc finger (ZnF) domains of the yeast Luc7 protein in order to understand their roles in splice site selection using reporter assays, transcriptome analysis, and genetic interactions. Although we were unable to determine a function for the first ZnF domain, humanization of the second ZnF domain to mirror that found in LUC7L or LUC7L2 resulted in altered usage of nonconsensus 5′ss. In contrast, the corresponding ZnF domain of LUC7L3 could not support yeast viability. Further, humanization of Luc7 can suppress mutation of the ATPase Prp28, which is involved in U1 release and exchange for U6 at the 5′ss. Our work reveals a role for the second ZnF of Luc7 in splice site selection and suggests that different ZnF domains may have different ATPase requirements for release by Prp28.
Keywords
INTRODUCTION
Eukaryotic pre-messenger RNA (pre-mRNA) is modified to remove internal sequences called introns by pre-mRNA splicing. Splicing is a highly conserved process across eukaryotes, and it is carried out by the large macromolecular machine known as the spliceosome. Spliceosomes are composed of five U-rich small nuclear ribonucleoprotein (snRNP) complexes (U1, U2, U4, U5, and U6) and dozens of auxiliary proteins (Wilkinson et al. 2020). These factors assemble de novo on every intron through the recognition of splice sites in order to form active spliceosomes and catalyze intron removal. Defects in this process often lead to aberrantly spliced RNA products and can be causative for human disease (Love et al. 2023).
Splicing often begins with base-pairing of the 5′ end of the U1 snRNA to the intron 5′ splice site (5′ss) at the exon–intron boundary to form the spliceosome E complex (Ruby and Abelson 1988; Shcherbakova et al. 2013; Fica 2020). U1 remains associated with the 5′ss during spliceosome assembly but must be released by the ATPase Prp28 during the pre-B to B complex transition to allow for U6 snRNA pairing to the 5′ss (Staley and Guthrie 1999). The sequence of the 5′ss is highly conserved in Saccharomyces cerevisiae (hereafter yeast), whereas human 5′ss are more divergent (Spingola et al. 1999; Roca et al. 2013). In addition, human splicing often requires auxiliary splicing factors to direct U1 recruitment and stabilize U1 on the pre-mRNA (Matlin et al. 2005; Espinosa et al. 2023). In addition to RNA base-pairing, the U1 snRNA:5′ss duplex is also stabilized by protein components of the U1 snRNP. In yeast, the U1 snRNP proteins Yhc1 and Luc7 flank the duplex (Fig. 1A). These proteins are conserved in humans; however, the human homolog of Yhc1 (U1-C) is considered to be a core component of the U1 snRNP, whereas the human homologs of Luc7 are auxiliary splicing factors. Cryo-electron microscopy (cryo-EM) has revealed the architecture of both isolated yeast U1 snRNP and of the spliceosome A complex (which contains the U1 and U2 snRNPs) and indicated that Yhc1 and Luc7 interact with U1 snRNA:5′ss duplex through their zinc finger (ZnF) domains (Li et al. 2017, 2019; Plaschka et al. 2018). Alterations in the Yhc1 and Luc7 ZnF domains have been shown to impair splicing in vivo and can be lethal (Schwer and Shuman 2014; Agarwal et al. 2016).
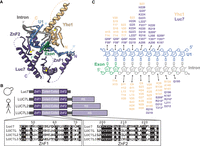
The U1:5′ss duplex contacts Yhc1/U1-C and Luc7/LUC7L. (A) U1:5′ss base-pairing is stabilized by the Yhc1 and Luc7 ZnF domains in the structure of the yeast prespliceosome (PDB 6G90). Luc7 ZnF2 contacts the 5′ss duplex, whereas ZnF1 lies in the direction of exon 1, which are both partially unresolved. (B) Schematic showing LUC7 and the three human paralogs. A sequence alignment shows the similarity of the ZnF domains. (C) Schematic showing Yhc1 (beige) and Luc7 (purple) residues located within 6 Å of the U1:5′ss duplex (blue/gray). Asterisks indicate residues that are not conserved between yeast and human Luc7 proteins.
Luc7 is highly conserved among eukaryotes, and vertebrates have three Luc7 paralogs: LUC7L, LUC7L2, and LUC7L3 (Fig. 1B; Fortes et al. 1999). Each paralog is proposed to control a distinct subset of alternative splicing events (Daniels et al. 2021; Jourdain et al. 2021). Loss of LUC7L2 has been implicated in myelodysplastic syndromes and related neoplasms and can lead to changes in glycolysis and metabolic reprogramming (Daniels et al. 2021; Jourdain et al. 2021). Specificities of the LUC7L paralogs for different 5′ss have been proposed to be due to differential recruitment to target RNAs and/or different sequence preferences (Daniels et al. 2021; Kenny et al. 2022). Understanding the molecular basis for specificity and function of the LUC7L proteins is important to establish how their loss can lead to disease or changes in metabolism.
Here, we examine differences among human LUC7L paralogs using a yeast model system. We generated yeast and human chimeric Luc7 proteins, focusing on the ZnF domains (ZnF1 and ZnF2), and assayed them for differences in splicing and splice site usage. We were unable to detect changes in the splicing of a reporter pre-mRNA due to changes in Luc7 ZnF1. In contrast, we find that the humanization of yeast Luc7 ZnF2 to mirror that in human LUC7L and LUC7L2 improves the growth of yeast expressing splicing reporters containing nonconsensus 5′ss, but yeast are inviable if ZnF2 is humanized to resemble LUC7L3 ZnF2. Consistent with reporter substrate data, we identify a subset of yeast transcripts that show altered processing upon humanization of Luc7 in vivo. Finally, we show that humanized Luc7 does not bypass the requirement for Prp28 in splicing but does suppress cold-sensitive (cs) phenotypes observed with Prp28 mutants. Taken together, these data show that the Luc7 ZnF2 domain facilitates 5′ss selection.
RESULTS
Humanized Luc7 ZnF proteins support yeast viability except for LUC7L3 ZnF2
To investigate the human Luc7 homologs, we generated a yeast “shuffle” system to introduce Luc7 mutants in yeast. We deleted the chromosomal LUC7 gene while maintaining yeast viability by expression of wild type (WT) Luc7 from a low-copy, URA3/CEN6-containing plasmid. Plasmid expression of Luc7 had no effect on yeast viability in standard growth conditions (Supplemental Fig. S1A). To introduce human Luc7 homologs in yeast, we inserted yeast codon-optimized gene fragments encoding truncations of the open reading frames of LUC7L, LUC7L2, or LUC7L3 into a low-copy, TRP1/CEN6-containing plasmid containing the native LUC7 promoter and terminator sequences. These truncations coded for the predicted structured regions of the proteins but did not include the C-terminal RS domains. Each of the constructs also included a 3 × HA epitope tag at the protein C terminus.
After the transformation of yeast with these plasmids and subsequent 5-FOA selection, we found that none of the human homologs was able to support yeast growth as the sole copy of Luc7 (Supplemental Fig. S1B). We confirmed the expression of the proteins by western blotting. Although each protein was expressed, LUC7L and LUC7L2 were less abundant than either Luc7 or LUC7L3 (Supplemental Fig. S1C). In addition, we were unable to rescue viability when these proteins were expressed under the control of a strong TDH3 promoter (OE; Supplemental Fig. S1B). The inability of the human LUC7L proteins to fully compensate for yeast Luc7L has also been recently reported (Chalivendra et al. 2024). Based on these results, we instead focused on targeted mutations to humanize Luc7.
We analyzed the cryo-EM structure of the yeast spliceosome A complex to identify Luc7 amino acids located within 6 Å of the U1 snRNA/5′ss duplex (Fig. 1A,C; Plaschka et al. 2018). The amino acids predominantly come from ZnF2; however, we also decided to study ZnF1 (which is likely located further from the RNA duplex) because previous work showed genetic interactions between ZnF1 mutations and a number of splicing factors (Agarwal et al. 2016). We identified the corresponding amino acids in ZnF1 and ZnF2 of the human LUC7L paralogs based on a multiple sequence alignment generated by Clustal Omega (Fig. 1B; Sievers and Higgins 2014) and humanized yeast Luc7 accordingly by site-directed mutagenesis of the nonconserved amino acids (amino acid substitutions listed in Supplemental Table S1). We then expressed the humanized, C-terminal 3 × HA-tagged Luc7 variants in yeast on plasmids and under the control of the native Luc7 promoter. We refer to mutants of ZnF1 as ZnF1_L, L2, or L3 corresponding to ZnF1 from LUC7L, LUC7L2, or LUC7L3. The mutations needed to humanize ZnF2 for LUC7L and LUC7L2 are identical. We refer to mutants of ZnF2 as ZnF2_L/L2 or L3.
Luc7 chimeric proteins containing one or both ZnFs from LUC7L or LUC7L2 were viable (Fig. 2). Although a Luc7 chimera of ZnF1_L3 was also viable, a chimera containing ZnF2_L3 was not even though it was well-expressed in yeast (Supplemental Fig. S1D). ZnF2_L3 did not support viability even when ZnF1 was also humanized to that from LUC7L3 or any other LUC7L paralog. From these experiments, we conclude that although most ZnF modules from human Luc7 homologs can support yeast splicing, ZnF2_L3 is not functionally equivalent to either ZnF2 from Luc7 or LUC7L/L2.

Yeast are viable with humanized Luc7 ZnF1 and ZnF2. (A) Luc7 ZnF1 mutants are viable in yeast. The ZnF2_L/L2 mutant is also viable, whereas ZnF2_L3 is lethal. (B) Expressing Luc7 with mutations in both ZnF1 and ZnF2 does not rescue the lethality associated with ZnF2_L3. “-W” and “-W + 5-FOA” refer to synthetic media lacking tryptophan and media lacking tryptophan and containing 5-fluoroorotic acid, respectively.
ZnF2 but not ZnF1 changes 5′ss usage of a yeast splicing reporter
We next used the ACT1-CUP1 reporter assay to probe how the humanization of the Luc7 ZnF domains influences 5′ss selection. In this assay, yeast growth in the presence of various concentrations of Cu2+ is correlated with splicing of the reporter and mRNA production (Fig. 3A; Lesser and Guthrie 1993a). In the case of ZnF1, we used ACT1-CUP1 reporters with substitutions from −3 to −1 on the exonic side of the 5′ss because this region is closest to the binding site of ZnF1 (Figs. 1 and 3A). The WT ACT1-CUP1 substrate normally only contains a single potential base pair with the U1 snRNA within the −3 to −1 region (-1G could pair with U1 snRNA C9; Fig. 1C). G-1A and G-1C substitutions in the ACT1-CUP1 should, therefore, disrupt all pairing within this region, whereas a U-2A or C-3A should strengthen the interaction by allowing pairing with either U10 or U11. To increase the sensitivity of the assay, we also incorporated mutations at the +2 (U2A) or +5 (G5A) positions because substitutions at −3 to −1 have no discernible effect on the splicing of a substrate with a consensus 5′ss.
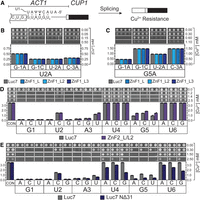
Mutation of the ZnF domains of Luc7 can have specific impacts on 5′ss usage. (A) Schematic representation of the ACT1-CUP1 reporter pre-mRNA and splicing assay. Proper processing of the splicing reporter confers resistance to Cu2+ in the growth media. (B) ACT1-CUP1 assays using a reporter with substitutions in the 5′ss sequence U2A (GaAUGU) and the indicated changes in the exon. ZnF1 mutants have no obvious effect on splicing. Dots represent the maximum [Cu2+] tolerated in two independent replicates. (C) Same as in B but using a reporter with the 5′ss G5A (GUAUaU). (D) ACT1-CUP1 assays using ZnF2_L/L2 to assay intronic positions of the 5′ss. Mutation of Luc7 ZnF2 improves splicing of A3U, G5A, G5C, and G5U but does not affect other reporters. (E) ACT1-CUP1 assays using the NΔ31 truncation mutant of Luc7.
None of the ZnF1 chimeras changed yeast tolerance to Cu2+ for any of the ACT1-CUP1 reporters harboring substitutions at −3 to −1 (Fig. 3B,C). No changes were observed even when ZnF2 was also modified to that from LUC7L/L2 (Supplemental Fig. S2). As expected, we also did not observe changes in splicing due to ZnF1 substitutions with reporters carrying substitutions at the +1 to +6 positions, likely because these substitutions are located distal to the ZnF1 interaction site (Supplemental Fig. S3). From these results, we conclude that paralog-specific contacts of ZnF1 made by these humanized Luc7 variants are unlikely to significantly influence splicing of the ACT1-CUP1 reporter in this assay.
For studies of ZnF2, we focused on using ACT1-CUP1 reporters harboring substitutions within the intronic portion of the 5′ss (+1 to +6) because ZnF2 contacts this region (Fig. 1C). When ZnF2 was replaced by ZnF2_L/L2, we observed increased Cu2+ tolerance of yeast with ACT1-CUP1 reporters containing substitutions at the +3 position (A3U) and +5 position (G5A, C, U) (Fig. 3D), in agreement with contacts between Luc7 and the snRNA/5′ss duplex. Because the high levels of Cu2+ tolerance observed with some reporters precluded analysis (i.e., U4 and U6 substitutions), we also carried out assays in which a second mutation was incorporated at the +2 position (U2A). We were unable to find any effect of ZnF2_L/L2 with these doubly substituted reporters for changes at the +4 or +6 positions (Supplemental Fig. S4). These results suggest that a function of ZnF2 in human LUC7L and LUC7L2 could be to promote splicing at weak 5′ss containing mismatches to the U1 snRNA at the +3 and +5 positions of the 5′ss.
Deletion of a Luc7/Sm ring interaction decreases usage of nonconsensus splice sites
We wondered whether other Luc7 mutants, not confined to the ZnF domains, would also show changes in splicing at +3 and +5. The Luc7 NΔ31 mutation abolishes contact between Luc7 and the U1 snRNP Sm protein ring and was previously shown to impair splicing of some pre-mRNAs, cause synthetic lethality with a number of other mutations, and bypass the need for Prp28 (Agarwal et al. 2016; Plaschka et al. 2018). Because this mutant was previously well-characterized, we carried out ACT1-CUP1 assays with Luc7 NΔ31-expressing yeast.
Unlike the ZnF2_L/L2 substitution, Luc7 NΔ31 caused a loss of yeast tolerance to Cu2+ using reporters with multiple substitutions at the +2 to +6 positions (Fig. 3E; Supplemental Fig. S4). The only position that was unaffected was +1. This suggests that pairing at this position and enforcing selection of the nearly invariant +1G at the 5′ss may be due to other snRNP components—likely Yhc1/U1-C, which makes extensive contacts with the duplex at this position (Fig. 1C; Kondo et al. 2015; Hansen et al. 2022). Surprisingly, Luc7 NΔ31 even decreased Cu2+ tolerance in yeast with U4A and U4G substitutions, which should result in increased pairing to Ψ5 in the snRNA. This would seem to indicate that WT Luc7 has evolved to function preferentially on substrates containing mismatches (i.e., a U) at the +4 position of the 5′ss. Interestingly, the +4 nucleotide is ultimately juxtaposed with A49 in the U6 snRNA during splicing catalysis, and +4U could form a canonical base pair with A49 (Kandels-Lewis and Séraphin 1993; Lesser and Guthrie 1993b). Luc7 may help the snRNP to tolerate a 5′ss/U1 snRNA mismatch that ultimately increases 5′ss base-pairing to U6.
Luc7 variants perturb splicing of endogenous yeast pre-mRNAs
5′ss sequences found in endogenous yeast introns are less diverse than the reporters we used in the ACT1-CUP1 assays. For example, we do not believe there are any naturally used 5′ss with adenosine located at the +5 position even though we observed increased Cu2+ tolerance with the G5A reporter and ZnF2_L/L2. We first tested two endogenous transcripts, SUS1 and RPL22B, for changes in splicing due to Luc7 ZnF2_L/L2 or Luc7 NΔ31. We chose these transcripts because changes in SUS1 splicing had previously been reported for Luc7 NΔ31 (Agarwal et al. 2016) and RPL22B has a cryptic, intronic 5′ss (GUUUGU) with an A3U substitution relative to the consensus (Kawashima et al. 2014). Because the splicing of ACT1-CUP1 reporters with A3U is stimulated by Luc7 ZnF2_L/L2 (Fig. 3D), we wondered if the humanized protein would also stimulate the use of the cryptic site in RPL22B.
We analyzed the splicing of SUS1 and RPL22B by RT-PCR using yeast strains expressing Luc7-ZnF2_L/L2 or Luc7 NΔ31. UPF1 was also deleted in these strains to block nonsense-mediated decay (NMD) and facilitate detection of RPL22B isoforms (Kawashima et al. 2014). Both Luc7 variants somewhat inhibited splicing of both SUS1 and RPL22B with Luc7 NΔ31 showing a greater splicing defect (Supplemental Fig. S5). We did not observe a substantial increase in the use of the cryptic 5′ss in RPL22B due to Luc7 ZnF2_L/L2. However, Luc7 NΔ31 accumulated unspliced RPL22B and had comparatively less usage of the cryptic 5′ss than either WT Luc7 or Luc7 ZnF2_L/L2. This is consistent with the ACT1-CUP1 assays and with Luc7 NΔ31 inhibiting splicing of weak, nonconsensus 5′ss, including A3U (Fig. 3E).
To further probe the effects of the Luc7 variants on the yeast transcriptome, we used total RNA-seq to analyze changes in splicing efficiency genome-wide. RNA-seq was performed in an upf1Δ genetic background deficient for NMD. The upf1Δ genetic background not only helps to stabilize unspliced mRNAs harboring premature termination codons but also facilitates a more direct analysis of splicing efficiency without confounding effects of differential isoforms stability (Sayani et al. 2008; Kawashima et al. 2014).
mRNA expression was globally unchanged when Luc7 ZnF2_L/L2 was expressed compared to WT, as no genes showed significant differential expression (P < 1 × 10−5). The Luc7 NΔ31 strain had nine differentially expressed genes (Supplemental Fig. S6A): YBL059W*, ATP5, IMD2, DGR2, YLR366W, RPS22B*, YML131W, HRB1*, and LYS9 (* indicates an intron-containing gene). HRB1 is interesting because of its reported role in the selective export of spliced mRNAs (Hackmann et al. 2014). Its modest upregulation (∼1.6-fold) is likely due to the accumulation of unspliced transcripts. It is worth noting that analysis of HRB1 RNAs also showed a significant fraction of unspliced reads in the control upf1Δ strain expressing WT Luc7 (∼1:1), indicative of suboptimal splicing even in the presence of the WT protein.
To calculate splicing efficiency, we quantified reads that could be unambiguously assigned as either spliced junctions or unspliced reads (i.e., spanning both exonic and intronic regions). For each intron annotation, we first calculated the ratio of unspliced reads to spliced reads. We then compared these ratios for each mutant relative to the WT control. Both Luc7-ZnF2_L/L2 and Luc7 NΔ31 had a negative impact on global splicing efficiency compared to WT (Fig. 4A). However, the effect was not universal as both mutants showed increased splicing efficiency of a select few transcripts. We grouped each intron by its change in splicing efficiency in each mutant compared to WT and analyzed splicing sequences for each group by generating sequence logos of the 5′ss, branch points, and 3′ss (Fig. 4A; Supplemental Fig. S6B).
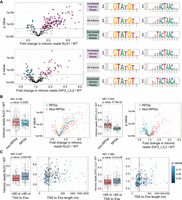
Analysis of RNA-seq data from cells expressing WT, NΔ31, or ZnF2_L/L2 Luc7 Proteins. (A) Plots showing changes in splicing efficiencies for each intron in Luc7 NΔ31 (top) and Luc7 ZnF2_L/L2 (bottom). p-values are the results of unequal variances t-tests using geometric means between three replicates. Introns with statistically significant changes in splicing efficiency were categorized into increased or decreased intronic reads in mutants over WT, p-value < 0.05 (dashed line). Sequence logos for 5′ss (middle) and BP (right) are shown for each category. (B) Changes in splicing efficiencies in Luc7 NΔ31 (left) and Luc7 ZnF2_L/L2 (right) versus WT for ribosomal protein genes (RPGs). Cyan points indicate RPGs. Histogram p-values are the results of unequal variances t-tests using geometric means. (C) Plots showing transcriptional start site to 5′ss distances versus changes in splicing efficiency for Luc7 NΔ31 (left) and Luc7 ZnF2_L/L2 (right) versus WT Luc7. Color indicates p-values. For plots in A–C, note that the x-axis in A and the y-axis in B and C represent fold changes. The black vertical line indicates 85 nt. (MD) Difference of geometric means.
We found that introns with increased splicing efficiency with Luc7 NΔ31 had near-perfect consensus 5′ss (GUAUGU). In line with the ACT1-CUP1 data, introns with non-“GUAUGU” 5′ss had an average of 45% more unspliced reads with Luc7 NΔ31 compared to WT (Supplemental Fig. S6C). The 5′ss sequence had surprisingly little effect with the Luc7 ZnF2_L/L2. Only one of the common, nonconsensus 5′ss showed a statistically significant change in splicing efficiency: Introns with a GUACGU 5′ss had 18% fewer unspliced reads on average (Supplemental Fig. S6C). This is consistent with the position of the ZnF2 region in contacting the U1 snRNA/5′ss duplex near the U4 position, but we did not observe a similar change at U4 substitutions in our ACT1-CUP1 assays (Fig. 3D; Supplemental Fig. S4).
The introns with increased splicing efficiency in both the Luc7 ZnF2_L/L2 and NΔ31-expressing strains showed a strong enrichment for the consensus U at position one of the branch point consensus (UACUAAC, underlined) as well as a more modest enrichment for the consensus C at position 7 (UACUAAC). Interestingly, the introns with increased splicing efficiency using the Luc7 ZnF2_L/L2 variant showed an enrichment for an A immediately downstream from the branch point (UACUAACA). An adenosine at this position would facilitate the formation of an additional base pair with the U2 snRNA at U2 nucleotide U33. Base-pairing between the U2 snRNA and intron is seen at this position in cryo-EM structures of the U2 snRNP (Plaschka et al. 2018; Zhang et al. 2024). This suggests that increased base-pairing between U2 and the branch point may compensate for the deleterious effects of the ZnF2 L/L2 variant.
We also found that the strain expressing Luc7 ZnF2_L/L2 was able to splice pre-mRNAs of RPGs protein pre-mRNAs with much higher efficiency than pre-mRNAs of non-RPGs, whereas the strain expressing Luc7 NΔ31 showed little difference between the two (Fig. 4B). In an attempt to explain this observation, we checked for correlations between intron length and splicing efficiency as well as between the transcription start site (TSS) to 5′ss distance and splicing efficiency, because introns found in RPGs are typically longer than those of non-RPGs (Spingola et al. 1999). Although both were statistically significant with the Luc7 ZnF2_L/L2 mutant, neither was a better explanatory variable for splicing efficiency than ribosomal versus non-RPG (Fig. 4C; Supplemental Fig. S6D). We do note, however, that the expression level of the RNAs could play a role in how 5′ss compete for U1 snRNP binding because RPGs are more highly expressed than most other intron-containing genes (Munding et al. 2013).
Although splicing efficiency in the Luc7 NΔ31–expressing strain was not significantly correlated with ribosomal versus non-RPG or intron length, it did show a significant correlation with the distance from TSS to the 5′ss (Fig. 4C) with longer distances from the TSS corresponding to generally lower splicing efficiency. Because our data (Figs. 3E and 4) and that by Agarwal et al. (2016) suggest that Luc7 NΔ31 is less able to stabilize the U1 snRNA/5′ss interaction, it would seem that this becomes even worse the further from the TSS and (possibly) the 5′ end of the nascent transcript. We do not know exactly what factors are responsible for this behavior, but Luc7 NΔ31 is synthetically lethal with deletion of the nuclear cap-binding complex (CBC) (Agarwal et al. 2016). It is possible that the ability of the CBC to stabilize U1 on the pre-mRNA (Görnemann et al. 2005; Larson and Hoskins 2017) and compensate for Luc7 NΔ31 is less effective at greater TSS to 5′ss distances. Similar RNA cap to 5′ss distance dependence for the function of Luc7 has been remarked upon previously with the presence of Luc7 favoring the selection of cap-proximal 5′ss (Puig et al. 2007).
Luc7 ZnF2_L/L2 alters the requirement for Prp28 during splicing
We next assayed the Luc7 mutants for an altered requirement of the DEAD-box ATPase Prp28 for U1/U6 base-pairing exchange at the 5′ss during spliceosome activation (Fig. 5A; Staley and Guthrie 1999). It has previously been shown that Luc7 NΔ31 can completely bypass the need for Prp28 activity and that the PRP28 gene is no longer essential when Luc7 NΔ31 is present (Agarwal et al. 2016). Unlike Luc7 NΔ31, Luc7 ZnF2_L/L2 does not bypass the requirement for the presence of Prp28 during splicing (Fig. 5B). We then tested whether Luc7 ZnF2_L/L2 could alter the demand for Prp28 activity using a Prp28 ATPase mutant (Prp28 R499A, a mutation in DEAD-box motif Va) that is proposed to impair ATP hydrolysis and leads to a cs growth phenotype (Fig. 5A). We predicted that if Luc7 ZnF2_L/L2 places increased demand on Prp28, then this Luc7 mutant should exacerbate the growth defects present with Prp28 R499A. Alternatively, if Luc7 ZnF2_L/L2 facilitates U1/U6 exchange by Prp28 R499A, it should result in suppression of the cs phenotype. Luc7 ZnF2_L/L2 not only suppresses the cs phenotype, it surpasses Luc7 NΔ31 in its ability to do so (Fig. 5C). Together, these data demonstrate that Luc7 ZnF2_L/L2 alters the requirement for ATP hydrolysis by Prp28 during splicing in a way that still requires a Prp28 presence and is distinctly different from Luc7 NΔ31. Although it is often assumed that Prp28 mutant suppression is due to weakening of U1/5′ss interactions, our results suggest that this can still occur even when a subset of U1/5′ss interactions are potentially strengthened as demonstrated by Luc7 ZnF2_L/L2 in the ACT1-CUP1 reporter assay (Fig. 3) and with endogenous 5′ss with U4C substitutions relative to the consensus (Fig. 4).

Luc7 ZnF2_L/L2 suppresses the cs phenotype of Prp28 R499A. (A) Schematic showing the impact of Prp28 alleles that modulate the U1 to U6 exchange during splicing. Truncation of Luc7 bypasses the requirement for Prp28 in splicing and allows splicing to proceed whereas Prp28 ATPase mutants inhibit this step and stall splicing. (B) Luc7 ZnF2_L/L2 does not bypass the requirement for Prp28 in splicing unlike Luc7 NΔ31. “-HW” and “-HW + FOA” refer to synthetic media lacking histidine and tryptophan in the absence or presence of 5-fluoroorotic acid, respectively. (C) Luc7 ZnF2_L/L2 suppresses cold sensitivity associated with Prp28 R499A.
DISCUSSION
In this work, we studied the roles of the yeast Luc7 ZnF domains in 5′ss selection using humanized variants in which the ZnF domains were mutated to resemble those found in the three human Luc7 paralogs. We were not able to detect an impact of ZnF1 substitutions on splicing of reporter substrates; however, substitutions in ZnF2 that mimic the corresponding domain in human LUC7L and LUC7L2 proteins increased usage of nonconsensus splice sites both in reporter assays and for endogenous yeast transcripts. A mutant with substitutions mimicking ZnF2 of LUC7L3, which is the most sequence divergent of the human paralogs with Luc7, was not viable. In contrast with the Luc7 ZnF2 variant, a Luc7 NΔ31 mutant generally decreased usage of nonconsensus 5′ss, including one endogenous cryptic splice site. We also noted that this variant also decreased usage of reporter pre-mRNAs with increased pairing to U1 snRNA (the U4A and U4G ACT1-CUP1 variants) and caused increased accumulation of unspliced transcripts with longer TSS to 5′ss distances. Despite differential impacts on 5′ss usage, Luc7 NΔ31 and the ZnF2 variant were both able to suppress a cs allele of Prp28, suggesting an impact on both U1 binding to pre-mRNAs and its release during U1/U6 exchange. Altogether, our data reveal how the Luc7 ZnF2 domain can tune 5′ss usage at multiple steps in splicing.
The molecular basis for human LUC7L, LUC7L2, and LUC7L3 promoting different splicing outcomes in human cells is not yet understood. Recently, it has been postulated that LUC7L paralog specificity may arise from differences in the number of potential base pairs on either side of the exon/intron junction (Kenny et al. 2022). LUC7L and LUC7L2 (which are more closely related to one another than either is to LUC7L3) may promote splicing primarily at 5′ss with higher levels of complementarity with U1 snRNA at the +3, +4, and +5 positions (CAG/GUAAGU) relative to the −3, −2, and −1 positions (CAG/GUAAGU). These were denoted as having a “right-handed” preference relative to the exon/intron junction (handedness used in this context is unrelated to chirality). In contrast, Luc7L3 appeared to have a preference for “left-handed” interactions with higher complementarity on the “exon side” of the junction. Based on the structure of yeast splicing complexes containing Luc7 (Fig. 1B,C), this would suggest that ZnF2 could play a role in preference for right-handed 5′ss, whereas ZnF1 could play a role in preference for left-handed sites. Differences found in the two ZnF domains between the human LUC7L variants and how the two domains collaborate with one another may contribute to 5′ss selectivity. Yeast 5′ss are much less diverse than their human counterparts with nearly all having extensive complementarity on the intron side (right-handed). Luc7 may function similarly to LUC7L and LUC7L2, and this may explain the lack of effect of ZnF1 changes on reporter and endogenous transcript splicing. In fact, we could find only four yeast genes (∼1% of all pre-mRNAs) with high complementarity within the exon (−3C, −2A, −1G relative to the exon/intron boundary).
Our results with yeast–human Luc7 chimeras also support the notion of a divergent function for LUC7L3. Yeast Luc7 proteins containing human LUC7L3 ZnF1 were viable but those containing LUC7L3 ZnF2 were not, regardless of the origin of ZnF1 (Fig. 2). This suggests that the function of yeast ZnF2 cannot be compensated for by the more divergent human LUC7L3 ZnF2. For human LUC7L3, this could indicate that although it may promote splicing of left-handed 5′ss, it is not able to do the same for those that are right-handed. This is in agreement with data from Kenny et al. (2022) showing that overexpression of LUC7L3 represses splicing of right-handed 5′ss.
We were not able to identify a phenotype associated with the replacement of yeast ZnF1 with that from any of the human homologs, even when using two different substrates with mutations that strengthened the left-handedness of the 5′ss (Fig 3B,C). We cannot conclude if this indicates that ZnF1 alone is not responsible for the left-handed preference for 5′ss by LUC7L3 or if this feature of the human splicing machinery cannot be recapitulated in our yeast system. Nonetheless, it suggests that further work is needed to define the role of ZnF1. Work from Agarwal et al. (2016) indicated that although yeast were viable with Luc7 containing a disrupted metal-binding site in ZnF1, several of these mutants possessed a ts phenotype and were synthetic lethal with mutations in a variety of other splicing factors. This indicates that ZnF1 does have a function in yeast splicing, but its role may be context-dependent.
One of the complexities in studying U1 snRNP-associated factors in vivo is that splicing outcomes (e.g., as measured by RNA-seq) are only correlated with U1 association. For splicing chemistry to occur, it is essential that U1 be released during the transition from the pre-B to B complex spliceosome. This release must occur so that 5′ss interactions with the U1 snRNA can be exchanged for those with the U6 snRNA. This normally requires the action of the DEAD-box ATPase Prp28. Consequently, inferring changes in U1 binding from measurements of mRNA production are difficult, and a convolution of U1 overall binding properties (on- and off-rates), U1 release by Prp28, and U6 binding among likely other influences. Our work shows that although the substitution of ZnF2 of Luc7 can result in relatively few overall changes in the transcriptome (Fig. 4), it can still perturb the ATPase-dependence of U1 release. Humanization of Luc7 ZnF2 does not bypass the need for Prp28 but does suppress the cs phenotype of a Prp28 ATPase site mutant (Fig. 5). This suggests that human LUC7L/L2 ZnF2 facilitates U1 release by Prp28. We do not know if this occurs by weakening the U1 snRNA/5′ss duplex at this stage and/or by stimulating Prp28 activity. However, it does provide additional evidence that the ZnFs play roles in both recognition of the 5′ss by U1 and its release during activation. Given the conservation of these factors, it is likely that this is also true in humans. Thus, understanding the function of the human Luc7 homologs should consider their impact on both U1 binding and release. Given that LUC7L2 promotes glycolysis at the expense of oxidative phosphorylation (Jourdain et al. 2021), it is tempting to speculate that different Luc7 homologs have different ATPase dependencies for the U1/U6 exchange that could correlate with cellular metabolic state.
MATERIALS AND METHODS
Saccharomyces cerevisiae strains used in these studies were derived from 46α (kind gift of David Brow) or a PRP28 and LUC7 double shuffle strain (gift of Beate Schwer) (Lesser and Guthrie 1993a; Agarwal et al. 2016). The open reading frame of LUC7 and the 500 bp upstream and downstream were amplified from yeast genomic DNA and cloned into the CEN6/ARS4 centromeric plasmids pRS416 and pRS414. Supplemental Tables S1 and S2 contain detailed lists of strains and plasmids used. Yeast transformation and growth were carried out using standard techniques and media (Amberg et al. 2005).
Site-directed mutagenesis
Point mutants were generated using inverse polymerase chain reaction (PCR) with Phusion DNA Polymerase (New England Biolabs) or Herculase II (Agilent). PCR was performed for 16 cycles using primers with the desired nucleotide changes incorporated at or near the 5′ ends. Template DNA was removed by treatment with DpnI (New England Biolabs), and the PCR products were subsequently 5′ phosphorylated using T4 polynucleotide kinase (New England Biolabs) and self-ligated by T4 DNA ligase (New England Biolabs) before being used to transform Top 10 competent cells (Thermo Fisher Scientific). Individual colonies were screened by Sanger sequencing to identify the desired changes.
Temperature growth assays
Yeast strains were grown to the mid-log phase in YPD supplemented with 0.003% (w/v) adenine hemisulfate (YPAD) or selective media, the OD600 was adjusted to 0.5, and equal volumes were spotted onto YPAD or selective plates. Plates were incubated at the indicated temperature and scored after 3 days of growth at 30°C or 3 days of growth at 23°C or 37°C.
ACT1-CUP1 copper assays
ACT1-CUP1 reporters and growth assays have been described previously (Lesser and Guthrie 1993a; Carrocci et al. 2017). Briefly, yeast strains expressing WT or mutant proteins and ACT1-CUP1 reporters were grown to mid-log phase in SC-LEU media to maintain selection for the reporter plasmids and adjusted to OD600 = 0.5, and equal volumes were spotted onto plates containing 0, 0.025, 0.05, 0.075, 0.1, 0.15, 0.2, 0.25, 0.3, 0.4, 0.5, 0.6, 0.7, 0.8, 0.9, 1.0, 1.1, 1.2, 1.3, 1.4, 1.5, 1.6, 1.7, 1.8, 1.9, 2.0, 2.25, 2.5, 3.0, or 3.5 mM CuSO4. Plates were scored after 3 days of growth at 30°C. Assays were performed at least two or more times.
RT-PCR RNA analysis
Yeast were grown in YPAD media until OD600 reached 0.5–0.8. Cells (8 OD600 units) were harvested by centrifugation and washed with water, and total cellular RNA was isolated using a MasterPure Yeast RNA Purification Kit (Lucigen) according to the vendor's instructions. DNase I–treated total RNA (4 µg) was reverse transcribed using PrimeScript Reverse Transcriptase (Takara Bio) and random hexamers (Thermo Fisher Scientific). Assembled reactions were incubated at 30°C for 10 min, followed by 1 h at 42°C for complete extension. The RT was heat inactivated by treatment at 70°C for 15 min, and reactions were diluted 1:20 and used in PCR without further purification. PCR reactions were carried out using Taq polymerase (New England Biolabs) and contained 2.5 µM gene-specific primers that flanked the intron. One of the primers was also labeled at the 5′ end with Cy5 to facilitate fluorescence imaging. Products were separated using 2.5% (w/v) metaphor agarose (Lonza) in 1 × TBE, and the gel was subsequently imaged using an iBright imager (Thermo Fisher Scientific). Band intensities were quantified using ImageJ.
RNA extraction and RNA sequencing
RNA was isolated as previously described (Wang et al. 2020). Briefly, strains expressing either LUC7-WT or mutants luc7-nΔ31 and luc7-znf2 and with UPF1 deleted were grown overnight in YPD at 30°C. The following morning, the cultures were diluted in fresh YPD to OD600 = 0.1 and grown at 30°C for two doublings; after which cells were collected and flash frozen in liquid nitrogen. Total RNA was extracted using hot phenol:chloroform followed by ethanol precipitation. Total RNA (40 µg) from each condition was then treated with DNase I (Invitrogen) to remove genomic DNA. Samples were ribo-depleted using the Ribocop for Yeast kit (Lexogen), and libraries were prepared using the CORALL Total RNA-seq V2 kit (Lexogen). Barcoding was carried out using the UDI 12 nt Set B1 (Lexogen), and amplification was done using 15 cycles of PCR. Sequencing was performed on a NovaSeq PE150 by Novogene, which also carried out sample demultiplexing.
Read quality control and mapping
UMI-tools were used to extract unique molecular identifiers (UMIs) from reads (Smith et al. 2017). Each UMI was 12 nt long (‐‐bc-pattern = NNNNNNNNNNNN). Cutadapt was used for quality control and to remove adaptors and poly(A) sequences (Martin 2011). Terminal “N”s were removed from each read. Adapters -g “T{100}” -a AGATCGGAAGAGC -A AGATCGGAAGAGC -A “A{100}.” A minimum overlap of 2 nt was required for each adapter and a maximum of 2 errors was allowed in each adapter sequence. Bases with quality scores <20 were removed from both ends of each read. A minimum read length of 50 nt was required. Any read with more than four ambiguous bases (N) after filtering was removed. Reads were aligned to the Saccharomyces genome (S288C version R64-3-1) downloaded using HISAT2 (version: 2.2.1) (Cherry et al. 2012; Engel et al. 2014; Kim et al. 2019). Known splice site annotations were acquired from the SGD. For noncanonical splicing events, a minimum intron length of 20 nt and a maximum intron length of 1000 nt were required. A penalty of 0 was set for noncanonical splice site alignment. PCR duplicates were removed using umi_tools extract before counting.
Counting of mRNAs, spliced reads, and unspliced reads
Genome annotation files were acquired from the SGD. Counting was done using Rsubread featureCounts (Liao et al. 2019). Each read in a pair was counted individually, and multiple overlapping was allowed. DESeq2 was used to estimate differential gene expression (Love et al. 2014). To count unambiguously unspliced reads, both sides of each intron were counted separately, and the results were averaged. The annotation file was 1 nt outside of the intron and 1 nt inside the intron; a read needed to be mapped to both positions to be counted. Only nonsplit reads were counted, and each read in a pair was counted individually with multiple overlapping allowed. To count unambiguously spliced reads, both sides of each intron were counted separately, and the results were averaged. The annotated file was 5 nt outside of the intron, and a minimum overlap of 4 nt was required. Only split alignments were counted. In some cases, alternative splice sites were counted as spliced reads; however, this was a small minority of reads. Each read in a pair was counted individually, and multiple overlapping was allowed. Any introns with no spliced read in any replicate were removed from further analysis. The implementation can be found in the counting script (https://github.com/SamDeMario-lab/Luc7_splicing).
Splicing efficiency calculations
Splicing efficiency was calculated as the ratio of reads which were unambiguously unspliced over unambiguously spliced for each intron. This ratio was calculated individually for each replicate, and the geometric mean was used as the splicing efficiency for downstream analysis. We note that because of PCR biases, our splicing efficiency calculations are unlikely to be accurate representations of the true levels of spliced and unspliced transcripts. However, all of our analysis is based on relative changes in splicing efficiency and, therefore, the biases are uniform across all samples. The “Intronic reads in mutant/WT” ratio was calculated as the splicing efficiency of the mutant divided by the splicing efficiency of strains expressing WT Luc7.
Data processing and plot generation
Data were processed and visualized in R version 4.2.2 (R Core Development Team 2022). DESeq2 was used to test differential expression (Love et al. 2014). Plots were generated using ggplot2 (Wickham 2016), ggforce was used to create reversed log transformed axes (Pedersen 2020), and ggseqlogo was used to make sequence logos (Wagih 2017). RColorBrewer was used to select color pallets for most panels (Neuwirth 2022). Figures were arranged using gridExtra (Auguie and Antonov 2017). BSgenome.Scerevisiae.UCSC.sacCer3, biostrings, and biomartr were used to extract reference sequences from genomic coordinates (Team TBD 2014; Drost and Paszkowski 2017; Pagès et al. 2024). Likely branch point sequences were acquired from the Ares laboratory Yeast Intron Database (Grate and Ares 2002). Any intron without a predicted branchpoint was excluded from the branch point analysis.
DATA DEPOSITION
All scripts used to process data and count tables and annotation files are available on GitHub (https://github.com/SamDeMario-lab/Luc7_splicing). All data generated during this study are available on the GEO (https://www.ncbi.nlm.nih.gov/geo/) at accession number PRJNA1022512.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
A.A.H. is a member of the scientific advisory board and carrying out sponsored research for Remix Therapeutics.
ACKNOWLEDGMENTS
We thank members of the Hoskins laboratory for their helpful discussions. We thank Dave Brow, Charles Query, and Beate Schwer for the strains and plasmids used in this study. This work was supported by grants from the National Institutes of Health (R35 GM136261 to A.A.H. and GM130370 to G.F.C.) with additional support from a Research Forward grant award from the Wisconsin Alumni Research Foundation.
Footnotes
-
Handling editor: Erik Sontheimer
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079956.124.
- Received January 21, 2024.
- Accepted April 14, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








