
High-resolution reconstruction of a C. elegans ribosome sheds light on evolutionary dynamics and tissue specificity
- Enisha Sehgal1,6,
- Chloe Wohlenberg1,
- Evan M. Soukup1,
- Marcus J. Viscardi1,
- Vitor Hugo Balasco Serrão2,3 and
- Joshua A. Arribere1,4,5
- 1Department of MCD Biology, University of California at Santa Cruz, Santa Cruz, California 95064, USA
- 2Department of Chemistry and Biochemistry, University of California at Santa Cruz, Santa Cruz, California 95064, USA
- 3Biomolecular Cryoelectron Microscopy Facility, University of California at Santa Cruz, Santa Cruz, California 95064, USA
- 4RNA Center, University of California at Santa Cruz, Santa Cruz, California 95064, USA
- 5Genomics Institute, University of California at Santa Cruz, Santa Cruz, California 95064, USA
- Corresponding author: jarriber{at}ucsc.edu
-
Handling editor: Marina Rodnina
Abstract
Caenorhabditis elegans is an important model organism for human health and disease, with foundational contributions to the understanding of gene expression and tissue patterning in animals. An invaluable tool in modern gene expression research is the presence of a high-resolution ribosome structure, though no such structure exists for C. elegans. Here, we present a high-resolution single-particle cryogenic electron microscopy (cryo-EM) reconstruction and molecular model of a C. elegans ribosome, revealing a significantly streamlined animal ribosome. Many facets of ribosome structure are conserved in C. elegans, including overall ribosomal architecture and the mechanism of cycloheximide, whereas other facets, such as expansion segments and eL28, are rapidly evolving. We identify uL5 and uL23 as two instances of tissue-specific ribosomal protein paralog expression conserved in Caenorhabditis, suggesting that C. elegans ribosomes vary across tissues. The C. elegans ribosome structure will provide a basis for future structural, biochemical, and genetic studies of translation in this important animal system.
Keywords
INTRODUCTION
The ribosome is a fundamental feature of cellular life that decodes and regulates cellular information. Studies of protein synthesis across organisms continue to discover how ribosomes move across mRNAs, how ribosomes associate with and recruit machinery to RNAs, and how ribosomal functions are altered in disease states (for example, see Choi et al. 2018; Knight et al. 2020; Brito Querido et al. 2024). A powerful modern tool in the study of protein synthesis is the structure of the ribosome, yielding insight into the molecular surfaces and dynamics that underlie protein synthesis. While the core functions of the ribosome are conserved across life, sequence divergence in the associated machinery can confound comparisons across organisms. Thus, a given organism's ribosome structure represents an invaluable tool in the study of translation in that organism, and it also enables comparative translational analyses across organisms.
Caenorhabditis elegans is a powerful model organism with rich contributions to the study of translation and posttranscriptional gene regulation. As an intact animal with a fast generation time, it represents an opportunity to examine the contributions of translation and ribosomes to organismal development, reproduction, and health and fitness. Seminal work in C. elegans significantly and positively impacted the fields of microRNAs, translational surveillance, translational regulation and signaling, and other pathways (e.g., Hodgkin et al. 1989; Evans et al. 1994; Ambros 2008). C. elegans also contains several interesting mRNA and translational phenomena that merit investigation, such as spliced mRNA leaders, trimethyl-guanosine mRNA 5′ caps proximal to RNA hairpins, and surprisingly short poly(A) tails (Liou and Blumenthal 1990; Blumenthal 2012; Lima et al. 2017; Bernard et al. 2023).
Most commonly used model organisms boast high-resolution ribosome structures, but C. elegans does not, hampering a molecular understanding of C. elegans’ translational processes. The current best C. elegans ribosome model is 6.9 Å and is from in situ cryo-electron tomography (cryo-ET) studies using subtomogram averaging (Schiøtz et al. 2023), insufficient for high-resolution molecular understanding. Here we report the first high-resolution single-particle cryo-electron microscopy (cryo-EM) map of an endogenous C. elegans ribosome at an overall resolution (FSC0.143) of 2.63 Å. The map represents the highest resolution map reported to date from whole animals, and it will serve as a launching point for future translational studies in this organism. We describe unique features of the C. elegans ribosome, including evidence for tissue-specific ribosome heterogeneity.
RESULTS
Map and structure overview
We purified ribosomes from C. elegans via sucrose gradient fractionation. Given our interest in active protein synthesis, we used conditions that would enrich for actively translating ribosomes: we stabilized actively elongating ribosomes with the translation elongation inhibitor cycloheximide, and we also used buffer conditions that dissociate empty ribosomes (see Materials and Methods). We identified 286,689 ribosomal particles and obtained three classes via 3D Classification (Supplemental Fig. S1). All three classes yielded high-resolution consensus maps with tRNA density (2.59–2.80 Å), showing the success of our purification. We visually inspected the three maps, selected one with the most complete small subunit density, and proceeded with model building. The overall resolution (FSC0.143) of the map was 2.63 Å, with more than half of the map showing a local resolution better than 3.00 Å (Supplemental Fig. S2; Materials and Methods).
Using the map, we successfully modeled a majority of the C. elegans ribosome (Fig. 1; Supplemental Fig. S3). We started with the Drosophila melanogaster ribosome structure given its relative evolutionary distance (Supplemental Fig. S1; Anger et al. 2013). The model includes the 5S, 5.8S, 18S, and 28S ribosomal RNAs (rRNAs). The model covers ∼85% of all rRNA bases, encompassing the core of the eukaryotic ribosome, with the remaining ∼15% being the P- and L-stalks as well as some rRNA expansion segments. We also modeled the ribosomal proteins, with the exceptions of those bound to the P- and L-stalks (uL10/P0, P1, P2, uL1, uL11), eL28, and RACK-1 (Fig. 1A; Table 1; Supplemental Table S1). The latter two were conspicuously absent on the map (see below). For modeled ribosomal proteins, we observed density for an average of 90% of all amino acids, with flexible N- and C-termini being less commonly seen in our map. Lastly, we observed density that allowed us to model A- and P-site tRNAs, mRNA in the A-, P-, and E-sites, as well as cycloheximide and spermidine near the 60S E-site (Fig. 1B,C).
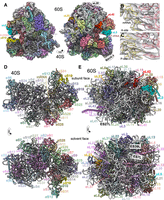
High-resolution structure of a C. elegans ribosome. (A) Segmentation of the reconstructed map of the C. elegans ribosome. 28S rRNA in white, 18S rRNA in gray, A- and P-site tRNAs in pink, and ribosomal proteins are colored. Highlighted in brighter colors and proteins noted: uS19 (yellow), uL5 (blue), uL23 (orange), eL42 (red). RACK-1 and eL28 were not observed, but their approximate locations are indicated. Panels B and C show the zoom of map (light gray) and model (mRNA in yellow and tRNA in pink) for the A- and P-sites, respectively. Note that map heterogeneity is expected in the A- and P-sites as ribosomes were prepared from whole C. elegans and would be expected to contain a diversity of sequences. (D) Atomic model of the C. elegans 40S, with coloring as in A and ribosomal features indicated. Top panel shows the 60S-facing side, with mRNA in light yellow. Bottom panel shows the solvent-facing side. tRNAs hidden for clarity. (E) Atomic model of the C. elegans 60S, with coloring as in A. Top and bottom panels show 40S- and solvent-facing sides, respectively. Expansion segments (ES) 27L, 7L, and 39L are highlighted. tRNAs were omitted from (D) and (E).
CryoEM data collection, single-particle reconstruction maps, and model statistics
The C. elegans ribosome is exceptional in its small size among animal ribosomes. The C. elegans ribosome has an overall mass of ∼3.2 MDa, substantially less than either D. melanogaster (3.7 MDa) or humans (4.0 MDa) (Fig. 2A,B). The C. elegans ribosome is smaller due to loss of mass in its proteins and rRNAs (Fig. 2B). The compaction of the C. elegans ribosome is especially noticeable in the 28S rRNA, which is ∼150 kDa and ∼500 kDa lighter in C. elegans than D. melanogaster and humans, respectively. The 28S rRNA loss mostly occurs in expansion segments, where 7–639 nt of rRNA was lost per segment (Fig. 2C).
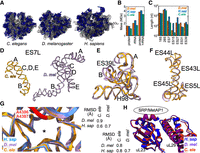
The C. elegans ribosome is streamlined but conserved. (A) Ribosomes from the indicated organisms are shown, with ribosomal proteins in blue and ribosome RNA in gray. D. mel and human structures from Anger et al. (2013), with eEF2 removed. (B) Sizes of the protein, rRNA, and both components of the ribosome in the respective organisms. We included masses for eL28, RACK-1, and the stalk proteins in C. elegans. For rRNA, we included masses for the expansion segments, regardless of whether they were present in the respective models. Similarly, we report the mass for full-length ribosomal proteins, regardless of the presence or absence of N- and C-termini in the maps. (C) Lengths in nucleotides of the indicated rRNAs or 28S expansion segments. As in B, we report the length of the entire expansion segment, not just the segments in the respective models. (D) ES7L from C. elegans (orange) and D. melanogaster (purple). The A, B, C, D, and E arms of ES7L are indicated. (E) ES39L from C. elegans (orange) and D. melanogaster (purple), aligned in three-dimensional space, with A and B arms and Helix 98 indicated. (F) Same as E for ES5L and ES43-45L. (G) Alignment of 28S rRNA within 20 Å of the peptidyl transfer center of the human (blue), Drosophila (purple), and C. elegans (orange) ribosomes. Asterisk indicates the location of the 3′ end of the P-site tRNA. Two universally conserved adenosines (human A4396 and A4397) are highlighted in red. RMSD table measuring 3D similarity for the PTC from the indicated species shown at right. (H) 3D alignment of uL23 and uL29, with the binding site of protein maturation and secretion machineries shown. Color scheme and RMSD table as in G.
Inspection of the reduced regions reveals a streamlined ribosome with a loss of expansion segments and associated ribosomal protein regions. One striking example is the 28S rRNA expansion segment 7L (ES7L). The C, D, and E arms of ES7L are reduced to a mere ∼22 nt in C. elegans, compared with ∼90 nt in D. melanogaster and ∼310 nt in humans (Fig. 2D; Supplemental Fig. S4). Ribosomal protein regions that bind ES7L are also reduced in C. elegans: the uL4 C-terminus and eL6 N-terminus are shorter (Supplemental Fig. S5A,B). Another example is a bulge in ES39L that is lost in C. elegans; the associated eL14 C-terminus is lost as well (Supplemental Fig. S5C). Still other ribosomal proteins were pared down without an obvious change in their nearby environment, for example, eL29, the C-terminus of which is 14 and 97 amino acids shorter in C. elegans compared to D. melanogaster and humans, respectively (Supplemental Fig. S5D). A handful of ribosomal proteins (uS19, eS21, eS31) gained unstructured additions of a few amino acids near their termini, and one protein (uL5) gained an alpha helix at its N-terminus (Supplemental Fig. S6).
Despite reductions, key expansion segment features are conserved. For example, ES27L, important for protein secretion, fidelity, and Ebp1-association, is conserved in C. elegans (182 nt), albeit shorter than that of D. melanogaster (234 nt) (Fujii et al. 2018; Shankar et al. 2020; Wild et al. 2020). ES39L, which binds to SRP and is important for protein secretion, is reduced in size in C. elegans (141 nt) compared to Drosophila (185 nt) despite occupying a similar 3D space (root mean square deviation [RMSD] 1.3 Å) (Fig. 2E; Halic et al. 2004). Similarly, another cluster of helixes (ES5L, ES45L, ES43L, ES44L) important for ribosomal stalling recognition by Gcn1 are conserved despite being seven nucleotides shorter (RMSD 1.3 Å) (Fig. 2F; Pochopien et al. 2021).
Many other structural characteristics of the C. elegans ribosome are also highly conserved. The overall structure of the peptidyl transferase center, the mRNA binding cleft, and the peptide exit tunnel are all highly conserved (Fig. 2G; Supplemental Fig. S7; Jomaa et al. 2022; Samatova et al. 2024). Interaction interfaces with protein secretion machinery are also conserved (Fig. 2H; Voorhees and Hegde 2015; Gamerdinger et al. 2023). Thus, despite variation and some reduction, many of the core structural features of the ribosome that pertain to protein synthesis are conserved in C. elegans.
Altogether, we present the first, high-resolution reconstructed model of a C. elegans ribosome, revealing a streamlined yet conserved animal ribosome. The structure will be a useful starting point for future structure-based ribosomal studies in this organism.
Conservation of cycloheximide mechanism
Cycloheximide binds eukaryotic ribosomes in the E-site and blocks hybrid formation and translocation (Garreau de Loubresse et al. 2014; Myasnikov et al. 2016; Shen et al. 2021). We examined our maps to better understand the C. elegans ribosome structure in the presence of cycloheximide.
Our heterogeneous refinement identified three states (Supplemental Fig. S1), two of which were intact 80S ribosomes, and the last of which contained poor SSU density and will not be discussed further. The two 80S classes fit clearly into two states (Supplemental Fig. S1). The first of these (State 1) encompassed 50.9% of particles and contained cycloheximide in the E-site, peptidyl-tRNA in the P-site, and an empty A-site (Fig. 3A,B). The second state (State 2) encompassed 34.2% of particles and contained cycloheximide in the E-site, a deacylated tRNA in the P-site, and a peptidyl-tRNA in the A-site (Fig. 3C,D). Both State 1 and State 2 ribosomes are in the classical PRE conformation (Fig. 3E). State 2 is most similar to a published cycloheximide-bound Neurospora crassa ribosome structure, with the A- and P-site tRNAs superimposible (RMSD < 1.0 Å, Supplemental Fig. S8). The existence of both States 1 and 2 supports the idea that cycloheximide inhibits tRNA translocation rather than A-site reactivity, as put forth previously (Shen et al. 2021).
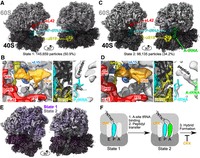
Cycloheximide stabilizes classical PRE ribosomes in C. elegans. (A) Single-particle cryoEM reconstructed map of State 1, with indicated features highlighted; large subunit in light gray and small subunit in dark gray. (B) Highlight of indicated ribosome features. (Left) Cycloheximide (CHX, orange) and spermidine (SPD, yellow) bound near the E-site at eL42 (red) and uL15 (blue). (Middle) uS19 C-terminal tail (yellow) in between the P- and A-sites. (Right) Peptidyl transferase center with 3′OH of P-tRNA in blue and path of the nascent peptide indicated. (C) Single-particle cryoEM reconstructed map of State 2, with indicated features highlighted. (D) As in B, but for State 2. (E) Overlay of State 1 (purple) and State 2 (gray), showing high similarity throughout the map. State 1 large subunit in light purple, and small subunit in dark purple. (F) Model of ribosomal states obtained via single-particle cryo-EM. CHX, cycloheximide. P-site tRNA is in cyan, A-site tRNA is in green, and the peptide is attached as indicated.
We noted the conservation of several aspects of the cycloheximide-bound ribosome. Cycloheximide is bound in a small pocket made by eL42 and uL15 (Fig. 3B,D; Garreau de Loubresse et al. 2014; Myasnikov et al. 2016; Shen et al. 2021). Within this pocket, we also noticed density consistent with spermidine (Fig. 3B,D), as noted in N. crassa (Bhaskar et al. 2020; Shen et al. 2021). In the State 2 structure, we noticed the C-terminal tail of uS19 reaching between the P- and A-site tRNAs in the vicinity of the mRNA, consistent with the idea that uS19's tail stabilizes the P-site tRNA (Fig. 3D; Bhaskar et al. 2020; Shen et al. 2021). The uS19 tail was disordered in the State 1 structure (Fig. 3B). As in the cycloheximide-bound ribosome from N. crassa and humans, the L1 and P1 stalks were disordered, supporting the idea that cycloheximide fails to stabilize the stalks in a rigid conformation (Supplemental Fig. S9; Fujii et al. 2018; Shankar et al. 2020). For this reason, we expect that future work without cycloheximide will more easily yield structures of the stalks and associated proteins uL10/P0, P1, P2, uL1, and uL11.
Generally, our analysis of the C. elegans cycloheximide-bound structure shows that the binding and mechanism of cycloheximide is highly similar to other eukaryotes and that uS19 dynamics during elongation are conserved between C. elegans, N. crassa, and humans (Garreau de Loubresse et al. 2014; Myasnikov et al. 2016; Bhaskar et al. 2020; Shen et al. 2021).
RACK-1 and eL28
We noted a conspicuous absence of density for two ribosomal proteins: eL28 and RACK-1 (Fig. 1A). Our analyses support the idea that homologs of both proteins exist in C. elegans, but that they dissociated from the ribosome during complex preparation.
In eukaryotes, eL28 stabilizes a helix of ES7L of the large subunit and is adjacent to eL18, eL32, and uL4. eL18, eL32, and uL4 were well-ordered in our map, though the portion of ES7L that binds to eL28 was not (ES7LA, 445–513 nt, Fig. 4A). While we identified a C. elegans homolog for eL28 (rpl-28), it is notable in that it has a low sequence identity to homologs in D. melanogaster and humans (Fig. 4B). C. elegans’ rpl-28 mRNA is expressed at a similar level to other ribosomal protein genes (Fig. 4C,D). Thus, C. elegans expresses a substantially diverged eL28 homolog.
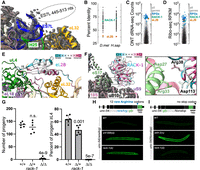
eL28 and RACK-1 are missing from the map, but exist in C. elegans. (A) Nt 445–513 of ES7L are poorly structured. The model is shown in colors, and the map density is in gray mesh. The approximate position of the missing segment of ES7L is indicated. In other organisms, this segment of ES7L binds eL28. (B) Conservation of ribosomal protein genes between C. elegans, D. melanogaster, and humans. The percent identity of each C. elegans ribosomal protein homolog is shown, with eL28 and RACK-1 highlighted. (C) mRNA expression of all genes (gray), ribosomal protein genes (blue), RACK-1 (green), and eL28 (red). mRNA expression was determined by Oxford Nanopore direct RNA-seq using published data (Viscardi and Arribere 2022). (D) mRNA translation of the indicated genes; same colors as C. mRNA translation was determined by ribosome footprint profiling (Ribo-seq) using published data (Stadler and Fire 2011). (E) eL28 conservation overlaid on eL28 binding site. eL28 was positioned in the ribosome using D. melanogaster as a model. eL28 protein model from AlphaFold (Varadi et al. 2022). Conservation data from consurf (Yariv et al. 2023), with more conserved residues in red and less conserved residues in blue. Note that ES7L would lay across the top of eL28, contacting the less-conserved regions of the protein. (F) Atomic model for RACK-1 binding in C. elegans, with map in gray mesh and model in colors. RACK-1 model from AlphaFold, with conservation colored as with eL28. Inset shows two salt bridges conserved in C. elegans that are known to be important for RACK-1::ribosome association in Saccharomyces cerevisiae. (G) Viability and developmental timing of the indicated strains. P-values are from Welch's t-test and are for comparison with wild-type (WT) animals; n.s., not significant. (H) unc-54(rareArg) reporter diagram and its expression in the indicated strains; the internal rareArg stretch stalls ribosomes and elicits mRNA decay (blue triangle). znf-598 is a positive control that is required for repression of the unc-54(rareArg) reporter; WT. (I) unc-54(Nonstop) reporter diagram and its expression in the indicated strains; the lack of an in-frame stop codon leads to ribosomal stalling at the 3′ end (triangle), eliciting mRNA repression, and causing low GFP levels. skih-2 is a positive control that is required for the repression of the unc-54(Nonstop) reporter.
To determine if C. elegans RPL-28 could plausibly bind to the ribosome, we placed an AlphaFold structure at the eL28 binding site, and overlaid conservation data (Fig. 4E; Varadi et al. 2022; Yariv et al. 2023). These data revealed that the portions of RPL-28 that would contact eL32, eL18, and uL4 are conserved, while the more solvent-exposed and ES7L-facing side is not. Taken together, these results support the idea that C. elegans produces a protein homologous to eL28, but that its association with ribosomes is unstable under the conditions examined. The reasons for this are unclear, though we note that ES7L is known to be highly variable (Wang et al. 2021; Fan et al. 2022), other ribosomal proteins that contact ES7L diverge in C. elegans (Supplemental Fig. S5A,B), and that eL28 is not universal across eukaryotes (Matzov et al. 2020). As ES7L has roles in ribosome biogenesis and selenocysteine incorporation in other organisms (Jeeninga et al. 1997; Ramesh and Woolford 2016; Wang et al. 2021), these processes may be impacted in C. elegans.
Another ribosomal protein conspicuously absent from our map is the small subunit protein RACK-1. RACK-1 is well-known to exchange with solution, and prior cryoEM ribosome structures in some protozoan eukaryotes failed to observe density for RACK-1 (Wong et al. 2014b; Eiler et al. 2024). C. elegans contains an obvious and robustly expressed homolog of RACK-1 (Fig. 4B–D) that conserves sites important for RACK-1::ribosome binding in S. cerevisiae (Fig. 4F; Thompson et al. 2016).
As with eL28, we entertained the idea that RACK-1 is produced and normally binds ribosomes, but that its ribosomal association is unstable under the conditions used. To test this idea, we analyzed in vivo translational processes that, in other organisms, require RACK-1. RACK-1 is functionally important for some translational surveillance pathways, and it performs a structural role at the interface between collided ribosomes (Ikeuchi and Inada 2016; Sundaramoorthy et al. 2017; Juszkiewicz et al. 2018). Interestingly, while loss of many ribosomal proteins is inviable in C. elegans, loss of RACK-1 is viable though we did note developmental delays and decreased fecundity (Fig. 4G; Chu et al. 2014; Marudhupandiyan et al. 2017; Cenik et al. 2019). The availability of a loss-of-function rack-1 mutant enabled us to test RACK-1's requirement in translational surveillance pathways, thus inferring its presence on ribosomes in vivo.
Saccharomyces cerevisiae Asc1p/Rack1p is required for mRNA cleavages in response to ribosomal stalls (Ikeuchi and Inada 2016; Sundaramoorthy et al. 2017; Juszkiewicz et al. 2018). We therefore examined a C. elegans reporter [unc-54(rareArg)] that stalls ribosomes at an internal poly-arginine stretch (Monem et al. 2023). Ribosomes that stall at the poly-arginine stretch trigger No-Go mRNA Decay; mutant backgrounds that fail to efficiently recognize ribosome stalls exhibit higher GFP as ribosomes have more time to translate through the stall and produce downstream GFP. Thus, No-Go mRNA Decay activity is inversely related to GFP levels in the unc-54(rareArg) reporter background. rack-1 animals exhibited substantial unc-54(rareArg) de-repression, consistent with a deficiency in the cellular response to ribosome collisions (Fig. 4H). A second reporter [unc-54(Nonstop)], which measures repression of mRNAs without stop codons (Arribere and Fire 2018; Glover et al. 2020), was efficiently repressed in rack-1 animals (Fig. 4I). Thus, C. elegans RACK-1 has specific effects at internal, translation elongation stalls, but not ribosomal stalls at the 3′ ends of mRNAs.
The conservation, expression, and functional data support the idea that RACK-1 is functionally bound to ribosomes in vivo, but that its association is insufficient to survive our ribosome preparation protocol.
Conservation and divergence in ribosomal protein paralogs
In the course of building the C. elegans ribosome, we noticed five cases where C. elegans contains a pair of ribosomal protein paralogs, corresponding to P2, uL5, uL23, eL37, and eL41. Our map contained well-defined density for the last four. We describe our observations surrounding each here, from most to least conserved.
C. elegans eL41 is encoded by two tandemly duplicated genes on chromosome one, rpl-41.1 and rpl-41.2. Since they encode an identical protein, neither can be definitively identified as present in our map over the other. However, nucleotide divergence enabled us to quantify mRNA expression, which showed that rpl-41.2 is >10-fold more highly expressed than rpl-41.1 (Fig. 5A,B). We also noticed a report of tandem duplication of the region containing rpl-41.1 and rpl-41.2, indicating its genomic neighborhood may be prone to duplication (Thompson et al. 2013).
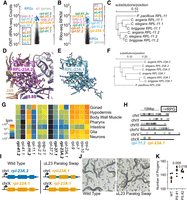
C. elegans uL5 and uL23 are encoded by tissue-specific paralogs. (A and B) Expression data as in Figure 4C,D, re-colored to indicate the ribosomal proteins with paralogs. (C) Conservation of uL5 paralogs throughout Caenorhabditis. C. briggsae, C. angaria, and C. elegans are three representative Caenorhabditis species, and P. pacificus (a more distantly related nematode) is an outgroup. Phylogenetic tree made with CLUSTAL Omega. (D) RPL-23A.2 within the C. elegans ribosome structure, with all side chains shown, colored by heteroatom. (E) AlphaFold model of RPL-23A.1 with side chains shown in the same orientation as RPL-23A.2. Note the conservation of side chains that contact nearby rRNA and uL29 sites in the ribosome. (F) As in C, but with uL23 paralogs. (G) Heatmap of mRNA expression from single-cell RNA-seq from L2 animals, binned by tissue. Four constitutive, nonparalogous ribosomal proteins are shown for reference (rps-2, rps-10, rpl-7, rpl-9). The remaining ribosomal proteins are presented in pairs, with the bold gene encoding the protein most present in our map; tpm, transcripts per million. The low counts for rpl-37.1/2 and rpl-41.1/2 are likely an artifact of their high nucleotide identity, leading to multimapping reads that are filtered in the scRNA-seq pipeline (Cao et al. 2017). (H) Genomic location of ribosomal protein genes. Each gray dot represents the position of a single ribosomal protein gene in the genome. Soma-specific paralogs on the X-chromosome are indicated. (I) Schematic of paralog swap experiment. WT animals contain rpl-23A.2 (blue) on chromosome one and rpl-23A.1 (orange) on the X-chrosome. In “uL23 Paralog Swap” animals, the coding sequence of the rpl-23A.2 gene has been mutated to that of rpl-23A.1 (orange) while preserving the noncoding sequences of rpl-23A.2 (blue). (J) Photographs of WT and uL23 paralog swap animals. (K) Brood size analysis of WT and uL23 paralog swap (PS) animals. Two independently derived isolates of the rpl-23A.2 gene replacement were tested, “#1” and “#2.” Each dot represents a single animal; average of that strain is given by the red line. Note the y-axis scale starts at 100. P-value from Welch's t-test for comparison with WT.
eL37 has two sequence paralogs in C. elegans, RPL-37.1 and RPL-37.2. The two genes are highly similar: 97.8% and 98.6% identical at the amino acid and nucleotide levels, respectively. Despite the high identity, our map contained clear density supporting RPL-37.1 as the more abundant (Supplemental Fig. S10). mRNA expression data indicates that rpl-37.2 is expressed, albeit lower than rpl-37.1 (Fig. 5A,B); we also noticed that rpl-37.1 contains an intron, whereas rpl-37.2 does not. Searches for homologs in closely related Caenorhabditis identified a single protein most closely related to RPL-37.1. Based on these data, we speculate that rpl-37.2 is derived from a recent retrotransposition of rpl-37.1, and that rpl-37.2 is in the process of becoming a pseudogene of rpl-37.1.
uL5 is potentially encoded by two paralogs, RPL-11.1 and RPL-11.2, that are highly similar at the amino acid level (91% identity, 96% similarity). Similarity between the two is lower at the nucleotide level (84.1% identity), with a number of synonymous substitutions, indicating coding conservation of both homologs. Expression analysis detected both rpl-11.1 and rpl-11.2 at the mRNA and translational level (Fig. 5A,B), and knockdown of either genes’ mRNA is reported to be lethal (Rual et al. 2004; Sönnichsen et al. 2005). Sequence homologs of both RPL-11.1 and RPL-11.2 are present throughout the Caenorhabditis lineage (Fig. 5C), indicating the conservation of the two paralog structure. Despite the high degree of sequence similarity, the handful of different amino acids allowed us to identify RPL-11.2 as being most present in our maps (Supplemental Fig. S10). Because the handful of RPL-11.1 positions that differ from RPL-11.2 are conservative substitutions, the rpl-11.1 mRNA is well-expressed, and the RPL-11.1 protein is conserved in Caenorhabditis, we hypothesize that RPL-11.1 could also function in the ribosome.
uL23 encodes a large subunit protein that binds near the peptide exit tunnel and facilitates protein maturation and trafficking (Voorhees and Hegde 2015; Denks et al. 2017). C. elegans contains two uL23 paralogs: RPL-23A.1 and RPL-23A.2, which are 70% identical (79% similar). RPL-23A.2 is most present in our map, though we also observed less abundant density in support of RPL-23A.1 (Supplemental Fig. S10). Sites that interface with the ribosome and the signal recognition particle are conserved in both paralogs (Fig. 5D,E), RPL-23A.1/2 paralog pairs are conserved within the Caenorhabditis lineage (Fig. 5F), and knockdown of either genes’ mRNA is lethal (Ceron et al. 2007; Xiao et al. 2022). These results suggest that RPL-23A.1 and RPL-23A.2 have distinct and nonoverlapping roles within Caenorhabditis, and that both are likely required for normal translation.
We also examined single-cell transcriptomics to determine if the composition of the C. elegans ribosome varies across tissues (Cao et al. 2017). Paralogous ribosomal protein mRNA expression at the L2 larval stage showed similar trends to the RNA-seq and Ribo-seq data from the L4 stage (Fig. 5G). For both uL5 (RPL-11.2) and eL37 (RPL-37.1), the paralog present in our map was also more highly expressed at the mRNA level across tissues. However, we noticed substantial variability across tissues for uL23 (rpl-23A.1 and rpl-23A.2) and uL5 (rpl-11.1 and rpl-11.2). For example, rpl-23A.2 mRNA is 18-fold more abundant than rpl-23A.1 in the gonad, while rpl-23A.1 mRNA is greater than twofold more abundant than rpl-23A.2 mRNA in the pharynx. Similarly, rpl-11.1 is highly biased for gonad-specific expression, whereas rpl-11.2 is somatic. As the majority of cells in a C. elegans population are somatic, we expect that our ribosomal map is mostly somatic ribosomes, explaining the predominance of RPL-11.2 and RPL-23A.2 in our maps.
We hypothesized that the differences in gonad-specific expression may result from differences in the genes’ chromosomal locations. We therefore examined the location of all C. elegans ribosomal protein genes across the genome. Ribosomal protein genes were almost exclusively autosomal. Two exceptions were rpl-11.2 and rpl-23A.1; both are on the X-chromosome. Since the X-chromosome is repressed in the C. elegans gonad (Strome et al. 2014), gonad expression is driven from the corresponding autosomally-located paralogs, rpl-11.1 and rpl-23A.2, respectively.
We next asked the question of whether C. elegans’ ribosomal paralogs could functionally substitute for one other. Given their sequence divergence and divergent gonad expression, we focused on uL23. We used CRISPR/Cas9 to insert a synthetic version of the rpl-23A.2 gene (Fig. 5I; Supplemental Fig. S11). The synthetic rpl-23A.2 mutant contained WT rpl-23A.2 noncoding sequences (promoter, 5′/3′ UnTranslated Regions, and introns), so as to preserve the expression of the encoded protein. The synthetic rpl-23A.2 mutant's coding region was mutated to that of rpl-23A.1, so as to express the RPL-23A.1 protein. The resulting animals would be expected to express a single uL23 paralog, namely that encoded by the rpl-23A.1 locus. The resulting animals did not exhibit substantial phenotypic abnormalities and appeared grossly WT (Fig. 5J). However, the mutant exhibited a slight increase in brood size during the first day of adulthood. These results show that the RPL-23A.1 protein is sufficient to support animal life in the absence of RPL-23A.2, and also raise the possibility that there are functional differences between the RPL-23A.1/2 proteins.
Altogether, our analyses revealed two cases where paralogous ribosomal proteins diverged and acquired distinct expression profiles, and raise interesting questions about the functional consequences of distinct ribosomal protein paralogs.
DISCUSSION
The C. elegans cycloheximide-bound ribosome shows the conservation of the cycloheximide mechanism from fungi to animals. A cycloheximide structure from N. crassa captured ribosomes predominantly in a state resembling our State 2 (ribosomes with deacylated P-site tRNA and a peptidyl A-site tRNA) (Shen et al. 2021). In addition to State 2, we observed a distinct State 1 representing ribosomes with only a peptidyl P-site tRNA. The difference likely lies in preparation conditions: as the N. crassa study used cycloheximide in vivo, ribosomes in the cell would have an opportunity to accommodate an aminoacylated tRNA into the A-site prior to stalling. Here we added cycloheximide ex vivo, and we reason that the large fold dilution of translational components in our lysates prevented an additional peptide bond from forming, yielding the peptidyl-P-site tRNA State 1. Our work, together with other studies (Garreau de Loubresse et al. 2014; Myasnikov et al. 2016; Shen et al. 2021), clearly supports the idea that cycloheximide does not block A-site reactivity.
The C. elegans ribosome is noticeably streamlined compared to other animal ribosomes. This compaction is seen in ribosomal proteins as well as the rRNAs. The greatest loss by mass occurs in the 28S rRNA expansion segments. The reasons for its compaction are not clear; it may be due to selection for efficient and rapid growth. Expansion segments are known to affect ribosome biogenesis and protein synthesis (Rauscher and Polacek 2024), and so we expect that there are interesting translational repercussions from C. elegans’ comparatively smaller ribosomes. One associated effect may be the divergence of eL28, which appears to be rapidly evolving in line with the shortening of its binding site within ES7. Despite divergence within the expansion segments, we note that the core of the C. elegans ribosome is highly similar to those of other animals.
We noticed that RACK-1's association with ribosomes is labile in C. elegans, echoing similar reports across eukaryotes (Wong et al. 2014b; Eiler et al. 2024). This observation has implications for translational studies: our ribosome preparation protocol is similar to those used for ribosome footprint profiling (Ribo-seq), raising the possibility that a subset of ribosomal species may not be well-represented in the resultant sequencing libraries. In particular, ribosomal structures that require RACK-1 (e.g., translational surveillance intermediates) may not be captured via the technique. We recommend that care be taken to ensure the integrity of ribosomes generally, esp. RACK-1 association, when analyzing ribosome purifications via Ribo-seq and related polysome-sedimentation techniques.
A surprising finding from our work is the extent to which C. elegans ribosomes may vary across tissues. We prepared ribosomes from asynchronously grown whole animals, which contain an amalgamation of all C. elegans tissues’ ribosomes. Our transcriptomic analyses of L2-staged animals (Fig. 5G) suggest ribosomal protein paralog mRNAs vary across tissues, though it remains possible that these may not translate into protein-level differences. While we noted map heterogeneity at paralog-specific sites (Supplemental Fig. S10), it is unclear whether these sites vary by tissue. Thus, future work will need to be done to determine whether C. elegans ribosomes vary across tissues, and if so, what are the associated functional consequences.
Our analysis of ribosomal proteins revealed two clear cases where Caenorhabditis conserves a pair of paralogs: uL5 and uL23. A simple explanation for the conserved paralogs is that each member of the pair took on a distinct expression pattern, requiring the animal to conserve both paralogs to produce ribosomes across tissues and/or developmental times. Consistent with this, both uL5 and uL23 paralog mRNAs exhibit distinct tissue-specific expression patterns (Fig. 5G) (see caveats in prior paragraph). Also consistent is the observation that both uL23 paralogs are essential, but that replacement of one paralog with the other allows for grossly normal animals to develop (Fig. 5I,J).
Another, nonmutually exclusive possibility is that uL5 and uL23 paralogs acquired novel functional roles. Given uL23's direct contact with protein maturation and trafficking machinery, one could imagine polymorphisms in rpl-23A.1 or rpl-23A.2 impacting protein synthesis. Similarly, polymorphisms in rpl-11.1 and rpl-11.2 could impact tRNA dynamics given uL5's proximity to the tRNA channel. Combined with tissue-specific expression patterns, polymorphisms could lead to meaningful functional differences in translation across the animal. C. elegans is not unique in this regard: mammalian paralogs of several ribosomal proteins acquired tissue-specific expression patterns, and some paralogs exhibit striking tissue-specific and disease-specific effects (Nadano et al. 2002; Lopes et al. 2010; Sugihara et al. 2010; Wong et al. 2014a; Zhang et al. 2017; Shiraishi et al. 2023; Weinstein et al. 2024). The genetic tractability of C. elegans, coupled with the structural insights from high-resolution ribosome structures, hold promise to shed light on the molecular consequences of ribosomal protein paralog divergence and function in animals.
MATERIALS AND METHODS
C. elegans ribosome purification
Asynchronous C. elegans grown at 22.5°C on NGM plates seeded with OP50-1 were pelleted through a 5% sucrose cushion in N50 (50 mM NaCl), washed repeatedly in N50, and flash frozen in liquid nitrogen as ∼100 µL pellets. Four such pellets were ground via mortar and pestle in liquid nitrogen, and the resulting powder was split among three tubes. To each tube, we added 1 mL of ice-cold polysome lysis buffer (20 mM Tris-HCl pH 8.0, 140 mM KCl, 1.5 mM MgCl2, 1% Triton, 100 µg/mL cycloheximide). The polysome lysis buffer is based on similar formulations from published C. elegans translational studies (e.g., Stadler and Fire 2011; Aeschimann et al. 2015). The three lysate tubes were clarified via a 10,000 rcf spin at 4°C for 10′, combined, and loaded onto an SW32 10%–50% sucrose gradient in sucrose gradient buffer (15 mM Tris-HCl pH 7.5, 300 mM NaCl, 15 mM MgCl2, 100 µg/mL cycloheximide). SW32 gradients were run for 17 h at 22,500 rpm, fractionated, and 80S monosomes were harvested.
Approximately 500 µL of 80S monosome was concentrated on a concentrator column (Pierce Concentrator, PES, 50 kDa MWCo) by centrifuging at 15,000 rcf at 4°C. After the sample volume fell below 75 µL, 500 µL of cryo buffer (50 mM HEPES-KOH pH 7.4, 100 mM KOAc, 5 mM MgCl2, 1 mM DTT) was added, and the sample was again concentrated. The cryo buffer wash was repeated once more. Samples from two 80S monosome gradients were combined and concentrated to a final volume of 30 µL.
Single-particle cryo-EM sample preparation and data acquisition
A 3.5 µL aliquot of the purified endogenous C. elegans ribosome sample was deposited onto a Quantfoil R 2/1 + 2 nm extra carbon film 200 mesh grid (EM Sciences) following glow discharge using an Easy Pelco Glow-discharger. The excess sample was blotted for 2.5 sec with a force of −10% and 100% humidity at 22°C before being plunged frozen into liquid ethane using the Vitrobot Mark IV (Thermo Fisher) situated at the UCSC Biomolecular cryoEM facility. The frozen grids underwent screening in a Glacios microscope (Thermo Fisher) operating at 200 kV equipped with a Gatan K2 Summit direct detector. Top-selected grids were used for data collection at the Pacific Northwest CryoEM Center (PNCC proposal 160258, Oregon, USA).
Data collection took place on a fringe-free Thermo Fisher Scientific Titan Krios G3i operating at 300 kV coupled to a Gatan K3 direct detector coupled to a BioContinuum energy filter in super-resolution counting mode with a physical pixel size of 0.65 Å/pix (super-res 0.325 Å/pix). A total of 8337 movies were captured in a stack of 51 frames, with a cumulative dose of 40.94 e−/Å2.
The acquired movies underwent preprocessing with cryoSPARC v4.2.1 (Punjani et al. 2017), including patch motion correction and CTF correction. Following movie curation, which resulted in 8288 selected movies, 997,400 particles were blob-picked using circular blobs of 200–300 Å in diameter. After two rounds of 2D classification, the selected particles (465,852 particles) were used for ab initio reconstruction followed by 3D classification. Two observed states (State 1: single tRNA located in the P-site, and State 2: tRNAs in the A- and P-site) were observed. After splitting the particles, for each map, a final cycle of local CTF refinement and local refinement, utilizing the file containing the top-selected 98,135 (State 2) and 145,659 (State 1) particles for each state, was performed to enhance map quality. Overall “gold standard” resolution (FSC0.143) using the autogenerated mask was 2.63 Å and 2.59 Å, with a local resolution range estimation of 2.5–3.5 Å for the masked volume using cryoSPARC v4.2.1 implementation.
rRNA and protein sequences
rRNA-seq of 18S (rrn-1.1, 1754 nt), 28S (rrn-3.1, 3509 nt), 5S (rrn-4.1, 119 nt), and 5.8S (rrn-2.1, 153 nt) were obtained from WormBase (Davis et al. 2022). Sequences were queried against the VC2010 strain (Yoshimura et al. 2019) with BLASTN (Altschul et al. 1990), and hits were analyzed for sequence identity. Each of 18S, 5S, and 5.8S sequences from WormBase was identical to the VC2010 sequence. 28S rRNA contained a single adenosine (at 3173 nt) in WormBase that was guanosine in VC2010; this polymorphism was confirmed via visual inspection of RNA-seq data spanning the site (Arribere and Fire 2018) and the cryoEM map. Ribosomal protein homologs were determined by BLASTP searches of fly and human proteins against the C. elegans genome. Several C. elegans ribosomal proteins are currently being renamed, therefore Supplemental Table S1 includes both old and new names for all C. elegans ribosomal proteins and stable WBGene names; names in the text and figures are for the new Wormbase version WS293.
C. elegans ribosome model building
We started with the D. melanogaster ribosome structure PDB ID: 4V6W (Anger et al. 2013), which we docked into our map in UCSF Chimera-X (v1.3) (Meng et al. 2023) manually and with the automated “fit-in-map” tool. The Drosophila ribosome structure was manually mutated in Coot v0.9.8 (Emsley et al. 2010) starting with rRNA, using a pairwise alignment of D. melanogaster and C. elegans rRNAs as a guide, and performing realspace refinement on small sections of rRNA. AlphaFold (Varadi et al. 2022) predictions of the C. elegans ribosomal proteins were aligned in Chimera-X (v1.7), regions of no support were trimmed from the model, and realspace refinement was performed in Coot. Finally, tRNA, mRNA, cycloheximide, and spermidine were added in Chimera-X, and realspace refinement was performed in Coot. Realspace refinement of the entire model was performed in Phenix v1.20.1 (Liebschner et al. 2019), the model was visually inspected in Coot for Ramachandran outliers, and a final round of realspace refinement was performed in Phenix. A model for ES7L was made using a combination of our ribosome map, and computational tools including Unafold, RNA Central, and R2DT (Markham and Zuker 2008; RNAcentral Consortium 2021; Sweeney et al. 2021).
The maps and model were deposited in the Electron Microscopy Databank and Protein Data Bank, respectively, with accession numbers EMD-44533/EMD-45392 and PDB IDs 9BH5/9CAI. Data collection and model refinement parameters are reported in Table 1. Figures were generated using UCSF Chimera-X (v1.7). Throughout the work, we used CLUSTAL Omega (Sievers and Higgins 2018) for multiple sequence alignment using the EMBL-EBI server (Thakur et al. 2023), which provided important context for the conservation and divergence of sequences within and outside the Caenohabditis lineage. Phylogenetic trees as output from CLUSTAL Omega run-on sequence alignments are shown. RMSD calculations were performed in Chimera-X and restricted to aligned residues (using the “match” function).
Gene expression analyses
For mRNA expression, we analyzed previously published data sets. In the case of Ribo-seq, we used published data from L4 animals (SRR522883 within SRA055804 [Stadler and Fire 2011]), which we processed and analyzed as described previously (Kim et al. 2022). For Oxford Nanopore (ONT) data, we used published data from L4 animals (SRX15127206 through SRX15127210 within PRJNA834154 [Viscardi and Arribere 2022]), which were processed and analyzed as follows: Base-called reads were aligned to the C. elegans genome (Genome assembly WBCel235) using MiniMap2 (v2.17-r941) (Li 2018), with recommended settings for direct RNA-seq: “minimap2 -x splice -uf -k14.” Additionally, the parameter “‐‐junc-bed” was used with a bed genome annotation file to provide MiniMap2 with splice junction information. Aligned reads were then assigned to genes using FeatureCounts (v2.0.0) (Liao et al. 2014) with recommended settings for direct RNA-seq: “featureCounts ‐‐isLongRead” and annotations from WBCel235. Additionally, two options were used to minimize inaccurate read assignment: “‐‐largestOverlap ‐‐isStrandSpecific 1.”
C. elegans brood size and developmental assays
C. elegans strains were grown at 20°C. A rack-1 deletion allele [rack-1(ok3676)IV, strain VC3013, hereafter rack-1(Δ)] was obtained from the Caenorhabditis Genetics Center (CGC), which is supported by the National Institutes of Health, Office of Research Infrastructure Programs (P40 OD010440). Heterozygous rack-1(Δ)/rack-1(+) animals were maintained using the tmC5 balancer (Dejima et al. 2018). L4 animals of each strain were staged and allowed to develop into first-day adults for 24 h. Three first-day adults of each genotype were singled and allowed to lay eggs for 24 h. After 24 h, the adults were picked off, and the resulting progeny was allowed to develop for 48 h. After 48 h, progeny numbers and staging were recorded. Brood size and developmental assay were performed twice, yielding six observations per genotype. A Shapiro–Wilk test was performed to confirm data normality. A Welch's t-test was performed comparing WT [rack-1(+)/rack-1(+)] to rack-1(Δ)/rack-1(+) or rack-1(Δ)/rack-1(Δ) animals, and p-values are indicated on the figures.
C. elegans strain construction and fluorescent imaging
rack-1 mutant animals were crossed with previously constructed reporters at the unc-54 locus: unc-54(srf1004; unc-54::T2A::FLAG::rareArg12::GFP)I [also known as unc-54(rareArg)] and unc-54(cc4092; unc-54::GFP::T2A::nonstop)I [also known as unc-54(Nonstop)] (Arribere and Fire 2018; Glover et al. 2020; Monem et al. 2023). The znf-598 allele was srf2119, a large deletion made by CRISPR/Cas9 (Monem et al. 2023); the skih-2 allele was cc2854, a large deletion made by CRISPR/Cas9 (Arribere and Fire 2018). Strains were validated via PCR and phenotypic analysis.
Animals were grown at 20°C. Animals were placed on a microscope slide in 1 mM levamisole in EN50 (50 mM NaCl, 1 mM EDTA) with a 0.15 mm coverslip. Imaging was done on a Zeiss Axio Zoom microscope with 1.0× objective. Imaging parameters were 80× zoom, 50% shift, and 250 msec (GFP) exposure time. All compared animals bearing a given reporter were imaged during the same imaging session and processed in parallel using Fiji under the same parameters. Grayscale value ranges are either 12–99 or 10–139 for unc-54(RareArg) and unc-54(Nonstop) reporters, respectively.
For the uL23 paralog swap, CRISPR/Cas9 was performed using Cas9 mRNPs targeting the rpl-23A.2 locus using dpy-10 as a marker (Arribere et al. 2014), using a PCR-amplified synthetic gBlock (IDT). Two independent CRISPR/Cas9 events were recovered of the desired allele, WJA 7040 (isolate #1) and WJA 7041 (isolate #2).
DATA DEPOSITION
The CryoEM density map has been deposited in the Electron Microscopy Data Bank under accession codes EMD-44533 (State 2) and EMD-45392 (State 1), and coordinates have been deposited in the Protein Data Bank under accession codes PDB ID 9BH5 (State 2) and ID 9CAI (State 1). Raw and processed data are available at EMPIAR (EMPIAR-12219, https://doi.org/10.6019/EMPIAR-12219). Scripts used in the generation of figures are provided under the arriberelab github account (folder 240515).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Sarah Loerch, Melissa Jurica, and members of the Noller Laboratory (John Paul Donahue, Dustin Niblett, Gillian Rexroad, and Laura Lancaster) for helpful discussions and advice throughout the project. We thank Tim Schedl and Stavros Diamantakis for advice on ribosomal protein gene names. Technical support was provided by Benjamin Abrams, UCSC Life Sciences Microscopy Center, RRID: SCR_021135. We thank Dr. Rose Marie Haynes, Dr. Marzia Miletto, and the Pacific Northwest Center for CryoEM (PNCC) at Oregon Health & Science University for data collection and support on data processing. A portion of this research was supported by National Institutes of Health (NIH) grant U24GM129547 and performed at the PNCC at OHSU and accessed through EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research. We also thank Drs. Irina Novikova and Craig Yoshioka at PNCC for data processing training and support at PNCC Compute. The authors also acknowledge the Biomolecular cryo-Electron Microscopy Facility at the Department of Chemistry and Biochemistry of the University of California–Santa Cruz (RRID:SCR021755) for scientific and technical assistance (NIH High-End Instrumentation program, S10OD02509). Molecular graphics and analyses were performed with UCSF Chimera-X, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases. We thank Wormbase. This work was supported by an R01 from NIGMS (R01GM131012) awarded to J.A.A. and a T32 training grant awarded to C.W. (5T32GM133391). Additional funding and support for communal resources are indicated throughout.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080103.124.
- Received May 15, 2024.
- Accepted August 15, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Enisha Sehgal is the first author of this paper, “High-resolution reconstruction of a C. elegans ribosome sheds light on evolutionary dynamics and tissue specificity.” While conducting research for this paper, Enisha was a research specialist in Dr. Joshua Arribere's lab at UC Santa Cruz, studying translation surveillance mechanisms in Caenorhabditis elegans (C. elegans). Currently, she is a PhD student in the Molecular Engineering program at the University of Washington, pursuing her graduate research in Dr. David Baker's lab at the Institute for Protein Design. Her research focuses on designing de novo proteins that bind nucleic acids to modulate gene expression and uncover novel gene editing tools.
What are the major results described in your paper and how do they impact this branch of the field?
Our paper presents the first high-resolution cryo-EM structure of the C. elegans ribosome, revealing a ribosome that is streamlined relative to other animal ribosomes. While much of the C. elegans ribosome is conserved with other animals, it also has rapidly evolving expansion segments and tissue-specific ribosomal protein paralogs. These findings provide critical structural insights into translational processes in C. elegans and pave the way for future studies on ribosome heterogeneity and its functional consequences across tissues. This work will enhance comparative ribosomal studies and contribute to a deeper understanding of translation in this model organism.
What led you to study RNA or this aspect of RNA science?
During my undergraduate studies at UC Santa Cruz, I was captivated by the intricacies of the transcriptome through my biochemistry and molecular biology course work. This interest led me to join Dr. Joshua Arribere's lab, where I investigated translation surveillance mechanisms in C. elegans. My curiosity about the dynamic elements influencing RNA metabolism prompted me to delve deeper into the complexities of gene regulation. The newly available cryo-EM facility at UC Santa Cruz provided an exciting opportunity to expand the lab's research toolkit and allowed me to explore the world of structural biology. This ultimately culminated in our high-resolution ribosome structure that forms the basis of this paper. This experience not only deepened my understanding of RNA science but also ignited a passion for combining structural and functional approaches in RNA research.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
My journey into science was profoundly shaped by early experiences with my father, a wildlife filmmaker. Together, we traveled across the country, capturing landscapes and the stories of those dedicated to preserving our environment. Assisting him with documentaries on climate change—such as the impact of habitat destruction at Point Reyes National Seashore and rainforest deforestation in the Amazon caused by animal agriculture initiatives—instilled in me a lifelong passion for environmental conservation and sustainability.
This passion evolved as I explored documentary filmmaking and photography, eventually delving into wet plate photography, a historic technique of creating images of silver on glass. From perfecting exposure times to troubleshooting developer reagents, this hands-on experience sparked my interest in scientific experimentation and chemistry.
In college, I decided to study a discipline that combined my love for the natural world with the intricacies of its composition—molecular biology. My academic journey, enriched by courses spanning ecology to engineering, deepened my appreciation for interdisciplinary science. This path has led me to pursue a PhD in molecular engineering, driven by a mission to advance sustainability through biotechnology. My aim is to bridge the gap between molecular engineering and environmental conservation, through becoming a scientist and educator dedicated to discovering sustainable solutions.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My decision to pursue a career in science was profoundly influenced by the remarkable individuals I had the privilege of working with in the Arribere Lab. Dr. Arribere, who gave me the opportunity to begin my research career, mentored me in a way that had me constantly curious about how to push our understanding of our field of science. When experiments would fail, or provide inconclusive results, the motto he shared with me was “the universe is under no obligation to make sense to us.” While existentially stimulating, it also lit a fire within me to attempt to have it make sense. I was also fortunate to learn from talented graduate students like Dr. Parissa Monem, Matt Modena, and Marcus Viscardi, whose collaborative spirit and expertise highlighted the essential role of teamwork in research. Their collective influence, as well as other amazing members who have passed through this lab, has shaped my curiosity, humility, and perseverance as a scientist.
What are your subsequent near- or long-term career plans?
After departing from the Arribere Lab, I began my PhD in the fall of 2023 in Dr. David Baker's lab at the University of Washington. My current research focuses on leveraging deep learning tools to design de novo proteins that bind to DNA with sequence specificity. In the near term, my goals are to validate my designed proteins for modulating gene expression and enzymatic capabilities, such as targeted nuclease activity. I aspire to create a versatile synthetic biology toolbox, including de novo transcription factors, nucleases, polymerases and other protein-based machinery for nucleic acids. Looking further ahead, I aim to apply these innovations to sustainability challenges, such as metabolic engineering for biosynthesis pathways, improving crop resistance, cleaner biomanufacturing processes, as well as in the biomedical field for gene therapies.








