
Examining the capacity of human U1 snRNA variants to facilitate pre-mRNA splicing
- Department of Basic Medical Sciences, College of Medicine-Phoenix, University of Arizona, Phoenix, Arizona 85004, USA
- Corresponding author: shalinijs{at}arizona.edu
-
Handling editor: Javier Caceres
Abstract
The human U1 snRNA is encoded by a multigene family consisting of transcribed variants and defective pseudogenes. Many variant U1 (vU1) snRNAs have been demonstrated to not only be transcribed but also processed by the addition of a trimethylated guanosine cap, packaged into snRNPs, and assembled into spliceosomes; however, their capacity to facilitate pre-mRNA splicing has, so far, not been tested. A recent systematic analysis of the human snRNA genes identified 178 U1 snRNA genes that are present in the genome as either tandem arrays or single genes on multiple chromosomes. Of these, 15 were found to be expressed in human tissues and cell lines, although at significantly low levels from their endogenous loci, <0.001% of the canonical U1 snRNA. In this study, we found that placing the variants in the context of the regulatory elements of the RNU1-1 gene improves the expression of many variants to levels comparable to the canonical U1 snRNA. Application of a previously established HeLa cell-based minigene reporter assay to examine the capacity of the vU1 snRNAs to support pre-mRNA splicing revealed that even though the exogenously expressed variant snRNAs were enriched in the nucleus, only a few had a measurable effect on splicing.
Keywords
INTRODUCTION
The U1 small nuclear RNA (snRNA) is an abundant 164 nt long noncoding RNA. It is a critical component of the spliceosome, a dynamic macromolecular complex that catalyzes pre-mRNA splicing by removing noncoding introns and ligating coding exons (Will and Luhrmann 2011). The U1 snRNA associates with three U1-specific proteins (U1A, U1-70K, and U1C) and seven Sm proteins (SmB/B′, SmC, SmD1, SmD2, SmD3, SmF, and SmG) to form the U1 small nuclear ribonucleoprotein (snRNP) which, via the 5′-region of the snRNA, base pairs to 5′-splice site (5′-ss) sequences at exon–intron junctions in nascent pre-mRNAs. During the initial steps of spliceosome assembly, the U1 snRNA also interacts with the RNA helicase U2-associated protein 56 (UAP56, also known as DDX39B) via its stem–loop 3 (SL3) and with the U2 snRNP specific splicing factor 3A1 (SF3A1) via its stem–loop 4 (SL4) (Sharma et al. 2014; Martelly et al. 2021; de Vries et al. 2022). These cross-intron contacts have been found to promote the formation of U1 and U2 containing prespliceosomal A complex and pre-mRNA splicing in vitro (Martelly et al. 2021). Subsequently, binding of the U4–U6/U5 tri-snRNP and many auxiliary proteins to the A complex forms the spliceosome, which undergoes remodeling and activation that requires the release of U1 and U4 snRNPs, along with many non-snRNP proteins, before splicing catalysis (Wan et al. 2020; Wilkinson et al. 2020).
In addition to splicing, U1 is known to be involved in other steps in gene expression, including transcription and 3′-end processing. U1 snRNP binding to promoter regions has been reported to facilitate recruitment of the basal transcription machinery and enhance transcription (Furger et al. 2002; Damgaard et al. 2008). Through its association with the transcription factor TFIIH, the U1 snRNA has been shown to control transcription efficiency (Kwek et al. 2002). The binding of U1 to sequences in the 3′ untranslated region (UTR) of transcripts inhibits 3′-end processing (Ashe et al. 1997; Gunderson et al. 1998). Finally, binding of U1 to pre-mRNAs at cryptic 5′-ss in introns is required for the prevention of premature cleavage and polyadenylation (Berg et al. 2012; Venters et al. 2019). This activity, referred to as telescripting, adds a potential regulatory step in gene expression and has also been shown to modulate pathophysiological properties of cancer cells, including proliferation, migration, and invasiveness (Oh et al. 2020).
Almost four decades ago, several studies reported that the human U1 snRNA is encoded by a multigene family consisting of transcribed variants and defective pseudogenes (Denison et al. 1981; Denison and Weiner 1982; Lund and Dahlberg 1984; Bernstein et al. 1985). Since then, many variant U1 (vU1) snRNAs have been demonstrated to not only be transcribed but also capped with trimethylated guanosine, packaged into snRNPs, and assembled into spliceosomes (Kyriakopoulou et al. 2006; O'Reilly et al. 2013; Mabin et al. 2021). The most abundant canonical U1 snRNA is expressed by seven genes; of which five are located on Chromosome 1, and two are located on Chromosome 14. A recent systematic analysis of the human snRNA genes by Mabin et al. revealed a total of 178 U1 snRNA genes that are present as either tandem arrays or single genes on multiple chromosomes (Mabin et al. 2021). Of these, 30 were found to be expressed in cell lines by bioinformatic analysis of short noncoding RNA-sequencing data sets. In addition to the canonical U1 snRNA, 15 variants were detected in human adult and fetal tissues, and in model cell lines by RT-qPCR. The reciprocal pattern of canonical U1 and vU1 snRNA expression has been associated with the differentiation of human embryonic stem cells (hESCs) to monocytes and motor neurons, with levels of the canonical U1 snRNA increasing and levels of vU1s decreasing during differentiation (Vazquez-Arango et al. 2016). Additionally, the expression levels of vU1s were found to be higher in hESCs than in HeLa cells, and in induced pluripotent stem cells (iPSCs) than in skin fibroblasts; thus, leading to the suggestion that the variants might be involved in cellular differentiation (O'Reilly et al. 2013; Vazquez-Arango et al. 2016). Notably, vU1s are expressed in other metazoans, including mouse, frog, fly, moth, and sea urchin, and have been reported to be involved in development and differentiation (Forbes et al. 1984; Lund et al. 1985; Nash et al. 1989; Santiago and Marzluff 1989; Lo and Mount 1990; Sierra-Montes et al. 2005). A switch in the expression pattern of vU1s during embryogenesis has been reported in many species and it has been suggested that variant-specific functions may be important for developmental stage-associated patterns of gene expression (Santiago and Marzluff 1989).
Although vU1 snRNAs have been shown to be expressed and incorporated into snRNPs and spliceosomes, their ability to facilitate pre-mRNA splicing catalysis has not been tested so far. In this study, we applied a previously established HeLa cell-based minigene reporter assay to examine the capacity of vU1 snRNAs to support splicing (Sharma et al. 2014; Wong et al. 2021). The results revealed that even though many of the human vU1s could be exogenously expressed at levels comparable to the canonical U1 snRNA and were enriched in the nucleus, the ability to support pre-mRNA splicing of only a few was appreciable.
RESULTS
Low expression of variant U1 snRNAs in human cell lines
To test the capacity of the vU1 snRNAs to catalyze pre-mRNA splicing, we selected 15 variants (Fig. 1; Supplemental Table S1). Some of these were selected, including vU1.4 + 5, vU1.3 + 12, vU1.6, vU1.8, vU1.14, and vU1.15, because they have previously been shown to be expressed in HeLa and/or K562 cells. Although the levels of expression were significantly lower (0.001%–0.15%) than that of the canonical U1 snRNA, these six variants were also shown to be assembled into snRNPs and incorporated into spliceosomes (O'Reilly et al. 2013; Mabin et al. 2021). To compare expression patterns of these in HeLa, K562, and other cell lines, we also included a few previously untested variants: vU1.7 + 9, vU1.1 + 10, vU1.6, vU1.8, vU1.14, vU1.20, vU1A5, and vU1A6. The primer pairs that were designed for the vU1 snRNAs were tested and found to not amplify the canonical U1 snRNA (Supplemental Fig. S1). Our RT-qPCR analysis confirmed that the vU1 snRNAs are generally expressed at low levels and their expression patterns were variable. Four of the variants, including vU1.4 + 5, vU1.1 + 10, vU1.17, and vU1A6, were not detected in any of the cell lines tested: HeLa, HEK293T, K562, MV-4-11, and TF-1a (Fig. 2). Three variants (vU1.8, vU1.14, and vU1A7) were present in HeLa, HEK293T, K562, and TF-1a but not in MV-4-11. Notably, vU1.19 was detected only in K562 cells, and very low levels of vU1.15 were seen in MV-4-11 cells but not in other cell lines. Other variants, including vU1.7 + 9, vU1.3 + 12, vU1.6, vU1.20, and vU1A5, were expressed at ≤0.001% of the canonical U1 snRNA in the five tested cell lines (Fig. 2). In mammalian cells, the endogenous U1 snRNA is estimated to be present at about one million copies per cell (Steitz et al. 1988; Baserga and Steitz 1993). Calculation of the copies per cell of the vU1 snRNAs based on this number shows that MV-4-11 cells have the fewest copies (≤10/cell), whereas hundreds of copies are present in HEK293T and K562 cells, and thousands are present in HeLa and TF-1a cells (Supplemental Fig. S2). Thus, vU1 snRNAs exhibit some cell line-specific differences in expression from their endogenous loci, and levels of the snRNAs that can be detected are significantly low compared to the canonical U1 snRNA.
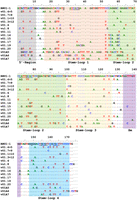
Sequence alignment of the canonical U1 snRNA with the vU1 snRNAs. Conserved bases are indicated with a dot and alterations are shown with base symbols. A dash indicates the absence of a base at that position. The 5′-region that base pairs with splice sites, stem–loops, and the Sm site of U1 snRNA are boxed and indicated below the aligned sequences. Nucleotides that have been reported to be involved in interactions of SL1 with U1-70K (Kondo et al. 2015; Gopan et al. 2022), SL2 with U1A (Kormos et al. 2011), and SL4 with SF3A1 (de Vries et al. 2022; Nameki et al. 2023) are underlined.
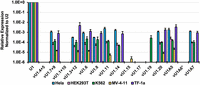
Low expression of U1 snRNAs from endogenous loci in human cell lines. Expression levels of snRNAs were measured by RT-qPCR. The expression values for vU1s were calculated relative to the canonical U1 snRNA and normalized to the U2 snRNA (n = 3) in HeLa, HEK293T, K562, MV-4-11, and TF-1a cells. vU1A6* was not tested in HEK293T cells. Sequences of primer sets used for RT-qPCR are provided in Supplemental Table S2.
RNU1-1 regulatory elements improve vU1 snRNA expression
Transcription of the major U1 snRNA gene, RNU1 (also referred to as HU1.1), is controlled by an enhancer-like distal sequence element (DSE) and an essential snRNA gene-specific proximal sequence element (PSE) (Hernandez 2001). Processing of the nascent transcript at the 3′-end requires the 3′-box that is located downstream from the coding sequence (Hernandez 1985). These regulatory elements differ considerably in the vU1 genes and could be one of the underlying reasons for their low expression levels (Guiro and O'Reilly 2015; Guiro and Murphy 2017). Additionally, the vU1s may not be appropriately recognized by nuclear 3′-end processing machinery and instead be targeted for degradation (Lardelli and Lykke-Andersen 2020). To see if the levels of the vU1 snRNAs could be increased by expressing them in the context of the regulatory elements of the canonical U1 gene, we introduced the vU1 sequences into the pNS6U1 plasmid that contains the essential upstream and downstream regulatory elements from the RNU1 locus. Previously, we have demonstrated efficient expression of the canonical and mutant U1 snRNAs from this plasmid in HeLa cells (Martelly et al. 2021). The expressed wildtype and mutant snRNAs were found to be processed, localized to the nucleus, and assembled into mature snRNPs. The exogenously expressed wildtype U1 and many of the mutants, carrying changes to SL3 and SL4, were also found to have the capacity to support splicing upon coexpression with a reporter in HeLa cells (Sharma et al. 2014; Martelly et al. 2021).
To examine if the presence of the regulatory elements improved the expression level of the vU1 snRNAs, we performed RT-qPCR assays 48-h post-transfection. This analysis showed that six variants, including vU1.4 + 5, vU1.1 + 10, vU1.15, vU1.17, vU1.19, and vU1A6, that were undetectable in untreated HeLa cells could be detected when expressed in the context of the regulatory elements from RNU1-1 (Fig. 3A). The achieved levels of vU1.4 + 5, vU1.15, vU1.17, and vU1.19 were similar to or higher than the endogenous U1 snRNA but those of vU1.1 + 10 and vU1A6 were significantly lower at 0.05- and 0.5-fold, respectively, relative to the endogenous U1 snRNA. Of the snRNAs that could be detected in untreated HeLa cells, including vU1.7 + 9, vU1.3 + 12, vU1.6, vU1.8, vU1.11, vU1.14, vU1.20, vU1A5, and vU1A7, there was little improvement in levels of vU1.6, vU1.8, vU1.14, and vU1A5, ranging from 0.05- to 0.5-fold relative to the endogenous U1 snRNA. Levels of exogenously expressed vU1.7 + 9, vU1.3 + 12, vU1.11, vU1.20, and vU1A7 were similar to or higher than the endogenous U1 snRNA (Fig. 3A). Agarose gel analysis of the products confirmed the expected amplicon sizes, low expression of the variants from their endogenous loci, and improved expression in HeLa cells upon transfection with the pNS6vU1 plasmids (Fig. 3B). These results suggest that the presence of the functional regulatory elements from the RNU1 gene supports robust expression of a few but not all U1 variants. Notably, the high expression levels of the exogenous vU1s did not have any observable effects on the proliferation of cells and also did not affect the yield of total RNA.
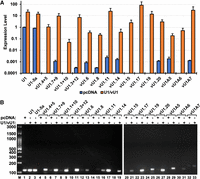
Context of the RNU1-1 gene regulatory elements improves the expression of vU1 snRNAs in HeLa cells. (A) Expression levels of vU1 snRNAs, calculated relative to the endogenous canonical U1 after normalization to the U2 snRNA (n = 3), are shown for HeLa cells transfected with either pcDNA or pNS6U1 plasmids harboring vU1 genes. (B) Agarose gel analysis of amplified RT-qPCR products for HeLa cells transfected with either pcDNA or pNS6U1 plasmids harboring vU1 genes. Sizes of fragments in a 100 bp DNA ladder are indicated. Sequences of primer sets used for RT-qPCR are provided in Supplemental Table S2.
Splicing activity of vU1 snRNAs
To test the capacity of the U1 snRNAs to catalyze pre-mRNA splicing, we applied the U1 genetic complementation assay that uses the 3-exon/2-intron Dup51p reporter (Fig. 4A; Sharma et al. 2014; Martelly et al. 2021; Wong et al. 2021; de Vries et al. 2022). Dup51p minigene carries mutations in the 5′-ss sequence of the second intron resulting in the skipping of exon 2 in the mature transcript in HeLa cells as measured by primer extension (Fig. 4C, lane 1). The effects of these 5′-ss mutations can be reversed by a compensatory U → A mutation at the fifth position in the 5′-region (nucleotides 1–11) of the U1 snRNA (U1-5a) that base pairs with the pre-mRNA (Fig. 4A,B). Coexpression of the U1-5a snRNA with the Dup51p reporter rescues exon 2 inclusion in the mature mRNA, whereas the canonical snRNA does not (Fig. 4C, lanes 2,3).
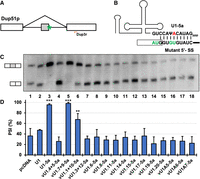
Primer extension analysis for measuring the capacity of vU1 snRNAs to facilitate splicing in HeLa cells. (A) Schematic diagram of the Dup51p minigene mRNA reporter indicating the location of the Dup3R primer that was used for primer extension. Dup51p pre-mRNA carries 5′-ss sequence mutations in intron 2 (indicated by the green asterisk) that cause exon 2 skipping. (B) Base-pairing of the 5′-region of the U1-5a snRNA to the 5′-ss of the Dup51p reporter. Changes to the intron 2 5′-ss sequence are shown in green. The U1-5a snRNA carries a compensatory U → A change at the fifth position (shown in red). (C) Primer extension analysis to monitor splicing of the Dup51p reporter pre-mRNA in HeLa cells coexpressing either pcDNA control or U1-5a plasmids expressing either the canonical or a vU1 snRNA. The full-length and exon 2 skipped Dup51p mRNA products are depicted to the left. (D) The percent spliced-in (PSI) value for exon 2 in the full-length Dup51 mRNA (±SD) is represented. Statistical significance was determined by comparisons to the wildtype control using t-test (lane 2). n = 3; (*) P < 0.05, (**) P < 0.01, (***) P < 0.001.
In the case of five vU1s used in this study, including vU1.7 + 9, vU1.1 + 10, vU1.6, vU1.14, and vU1.19, the 5′-region that base pairs with the pre-mRNA is identical to the canonical U1 (Fig. 1). Others carry an alteration in this region at either a single nucleotide (vU1.15, vU1.17, and vU1A7) or at two or more positions (vU1.4 + 5, vU1.3 + 12, vU1.8, vU1.11, vU1.20, vU1A5, and vU1A6) (Fig. 1; Supplemental Table S1). To facilitate binding of the variants to the Dup51p reporter transcript, we introduced the U → A mutation at the fifth position in vU1.7 + 9, vU1.1 + 10, vU1.6, vU1.14, and vU1.19. For the other vU1s, the 5′-region (nucleotides 1–11) was replaced with the sequence from U1-5a snRNA (Fig. 4B). These constructs were coexpressed with the Dup51p reporter in HeLa cells and assessed for their capacity to rescue exon 2 inclusion (Fig. 4C). Primer extension analysis revealed that only two of the vU1s, vU1.7 + 9-5a and vU1.1 + 10-5a, exhibited measurable activity and most variants were severely compromised in their ability to facilitate splicing catalysis. While vU1.7 + 9-5a rescued exon 2 inclusion similar to the level seen for the canonical U1-5a, vU1.1 + 10-5a was partially active (Fig. 4C, lanes 5,6). Notably, vU1.1 + 10 was expressed at a level significantly lower, at only 5% of the canonical U1 snRNA; however, it caused an increase in exon 2 inclusion from ∼36% to ∼67% (Figs. 3A and 4C, lane 6). All other variants failed to rescue exon 2 inclusion (Fig. 4C, lanes 7–18). In the case of vU1.6, vU1.8, vU1.14, vU1A5, and vU1A6, lack of activity may be due to the lower levels of expression (Fig. 3A). Although other variants, including vU1.4 + 5, vU1.3 + 12, vU1.11, vU1.15, vU1.17, vU1.19, vU1.20, and vU1A7, were expressed at levels similar to or higher than the endogenous U1 snRNA, they were unable to rescue splicing. Thus, these results show that despite changes in the 5′-region sequence to facilitate complementarity to the 5′-ss of the Dup51p reporter and the increased expression, many variant snRNAs do not exhibit the ability to support splicing activity. Notably, a previous study by Roca and Krainer that also utilized a splicing reporter-based assay showed the inability of vU1A7 to support splicing (Roca and Krainer 2009). It is likely that sequence alterations in regions other than the 5′-region (SL1, SL2, SL3, SL4, helix H, and the Sm site) may be affecting the structure and interactions for cellular processing to a mature snRNP and splicing catalysis (Fig. 1; Supplemental Table S1).
Variant U1-5a snRNAs exhibit nuclear localization
To determine the subcellular localization of the snRNAs, we performed nuclear–cytoplasmic fractionation of HeLa cells expressing vU1-5a snRNAs using a protocol that we have previously applied to demonstrate nuclear localization of U1-5a snRNAs carrying mutations in SL3 and SL4 (Martelly et al. 2021). Levels of snRNAs in the nuclear and cytoplasmic fractions were assessed by RT-qPCR (Fig. 5). For this analysis, the two variants, vU1.7 + 9 and vU1.1 + 10, that were found to facilitate pre-mRNA splicing and others that lacked this ability were selected. Of the inactive variants, vU1.15, vU1.17, vU1.19, and vU1A7 were chosen as upon transfection with the pNS6U1 constructs, they were found to be expressed at levels similar to or higher than the endogenous U1 snRNA in HeLa cells (Fig. 3A). These vU1 snRNAs were also reported to be processed and assembled into snRNPs and spliceosomes in previous studies (Kyriakopoulou et al. 2006; Mabin et al. 2021). Examination of snRNA levels in the total and nuclear RNA relative to the cytoplasmic fraction showed significant enrichment of the canonical U1, U1-5a, and the variant snRNAs in the nuclear fractions (Fig. 5A). Agarose gel analysis of the products confirmed higher levels of the canonical and variant snRNAs in the nuclear fractions relative to the cytoplasmic fractions from the transfected cells (Fig. 5B). Thus, these results indicate that a significant fraction of the exogenously expressed vU1 snRNAs is localized in the nucleus.
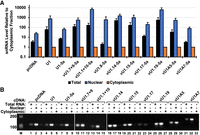
Analysis of snRNA nuclear localization in HeLa cells. (A) RT-qPCR analysis of canonical and variant U1 snRNAs in nuclear and cytoplasmic fractions from HeLa cells cotransfected with Dup51p reporter and either pcDNA or U1-5a constructs for canonical and variant U1 snRNA. The level of snRNAs in the total or nuclear fraction RNA was calculated relative to the cytoplasmic fraction (±SD; n = 3). (B) Agarose gel analysis of the RT-qPCR products from total, nuclear, and cytoplasmic RNA fractions. Sizes of fragments in a 100 bp DNA ladder are indicated. Sequences of primer sets used for RT-qPCR are provided in Supplemental Table S2.
A variant U1 with higher U1-70K binding
Of the two vU1 snRNAs that exhibited splicing activity, vU1.7 + 9 differs from the canonical U1 snRNA at a single nucleotide, that is, U150C. Previously, we have shown that U150 is not involved in the binding of the UBL domain of SF3A1 to SL4, and the U150C change does not affect the binding affinity of this interaction (de Vries et al. 2022). The second variant, vU1.1 + 10, which was found to be partially active, differs from the canonical U1 snRNA at 6 nt positions (Fig. 6A; Supplemental Table S1). To see if these changes in SL1, SL3, and SL4 would affect the binding of the stem–loops to their interacting proteins, we performed RNA-affinity purification (RAP) assays using biotinylated RNAs and HeLa nuclear extracts (Fig. 6B). The effect of C53A base change on the binding of SL2 to U1A was not tested as other studies have demonstrated the lack of C53's involvement in this interaction (Bach et al. 1990; Kormos et al. 2011). Western analysis of the RAP complexes showed that the canonical SL3 bound specifically to UAP56 and SL4 bound specifically to SF3A1 (Fig. 6B, lanes 7,11). However, compared to SL3 and SL4 of the canonical U1 snRNA, binding of vU1.1 + 10-SL3 and -SL4 to UAP56 and SF3A1, respectively, was significantly reduced (Fig. 6B, lanes 7,8 and 11,12, and Fig. 6C). Surprisingly, binding of U1-70K to vU1.1 + 10-SL1 was fourfold higher than to the canonical SL1 (Fig. 6B, lanes 3,4, and Fig. 6C). The SL1 of vU1.1 + 10 carries a C25G sequence change. The RAP analysis suggests that this single nucleotide alteration improves the binding of U1-70K to vU1.1 + 10-SL1.
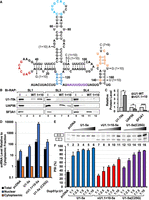
Analysis of vU1.1 + 10 associated sequence alterations on interactions with U1-specific proteins and splicing. (A) Secondary structure of U1 snRNA indicating sequence alterations in vU1.7 + 9 and vU1.1 + 10. (B) Western analysis of RAP complexes isolated using biotinylated SL1, SL3, and SL4 RNAs from canonical U1 and vU1.1 + 10. (C) Levels of U1-70K, UAP56, and SF3A1 in the vU1.1 + 10-SL1, -SL3, and -SL4 complexes were calculated relative to stem–loop complexes of the canonical U1 snRNA. n = 3; (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. (D) RT-qPCR analysis of canonical snRNAs in nuclear and cytoplasmic fractions from HeLa cells cotransfected with Dup51p reporter and either pcDNA or U1-5a constructs for canonical and variant U1 snRNA. The level of snRNAs in the total or nuclear fraction RNA was calculated relative to the cytoplasmic fraction (±SD; n = 3). (E) Primer extension analysis to monitor splicing of the Dup51p reporter pre-mRNA in HeLa cells coexpressing either pcDNA control or plasmids for either the canonical U1-5a, vU1.1 + 10-5a, or U1-5a(C25G) snRNA. PSI at increasing U1:Dup51p ratios were tested. The full-length and exon 2 skipped Dup51p mRNA products are depicted to the left. (F) PSI values for exon 2 in the full-length Dup51p mRNA (±SD) are represented (n = 3). Statistical significance was determined using t-test.
We next wanted to see if the C25G-associated higher binding of U1-70K to vU1.1 + 10-SL1 enhances the ability of a U1 snRNA carrying this alteration to facilitate pre-mRNA splicing. For this, we introduced C25G change in the U1-5a snRNA and tested its activity by cotransfecting with the Dup51p reporter in HeLa cells. Subcellular fractionation and RT-qPCR analysis confirmed nuclear enrichment of the U1-5a(C25G) snRNA (Fig. 6D). Primer extension analysis showed that the ability of U1-5a(C25G) to support splicing was better than that of vU1.1 + 10 but it was not higher than the canonical U1 (Fig. 6E,F, compare lanes 12–16 to 7–11 and 2–6). Thus, even though the C25G change increases U1-70K binding to SL1, the capacity of a U1 snRNA carrying this alteration to support pre-mRNA splicing is comparable but does not exceed that of the canonical U1 snRNA.
DISCUSSION
Expression of the vU1 snRNAs in the context of the functional regulatory elements from the RNU1 gene improved the expression levels of some but not all variants. Previous work has indicated the important contribution of transcription and RNA decay pathways in the maintenance of snRNA abundance (Shukla and Parker 2014; Lardelli and Lykke-Andersen 2020; Mabin et al. 2021). We found that even though expression of vU1.4 + 5, vU1.15, vU1.17, and vU1.19 was not detected from the genomic loci in HeLa cells, placing them in the context of the RNU1 regulatory elements significantly improved their expression levels to similar or greater than endogenous canonical U1. Although vU1.15 carries a U127C alteration in the Sm site, it has previously been shown to bind Sm proteins (O'Reilly et al. 2013; Mabin et al. 2021). Thus, the observed high levels of vU1.15 from the pNS6U1 construct could be resultant of its increased transcription. vU1A5 has also been shown to be capped with tri-methyl guanosine and bind Sm proteins in HeLa cells, although it harbors a G130A base change in the Sm site (Kyriakopoulou et al. 2006). However, the vU1A5 level achieved upon exogenous expression was not comparable to that of the endogenous U1 snRNA level (Fig. 3A). SL1 of the U1 snRNA and its interactions with U1-70K have been shown to be critical for the assembly of the Sm core and maintenance of U1 levels (Yong et al. 2002; So et al. 2016). Due to the SL1 sequence in vU1A5 being completely different from that of the canonical U1 snRNA, it is possible that the interaction of this variant with U1-70K is affected, and thus, preventing further maturation and stability (Fig. 1; Supplemental Table S1). Similarly, the large number of base changes in the SL1 of other variants, including vU1.6, vU1.8, and vU1A6, may affect their maturation and/or stability resulting in low levels in spite of the RNU1-1 gene context. Thus, different causes may contribute to the variable vU1 snRNA levels in HeLa and other cells.
Several of the vU1 snRNAs tested in this study, including vU1.15, vU1.17, vU1.19, and vU1A7, have been shown to be assembled into snRNPs and incorporated in spliceosomes (Mabin et al. 2021). However, other than vU1.7 + 9 and vU1.1 + 10, none of the vU1s tested exhibited the ability to support pre-mRNA splicing. This incapability, in our opinion, is most likely due to the many sequence alterations in the snRNA stem–loops that may be affecting their interactions with the U1-specific proteins and likely forming snRNPs with suboptimal capacity to interact with pre-mRNA and other spliceosome components. SL3 and SL4 do not bind a U1-specific protein but have been shown to interact with UAP56 and SF3A1, respectively, and it is likely that the various nucleotide changes can alter stem–loop structure and interactions (Sharma et al. 2014; Martelly et al. 2019, 2021; de Vries et al. 2022). Although the C25G base change did not result in an improvement in the splicing of the Dup51p reporter, the improvement in binding of U1-70K was significant. Modified U1 snRNAs carrying base changes to their 5′-regions are widely used for correction of adverse effects of mutant 5′-ss, and it may be worthwhile to test if the C25G base change in the engineered U1 snRNAs enhances splicing of other targets (Blázquez and Fortes 2015).
A role for the vU1 snRNA in regulating alternative pre-mRNA splicing has been suggested in several publications and it is possible that these alterations are resultant from reduced activity of the spliceosomes carrying vU1s. Another potential function of vU1 snRNAs may be regulation of gene expression via interaction with transcription factors in a manner mediated by the U1 snRNA–TFIIH complex (Kwek et al. 2002). The binding of U1 to promoter proximal 5′-ss has been found to increase recruitment of transcription factors and transcription initiation (Damgaard et al. 2008). Similarly, the binding of vU1 snRNAs to promoter regions may influence transcription. vU1.8 has been shown to inhibit polyadenylation and protect pre-mRNAs from premature cleavage and polyadenylation (O'Reilly et al. 2013). Other vU1s may have analogous telescripting functions, regulating the stability of a subset of nascent transcripts.
MATERIALS AND METHODS
Plasmid constructs
Plasmids for the Dup51p reporter and U1 snRNA expression (pNS6U1) have been described previously (Sharma et al. 2014; Wong et al. 2021). The pNS6U1 constructs expressing vU1 snRNAs were generated by in-fusion cloning (TaKaRa) using a linearized pNS6 plasmid and duplex fragments for vU1 snRNAs (Ultramer duplexes from Integrated DNA Technologies). Sequences for all vU1 plasmids were verified by Sanger sequencing.
Cell culture, transfection, and nuclear–cytoplasmic fractionation
HeLa cells were grown in DMEM containing 10% fetal bovine albumin and antibiotics (100 U/mL penicillin and 100 mg/mL streptomycin) and were periodically authenticated and tested for mycoplasma through the University of Arizona Genetics Core. For the U1 complementation assays, HeLa cells were seeded at 1.0 × 105 cells per well of a 12-well plate and incubated at 37°C overnight. The next day, Dup51p and pNS6U1/vU1 plasmids were cotransfected into HeLa cells at a ratio of 1:10 (0.2 µg of Dup51p and 2.0 µg of control pcDNA3.1 or pNS6U1/vU1 plasmid) and incubation was continued for an additional 48 h. Total RNA was extracted from the cells using the standard TRIzol–chloroform extraction protocol, treated with 10 units of DNase I per sample at RT for 20 min, and then re-extracted with phenol–chloroform (pH 4.5). For transfections with varying ratios of Dup51p:pNS6U1, pcDNA3.1 was added to make up the total amount of transfected DNA equal to 2.0 µg (Fig. 6E).
Subcellular fractionation of HeLa cells was performed using the previously described protocol (Gagnon et al. 2014; Martelly et al. 2021). HeLa cells were lifted from wells by trypsinization and pelleted by centrifugation at 300g for 5 min at RT. The cell pellets were resuspended with 1 mL of DMEM to deactivate trypsin. One-half of the pellet was used for extraction of total RNA using TRIzol, while the other half was resuspended in 300 µL of Igepal hypotonic lysis buffer (I-HLB) (10 mM Tris-HCl pH 7.5, 10 mM NaCl, 3 mM MgCl2, and 0.2% Igepal) and incubated on ice for 7.5 min. The nuclei were pelleted at 5000g for 10 min at 4°C, washed by resuspending in 100 µL of I-HLB, and the RNA was extracted using TRIzol. Supernatants containing the cytoplasmic fractions were treated with SDS/Proteinase K, and RNA was extracted using phenol:chloroform (pH 4.5). All RNA samples were treated with 10 units of DNase I, followed by cDNA synthesis and RT-qPCR, as mentioned above.
Primer extension and RT-qPCR
Primer extension to monitor splicing of the Dup51p reporter was performed using 32P-Dup3r primer (5′-AACAGCATCAGGAGTGGACAGATCCC-3′), as described previously (Sharma et al. 2014; Wong et al. 2021). The primer extension products were separated using a 10% urea–PAGE gel and visualized by the Typhoon FLA 9000 scanner. Densitometric scanning of the gel images was performed using the ImageQuant software, and PSI values for exon 2 were calculated using data obtained from three independent experiments. Statistical significance was determined by comparisons to the wildtype control using t-test (Fig. 4, lane 2) (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001).
For RT-qPCRs, cDNA was synthesized using 1.0 µg of RNA, random hexamers, and SuperScript III reverse transcriptase. RT-qPCR reactions were performed in triplicate using the SYBR Green Real-Time PCR master mixes and oligonucleotide pairs specific for the canonical U1, U2, or vU1 snRNAs (Supplemental Table S2) on a StepOnePlus Real-Time PCR System. RT-qPCR reactions were performed using 1.0 ng of cDNA, and oligonucleotide pairs specific for the canonical U1 or vU1 snRNAs and products were separated on 2.0% agarose gels and imaged by a Bio-Rad Gel Doc XR Imaging System.
RNA affinity purification and western analysis
Biotinylated stem‐loop RNAs (Bi-RNAs) were purchased from Integrated DNA Technologies. For RNA affinity purification (RAP), 2 nM of Bi-RNAs were prebound to 20 µL of Neutravidin beads at room temperature with end-over-end rotation for 1 h. The beads were washed four times with buffer DGN (20 mM HEPS-KOH pH = 7.9, 0.2 mM EDTA, 20% Glycerol, 80 mM potassium glutamate, 0.1 mM PMSF, 1 mM DTT, and 150 mM NaCl). The reaction mixture consisting of 50% HeLa cell nuclear extract, 2.2 mM MgCl2, and 10 units of RNaseOUT was added to the prebound beads, and incubation was continued at RT with rotation for an additional hour. The beads were again washed four times with buffer DGN and the bound protein was eluted by boiling at 95°C for 5 min in 1× SDS-PAGE loading buffer, separated on 10% SDS-PAGE gels, and transferred onto PVDF membranes that were probed with antibodies against U1-70K, UAP56, and SF3A1 (Sharma et al. 2014). The secondary anti-rabbit antibody conjugated to Cy5 fluorophore was purchased from Cytiva (catalog no. PA45011). The membranes were imaged with a GE Typhoon FLA 9000 Gel Scanner, and bands were quantified using ImageQuant.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by funds to S.S. from the National Institutes of General Medical Sciences (GM127464) and National Cancer Institute (P30CA023074) of the National Institutes of Health, the Valley Research Partnership Program (VRP: P1-4009 and VRP77), and the Arizona Biomedical Research Center (ABRC: 2022-010-30). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079892.123.
-
Freely available online through the RNA Open Access option.
- Received November 16, 2023.
- Accepted December 18, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








