
Disruption of the Aspergillus fumigatus RNA interference machinery alters the conidial transcriptome
- Abdulrahman A. Kelani1,
- Alexander Bruch1,
- Flora Rivieccio2,3,
- Corissa Visser2,3,
- Thomas Krüger2,
- Danielle Weaver4,
- Xiaoqing Pan1,
- Sascha Schäuble5,
- Gianni Panagiotou5,6,
- Olaf Kniemeyer2,
- Michael J. Bromley4,
- Paul Bowyer4,
- Amelia E. Barber7,
- Axel A. Brakhage2,3 and
- Matthew G. Blango1
- 1Junior Research Group RNA Biology of Fungal Infections, Leibniz Institute for Natural Product Research and Infection Biology—Hans Knöll Institute (Leibniz-HKI), 07745 Jena, Germany
- 2Department of Molecular and Applied Microbiology, Leibniz Institute for Natural Product Research and Infection Biology—Hans Knöll Institute (Leibniz-HKI), 07745 Jena, Germany
- 3Department of Microbiology and Molecular Biology, Institute of Microbiology, Friedrich Schiller University, 07743 Jena, Germany
- 4Manchester Fungal Infection Group, Division of Infection, Immunity and Respiratory Medicine, Faculty of Biology, Medicine and Health, University of Manchester, Manchester M13 9NT, United Kingdom
- 5Department of Microbiome Dynamics, Leibniz Institute for Natural Product Research and Infection Biology—Hans Knöll Institute (Leibniz-HKI), 07745 Jena, Germany
- 6Department of Medicine and State Key Laboratory of Pharmaceutical Biotechnology, University of Hong Kong, Hong Kong, China
- 7Junior Research Group Fungal Informatics, Friedrich Schiller University, 07745 Jena, Germany
- Corresponding author: matthew.blango{at}leibniz-hki.de
Abstract
The RNA interference (RNAi) pathway has evolved numerous functionalities in eukaryotes, with many on display in Kingdom Fungi. RNAi can regulate gene expression, facilitate drug resistance, or even be altogether lost to improve growth potential in some fungal pathogens. In the WHO fungal priority pathogen, Aspergillus fumigatus, the RNAi system is known to be intact and functional. To extend our limited understanding of A. fumigatus RNAi, we first investigated the genetic variation in RNAi-associated genes in a collection of 217 environmental and 83 clinical genomes, where we found that RNAi components are conserved even in clinical strains. Using endogenously expressed inverted-repeat transgenes complementary to a conditionally essential gene (pabA) or a nonessential gene (pksP), we determined that a subset of the RNAi componentry is active in inverted-repeat transgene silencing in conidia and mycelium. Analysis of mRNA-seq data from RNAi double-knockout strains linked the A. fumigatus dicer-like enzymes (DclA/B) and RNA-dependent RNA polymerases (RrpA/B) to regulation of conidial ribosome biogenesis genes; however, surprisingly few endogenous small RNAs were identified in conidia that could explain this broad change. Although RNAi was not clearly linked to growth or stress response defects in the RNAi knockouts, serial passaging of RNAi knockout strains for six generations resulted in lineages with diminished spore production over time, indicating that loss of RNAi can exert a fitness cost on the fungus. Cumulatively, A. fumigatus RNAi appears to play an active role in defense against double-stranded RNA species alongside a previously unappreciated housekeeping function in regulation of conidial ribosomal biogenesis genes.
Keywords
INTRODUCTION
RNA interference (RNAi) is a conserved molecular mechanism that was likely intact in the last common ancestor of modern eukaryotes (Shabalina and Koonin 2008; Torri et al. 2022). RNAi uses small RNA (sRNA) molecules to suppress gene expression either transcriptionally by heterochromatin recruitment or post-transcriptionally through sequence-specific messenger RNA (mRNA) degradation or translational interference (Moazed 2009; Dang et al. 2011; Torres-Martinez and Ruiz-Vazquez 2017). The first reported example of RNAi in fungi came from the model fungus Neurospora crassa as a process termed quelling, where introduction of repetitive transgenes induced post-transcriptional gene silencing (Romano and Macino 1992). Since then, several RNAi and small RNA biogenesis pathways have been characterized in fungi that are largely classified as Dicer-dependent (canonical) RNAi or Dicer-independent (noncanonical) RNAi (Ketting 2011; Ruiz-Vazquez et al. 2015; Torres-Martinez and Ruiz-Vazquez 2017).
Dicer is a type IV RNase III enzyme responsible for dsRNA cleavage and biogenesis of a variety of sRNA classes in different organisms (Nicolas et al. 2015; Torres-Martinez and Ruiz-Vazquez 2017). In fungi, the orthologous RNase III domain-containing enzymes are typically referred to as dicer-like (DCL) proteins due to subtle differences in domain structure. The generated sRNAs are then loaded into an Argonaute protein, the main component of the RNA-induced silencing complex that binds and processes sRNAs to guide selective destruction, translational repression, or transcriptional suppression of target mRNAs (Agrawal et al. 2003; Torres-Martinez and Ruiz-Vazquez 2017). In some instances, RNA-dependent RNA polymerases are required for RNAi pathways to amplify the silencing activity by generating additional dsRNA substrates using the single-stranded sRNAs as primers (Torres-Martinez and Ruiz-Vazquez 2017). While the core RNAi proteins are mostly conserved across eukaryotes, some gene losses and duplications have occurred, which further diversified the RNAi pathways and their functional specialization (Hutvagner and Simard 2008; Mukherjee et al. 2013; Torres-Martinez and Ruiz-Vazquez 2017). Loss of RNAi was observed to fuel hypervirulence in Cryptococcus due to higher transposon activity (Gusa et al. 2020), and killer virus-infected Saccharomyces cerevisiae strains are able to kill and therefore outcompete noninfected strains in the absence of RNAi (Bostian et al. 1980). The RNAi machinery have also been implicated in other functions, ranging from ribosome biogenesis to maintenance of cellular quiescence (Bernstein et al. 2012; Roche et al. 2016), further highlighting the importance of RNAi across the fungal kingdom.
In this study, we improve our understanding of RNAi in the human fungal pathogen, Aspergillus fumigatus. A. fumigatus is a ubiquitous, filamentous saprophyte and among the leading fungal killers (Latge 1999; Beauvais et al. 2013). It encodes a functional RNAi system consisting of two dicer-like proteins (DclA and DclB), two argonautes (PpdA and PpdB), and two RNA-dependent RNA Polymerases (RrpA and RrpB), together with several other, annotated peripheral factors (Fig. 1A; Mouyna et al. 2004; Hammond and Keller 2005; Hammond et al. 2008b). Here, we show that the A. fumigatus RNAi system is conserved among both clinical strains and environmental isolates. We then delineate the RNAi components necessary for inverted-repeat transgene (IRT) silencing and link the RNAi machinery to expression of conidial ribosome biogenesis genes and long-term fitness. Finally, we provide a brief characterization of the endogenous small RNA pool of A. fumigatus, which appears to be almost entirely separable from the observed differential expression and protein abundance in the knockout strains.
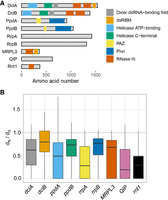
RNAi is conserved across 300 sequenced A. fumigatus strains. (A) Structural domains of A. fumigatus RNAi proteins were analyzed using UniProt (www.uniprot.org/). DclA and DclB both contain a DExH-helicase followed by a helicase-C, a Dicer-dsRNA binding fold that mediates binding with other proteins (Qin et al. 2010), a P-element-induced wimpy testis (PIWI)–Argonaute–Zwille (PAZ) domain for binding the 3′ end of the dsRNA, and two RNase III domains necessary for cleavage of bound dsRNA (Schauer et al. 2002; Mukherjee et al. 2013; Song and Rossi 2017). Both argonaute orthologs (PpdA/B) have the characteristic PAZ domain and a PIWI domain present in all eukaryotic argonautes (Jinek and Doudna 2009; Ender and Meister 2010). (B) Calculation of the ratio of nonsynonymous to synonymous base changes (dN/dS) for each of the core RNAi components and peripheral factors using 300 genomes from Barber et al. (2021). Ratios under one indicate that each gene is under neutral-to-purifying selection.
RESULTS
RNAi pathway genes are conserved in environmental and clinical isolates of A. fumigatus
The RNAi pathway has diversified extensively since its emergence around the time of the last eukaryotic common ancestor (Torri et al. 2022). Functions range from gene regulation to genome defense, but in some specific instances the machinery has been lost to provide a growth advantage (Billmyre et al. 2013). To determine whether the RNAi machinery is stably maintained in A. fumigatus, we explored the core RNAi components for genetic variation across 300 A. fumigatus genomes collected from diverse clinical and environmental niches (Barber et al. 2020, 2021). All the RNAi pathway genes were present as a single copy in the genomes, indicating a high degree of evolutionary conservation. While many strains contained genetic variation, such as missense substitutions or alterations to the 3′UTR (Supplemental Fig. S1; Supplemental Data Set S1), high impact variants like frameshifts or premature stops were rare and only observed in 1%–6% of samples (n = 4 for dclA, n = 1 for rrpA, n = 19 for rrpB). Of note, the rrpA and rrpB genes are thought to be dispensable for silencing in the related organism Aspergillus nidulans (Hammond and Keller 2005), whereas orthologs of the dclA gene contribute to the process of meiotic silencing of unpaired DNA during the sexual cycle in other organisms (Alexander et al. 2008). Calculation of the ratio of nonsynonymous to synonymous base changes (dN/dS) indicated that all members of the pathway are under purifying selection, often with one paralogous copy of the gene under a stronger degree of selection (Fig. 1B). Overall, we found a strong degree of genomic conservation among the A. fumigatus RNAi system, which together with the evidence of purifying selection suggests that the pathway likely plays a conserved, beneficial function across A. fumigatus strains and that loss does not strongly facilitate virulence in humans as has been hypothesized for other fungal pathogens (D'Souza et al. 2011; Billmyre et al. 2013).
Engineered loss of the RNAi machinery provides no growth advantage in stress or drug challenge
Genomic analysis suggested that the RNAi machinery is not frequently lost under clinical or environmental conditions, but we wanted to explicitly test the hypothesis that RNAi is dispensable for stress response and drug resistance. To do this, we created a series of knockouts in the core RNAi machinery, including single knockouts of dclA, dclB, ppdA, ppdB, rrpA, and rrpB (Supplemental Fig. S2). In addition, we made a series of pairwise knockouts in dclA/B, ppdA/B, and rrpA/B. None of the knockouts showed growth defects on minimal or rich media (Fig. 2A). Next, we examined the function of RNAi components in response to various stress conditions, including the cell wall stressors congo red, caffeine, and caspofungin; cell membrane stress agent sodium dodecyl sulfate (SDS); and oxidative stressor H2O2 (Supplemental Fig. S3). We also tested for sensitivity to azole drugs like voriconazole and tebuconazole, as well as thiabendazole (TBZ), which is a microtubule inhibitor previously used to link RNAi components to chromosome segregation in fission yeast (Hall et al. 2003). In each case, the RNAi knockout strains showed no obvious phenotypic difference in comparison with the wild type.
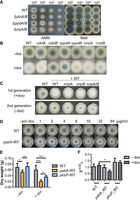
DclB and PpdA are required for IRT-mediated silencing in A. fumigatus. (A) Droplet growth assays for wild-type (WT), ΔdclA/B, ΔppdA/B, and ΔrrpA/B strains grown on AMM or Malt agar plates for 48 h. Image shown is representative of three independent experiments. (B) Wild-type and RNAi knockout strains transformed with IRT constructs were spot inoculated on AMM with and without inducer (10 µg/mL doxycycline [dox]) for 2 d. Image shown is representative of three independent experiments. (C) Wild-type and IRT-transformed strains were spot inoculated on AMM agar plates with inducer (10 µg/mL doxycycline) for 2 d. Spores were collected and the experiment was repeated without IRT induction. (D) The wild-type (CEA17ΔakuBKU80) strain was transformed with a plasmid (pSilent-pabA) expressing a dox-inducible IRT targeting pabA, a para-aminobenzoic acid synthetase-encoding gene. Wild-type and pabA-IRT-expressing strains were spot inoculated on AMM agar plates supplemented with different concentrations of dox. Images were taken after 2 d of incubation at 37°C. (E) Wild-type and pabA-IRT-expressing strains, as well as a control pksP-IRT-expressing strains were grown with and without dox induction for 24 h. Mycelia were collected and dried for 3 d at 60°C before measuring weight. Plot displays data from four biological replicates, with each replicate having three technical repeats. Significance was determined by two-way ANOVA with Bonferroni-corrected post hoc t-test comparing all columns. (F) Validation of silencing of pabA gene after induction of pabA-IRT using qRT-PCR. Plot shows standard error of the mean of three biological replicates performed in technical triplicates using the 2−ΔΔCT method (Livak and Schmittgen 2001). Significance was determined by one-way ANOVA followed by Bonferroni corrected post hoc t-test comparing all columns and assuming Gaussian distributions.
DclB and PpdA are necessary for inverted-repeat transgene silencing
The lack of phenotypic differences upon deletion of RNAi components led us to question the functionality and redundancy of the individual RNAi proteins in A. fumigatus and determine which core RNAi factors of A. fumigatus are required for silencing. It is known that exogenous delivery of RNA directly into the fungal cytoplasm is challenging due to a sequestration of negatively charged RNA by the cationic cell wall (Jochl et al. 2009). We first confirmed this finding in our own system using confocal microscopy of an exogenously delivered, 500-bp, fluorescein-labeled in vitro-transcribed dsRNA after incubation for 16 h at 37°C (Supplemental Fig. S4A). The dsRNA clearly accumulated in the cell wall of the fungus costained with calcofluor white as expected.
Next, we turned to heterologous expression of two inverted-repeat transgenes from the A. fumigatus chromosome to target a nonessential gene and a conditionally essential gene (Supplemental Fig. S4B). We reengineered a previously described system (Mouyna et al. 2004) using the pSilent-1 gene silencing vector (Nakayashiki et al. 2005) to produce a wild-type strain harboring a 500-bp, Tet-inducible pksP or pabA inverted-repeat transgene on the A. fumigatus chromosome at the pyrG locus. The nonessential pksP gene is required for dihydroxynaphthalene (DHN)-melanin biosynthesis, and silencing of pksP, results in an easy phenotypic readout via the production of pigmentless, white conidia (Langfelder et al. 1998). The second inverted-repeat transgene was complementary to the A. fumigatus pabA gene, which encodes a para-aminobenzoic acid synthetase (PabA). PabA catalyzes the biosynthesis of para-aminobenzoic acid (PABA), a precursor for folate biosynthesis in the fungus. Previous work has shown that deletion of pabA in A. fumigatus and A. nidulans results in loss of virulence and severe growth reduction, respectively, lending credence to pabA as a potential drug target (Brown et al. 2000). In fact, A. fumigatus pabA is conditionally essential, and only grows with appropriate supplementation (e.g., folate or p-aminobenzoic acid).
We first tested the pksP inverted-repeat transgene in the wild-type strain, where we observed white conidia after induction with doxycycline on AMM plates, indicative of successful post-transcriptional silencing of the pksP gene (Fig. 2B). Ultimately, we created two versions of these plasmids with different resistance cassettes so that we could also incorporate the construct onto the chromosome in each of our single and double knockouts. While similar silencing was observed in most transformed knockout strains, ΔdclB and ΔppdA remained able to produce melanized conidia, suggesting that both DclB and PpdA are essential for inverted-repeat transgene silencing and cannot be rescued by their respective paralogs, DclA and PpdB.
As mentioned, the RdRPs contribute to the RNAi pathway in some organisms by transcribing additional dsRNA substrates and amplifying the silencing response (Dang et al. 2011). To eliminate the possibility that the overexpression of our inverted-repeat transgene RNA negated the necessity of the amplification role of RdRPs in A. fumigatus, we also passaged the induced RdRP strains onto fresh plates lacking inducer. This experiment allowed us to determine whether the silencing persisted after the induction of the inverted-repeat transgene was removed. In the wild-type, ΔrrpA, and ΔrrpB strains silencing was quickly lost (Fig. 2C), indicating that these proteins likely do not contribute to amplification of silencing in A. fumigatus and in agreement with results observed previously in A. nidulans (Hammond and Keller 2005). A curious observation is that the loss of silencing occurs to a lower extent in the rrp knockouts than in wild type, perhaps suggesting an additional layer of complexity surrounding the RdRP proteins in A. fumigatus.
Using the in vitro-transcribed dsRNA with complementarity to the conditionally essential gene pabA, we observed no cessation of growth in our imaging experiments (Supplemental Fig. S4A), consistent with numerous previous studies showing that only minimal silencing can be achieved by the addition of exogenous RNA to Aspergillus species (Khatri and Rajam 2007; Abdel-Hadi et al. 2011; Kalleda et al. 2013; Mousavi et al. 2015; Nami et al. 2017). To test whether effective silencing of pabA could lead to growth repression, we again used a heterologously expressed dsRNA targeting the pabA gene from the chromosome of A. fumigatus as described above (Fig. 2D). We grew both the pabA-IRT-transformed strain and the wild-type strain with and without doxycycline induction on AMM agar plates and liquid cultures. In the absence of doxycycline, the pabA-IRT led to minor but significant growth inhibition, likely due to leaky expression (Fig. 2D,E). Upon induction with doxycycline, growth of the pabA-IRT-transformed strain was reduced on both AMM agar plates and liquid cultures when compared with the wild-type strain (Fig. 2D,E). Growth was more clearly inhibited in liquid culture than on solid media, likely due to more homogenous delivery of the doxycycline inducer. As a control, we also included the pksP-IRT strain in our liquid culture experiments and found that this construct also had a growth-inhibiting effect, albeit significantly less than the pabA-IRT strain (Fig. 2E). As a control, we confirmed by qRT-PCR that the pabA-IRT strain specifically targets pabA, resulting in a modest 43% decrease in message (Fig. 2F). Collectively, these results indicated that A. fumigatus RNAi can be leveraged to silence both a nonessential gene (pksP) and a conditionally essential gene (pabA) in a DclB- and PpdA-dependent manner.
mRNA-seq analysis suggests distinct contributions to conidial gene expression
The inverted-repeat transgene experiments suggested that only select components of the A. fumigatus RNAi machinery are required for transgene silencing. We were therefore interested to elucidate any additional roles of RNAi in A. fumigatus biology. To this end, we performed mRNA-seq on poly(A)-enriched RNA isolated from conidia and mycelium grown in liquid culture for 24 or 48 h (Supplemental Fig. S5), as RNAi is known to serve a role in asexual reproduction in other filamentous ascomycetes (Gaffar et al. 2019). We determined the differentially expressed genes (Supplemental Data Set S2) and found that each of the RNAi knockouts showed significant differences compared with wild type in the conidia samples but very few changes in expression in mycelium using this stringent cutoff (Fig. 3A). These findings were consistent with the lack of phenotypes observed in stress and growth assays. We performed Gene Ontology (GO)-slim analysis to improve our understanding of these changes in conidia and were surprised to see a clear link between the dcl and rrp genes with ribosome biogenesis and the nucleolus, where 85 genes associated with ribosome biogenesis were significantly down-regulated in the ΔdclA/B strain and 24 genes were down-regulated in the ΔrrpA/B strain compared with wild type (Fig. 3B; Supplemental Fig. S6; Supplemental Data Set S3). The argonaute proteins appeared to contribute to the conidial transcriptome differently, as we observed decreased expression of genes associated with transmembrane transport and the regulation of transcription, alongside up-regulation of genes associated with mRNA and tRNA metabolic processes.
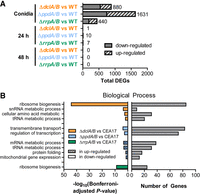
Deletion of RNAi components results in altered conidial transcriptome. (A) Genes that were identified as significantly differentially expressed in mRNA-seq. Differences were considered significant if q ≤ 0.05 and |log2 (MRN)| ≤ 1 by all four tools. The number next to the bar indicates the total number of differentially expressed genes (DEGs). (B) GO-Slim analysis (using FungiDB.org) for Biological Process generated using differentially expressed genes from conidia as input gene sets. Up- and down-regulated differentially expressed genes (left panel) as well as the number of differentially expressed genes (right panel) are indicated.
To expand on our mRNA-seq results, we performed LC-MS/MS-based proteomics on conidia and 48-h-old mycelium for each of the double knockouts and the wild type. Among the most abundant proteins detected in conidia were several histones (HhfA, Htb1), a Cu/Zn superoxide dismutase (SodC), and elongation factor 1-α (AFUA_1G06390) (Supplemental Data Set S4). At 48 h, we observed a high abundance of alcohol dehydrogenase (AFUA_7G01010), the same Cu/Zn superoxide dismutase (SodC) as in conidia, and several ubiquitin isoforms (AFUA_1G04040 and AFUA_3G11260); results that are consistent with the metabolic changes associated with carbon and iron starvation (Supplemental Data Set S5; Oberegger et al. 2000). The data set from conidia identified roughly 5810 proteins, likely making this one of the most thorough descriptions of the proteome of A. fumigatus conidia to date. For the 48-h mycelium, we identified 2380 proteins. Interestingly, from the top 200 most-abundant proteins from each data set, we only observed 26 in common, reiterating the massive changes that are undergone during the A. fumigatus asexual life cycle.
We next determined the differentially abundant proteins between knockout and wild type using a two-tailed Welch's t-test with Benjamini–Hochberg correction to control for multiple comparisons, which is known to be a stringent statistical test for proteomics analysis (Pascovici et al. 2016). The use of the Benjamini–Hochberg correction resulted in only three significant changes (adjusted P-value of ≤0.05), all in the conidia condition (Supplemental Data Set S4). Two proteins had lower abundance in the ΔdclA/B strain (AFUA_4G10100, AFUA_4G06250) and one protein (AFUA_7G08590) showed slightly increased abundance in the ΔppdA/B strain compared with wild type (Supplemental Data Set S4). The changes in AFUA_4G06250, a predicted ribosome biogenesis protein orthologous to Nop4, and AFUA_7G08590, a protein with a predicted BTB/POZ domain, were consistent with the change observed in our mRNA-seq analysis.
In many cases, controlling for multiple comparisons eliminates true hits from proteomics analyses, particularly in the context of low power associated with the standard three biological replicates (Pascovici et al. 2016). To more comprehensively analyze our data sets, we also reperformed the statistical analysis using only a two-tailed Welch's t-test, which revealed more widespread alterations to protein abundance in both conidia and 48-h-old mycelium as expected (Fig. 4; Supplemental Fig. S7; Supplemental Data Sets S4, S5). In conidia, we observed 164 proteins with at least twofold increase in abundance over wild-type in the ΔdclA/B strain and 113 proteins that significantly decreased in abundance. The ΔppdA/B and ΔrrpA/B strains showed fewer significantly altered proteins, with 49 proteins increased and 49 decreased in abundance compared with wild type for the ΔppdA/B strain and 37 proteins increased and 70 decreased for ΔrrpA/B (Supplemental Data Set S4). Interestingly, we observed very little agreement between the mRNA-seq and proteomics analysis, implying that the conidial transcriptome differs substantially from the conidial proteome. In 48-h-old mycelium, we again observed significantly different protein abundances using a two-tailed Welch's t-test. In particular, we found 35 proteins to be significantly more abundant (greater than twofold change) in the ΔdclA/B strain than wild type and 19 with decreased abundance, 61 proteins increased and 34 decreased in the ΔppdA/B strain compared with wild type, and 86 proteins increased and 36 decreased in abundance in ΔrrpA/B (Supplemental Data Set S5). Despite numerous cases of differential abundance, no clear pattern of regulation emerged from this analysis, potentially consistent with the idea that RNAi is pleotropic in its contribution to gene regulation. Collectively, these results suggest that the RNAi machinery in A. fumigatus influences fungal gene expression in multiple ways, with a particularly strong effect on genes associated with ribosome biogenesis in conidia.
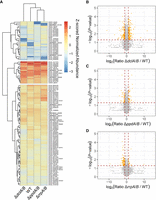
Conidia exhibit an altered proteome different from the transcriptome. (A) Clustered heat map depicting Z-scored normalized average abundance data from conidia of double knockouts and wild type for cases where proteins exhibited at least a log2(FC) = 2 and a P-value of ≤0.05 in one of the conditions. Normalized abundances were determined using area under the curve from label-free quantification. (B–D) Volcano plots showing expression ratio for double knockout compared with wild type for ΔdclA/B (B), ΔppdA/B (C), and ΔrrpA/B (D). The y-axis shows the −log10 transformed P-value, where higher values are more significant. Orange circles show proteins that are at least log2(FC) = 1 and with a P-value of ≤0.05.
RNAi genes exhibit growth stage-specific expression
The significant changes observed in conidia and mycelium led us to assess the expression of the different core and accessory RNAi components more closely in conidia versus mycelium for the wild-type and double-knockout strains using the data collected in the mRNA-seq experiment. The results clearly showed growth stage-specific expression for much of the RNAi machinery (Fig. 5). The dclA gene was more highly expressed in conidia than mycelium, whereas dclB increased with the age of the mycelium in a pattern similar to the mitochondrial ribosomal protein L3 (MRPL3) gene—known to encode an additional RNase III activity. The rnt1 gene, which encodes a nuclear RNase III enzyme that could potentially compensate for the dicer-like RNase III activity in some instances, showed higher normalized expression in conidia than mycelium like dclA. The ppdB gene was expressed in all stages, whereas ppdA was essentially absent in conidia, consistent with a predicted role in post-transcriptional gene silencing that is likely unnecessary in relatively transcriptionally dormant conidia. Instead, we observed high expression of the exonuclease QIP in conidia, and not in mycelium, possibly suggesting QIP contributes to asexual as well as sexual reproduction as in other systems (Xiao et al. 2010).
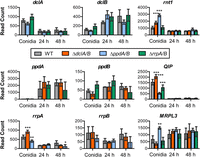
The RNAi genes are differentially expressed in conidia and mycelium. Median ratio normalization (MRN) expression indicated as read count of individual genes in conidia, 24-h and 48-h mycelium using the mRNA-seq experiment described in Figure 3. Significance was determined by two-way ANOVA with Bonferroni-corrected post hoc t-test, resulting in some minor differences from the data analyzed in Figure 3. (*) P ≤ 0.05, (**) P ≤ 0.01, (***) P ≤ 0.001.
Intriguingly, expression of many of the RNAi genes was significantly altered in the knockout strains as determined by two-way ANOVA with a Bonferroni posttest, with a few particularly notable examples. First, in the absence of the dcl or ppd genes, rnt1 levels were significantly increased in conidia (Fig. 5), perhaps suggesting some level of compensation by this alternative RNase III enzyme. The expression of QIP and rrpA increased in the dcl double knockout in line with a known interaction between QIP and rrpA, whereas expression of both decreased in the ppd double knockout. Finally, MRPL3 was greatly increased in conidia of the ΔppdA/B strain, potentially connecting the argonaute proteins to regulation of translation in mitochondria and consistent with the observation of an increase in GO-slim genes associated with mitochondrial gene expression.
RNAi contributes to fungal fitness over time
Next, to determine whether the differential expression and protein abundance observed in the mRNA-seq and proteomics experiments might influence the long-term fitness of A. fumigatus RNAi knockouts, we performed serial passaging of the ΔdclA/B, ΔppdA/B, and ΔrrpA/B double knockouts and a wild-type control strain for six generations on AMM agar plates as separate lineages. Spores were spotted in the center of the plate and allowed to grow at 37°C for 7 d with minimal disturbance to provide numerous rounds of starvation and sporulation to recapitulate the asexual lifecycle of A. fumigatus. One sublineage of each of the RNAi knockout strains ΔdclA/B and ΔppdA/B displayed subtle phenotypes that included white sectoring and altered production of conidia (Supplemental Fig. S8). Previous reports have indicated that these white colonies typically result from mutation of the pksP gene, which encodes a polyketide synthase enzyme essential for the initial step in the biosynthesis of DHN melanin (Langfelder et al. 1998; Gibbons et al. 2022). Conidia collected from the white sectors in our experiment remained white upon transfer to new plates and were confirmed by PCR to hold the appropriate resistance cassette of the knockout. We then counted spore concentrations after a final passaging and observed a decrease in spore abundance from several lineages of the ΔdclA/B and ΔppdA/B double knockouts (Fig. 6), suggesting that the ΔdclA/B and ΔppdA/B knockouts can become less reproductive over time.
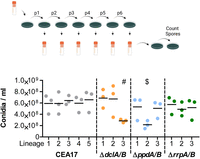
Deletion of RNAi components affects long-term fitness. (Top) Wild-type and double-knockout strains were grown as more than three lineages and passaged for six generations on AMM agar plates at 37°C for 7 d. Model created with Biorender.com. The evolved strains were then grown for an additional week and the spores were counted using a CASY counter as three biological replicates. The plot shows the values from each biological replicate of each lineage and the mean value of the biological replicates (black line). Significance was determined by one-way ANOVA with Bonferroni-corrected post hoc t-test comparing all columns. (#) P ≤ 0.05, ($) P ≤ 0.01 for all five comparisons with wild-type lineages.
A. fumigatus produces a limited pool of high-abundance endogenous small RNAs
Finally, we assessed the endogenous pool of small RNAs generated in A. fumigatus CEA10 wild type to uncover potential small RNAs with complementarity to the observed differentially expressed genes and proteins. We performed small RNA sequencing using RNA again isolated from resting conidia and mycelium grown for 24 or 48 h in liquid AMM culture. The RNA was processed with RNA 5′ pyrophosphohydrolase (RppH) to remove any 5′ triphosphates before library preparation to facilitate small RNA discovery. The resulting library concentrations were quite low for resting conidia and mycelium grown for 24 h, so we did not normalize the library input before sequencing, and instead sequenced all the available libraries to maximize sRNA discovery. Not surprisingly, we found the largest number of reads from the 48-h mycelium samples where we also observed the largest amount of small RNA in preparative analyses (Supplemental Table S1). We predicted small RNAs using Manatee, a web-based algorithm for identification and quantification of sRNAs (Handzlik et al. 2020). Three-hundred-twenty-seven sRNAs were initially predicted by the program for the three time points (conidia, 24-h mycelium, and 48-h mycelium) (Supplemental Data Set S6). Forty-two small RNA regions were identified in the conidia data set, 77 in the 24-h data, and 208 from the 48-h data. Removal of duplicated regions found in multiple samples resulted in 297 unique identifications, with 14 found in two conditions and eight found at all three time points (Supplemental Data Set S6). After applying a cutoff to remove transcripts with fewer than 200 total reads, we were left with a total of 23 high-abundance sRNA candidates (Table 1). These small RNA loci range from 19 to 25 nt in length, although the exact 5′ and 3′ nucleotides were not experimentally defined. The limited number of high-abundance candidates is consistent with previous work on A. fumigatus endogenous small RNAs (Ozkan et al. 2017).
A. fumigatus small RNAs identified by Manatee with over 200 reads
The most abundant small RNAs in conidia were antisense to AFUB_098420 (AFUA_4G04590), an uncharacterized gene predicted to be associated with ubiquinone biosynthesis, and AFUB_068210 (AFUA_4G11180), a predicted AAA family ATPase. As expected, we observed a higher abundance of small RNAs at 48 h, but these small RNAs also had limited complementarity to proteins with altered abundance in 48-h-old mycelium or conidia samples. Certainly, a more extensive survey of the small RNAs produced by A. fumigatus using the different RNAi knockout strains generated here will be required to ultimately tease apart the complex regulation across the asexual lifecycle.
DISCUSSION
The A. fumigatus RNAi pathway is functional in post-transcriptional gene silencing and involved in defense against mycoviruses (Mouyna et al. 2004; Ozkan et al. 2017). While work done in A. nidulans laid the groundwork for understanding further RNAi in A. fumigatus, distinct differences between the organisms, including truncations of the A. nidulans proteins (Hammond and Keller 2005; Hammond et al. 2008a,b), suggested that additional experimentation could be beneficial. We were able to show that the truncations of some core RNAi components in A. nidulans are not commonly repeated in A. fumigatus, as 300 sequenced genomes of A. fumigatus showed only rare truncation events (e.g., frameshifts) and relatively minor genetic variation. This relatively high level of conservation coupled with the observation that modest silencing can result in growth inhibition (Fig. 2F) suggests that we may even be able to exploit this machinery for therapeutic purposes if we can better facilitate RNA delivery in the future, as is being pursued with agricultural pests (Cai et al. 2019; Bruch et al. 2021).
Using a previously established inverted-repeat transgene approach (Mouyna et al. 2004), we created an inducible dsRNA against the pksP gene of A. fumigatus, which is required for production of DHN-melanin and the gray-green color of conidia. With this approach, we were able to determine that DclB and PpdA are required for inverted-repeat transgene silencing in A. fumigatus, whereas the other core components are dispensable for this form of RNA silencing. The finding that DclB and PpdA are essential for silencing matches previous work in N. crassa, where DCL-2 and QDE-2, orthologs of DclB and PpdA, were shown to be important for quelling (Dang et al. 2011). The result that A. fumigatus ppdA is necessary for silencing also matches results from A. nidulans with ortholog RsdA (Hammond and Keller 2005). Unlike N. crassa, DclA was not able to compensate in a ΔdclB deletion strain of A. fumigatus in our pksP-inverted-repeat transgene silencing experiments, suggesting discrete functions for DclA, perhaps during the poorly understood A. fumigatus sexual cycle. QDE-1 is important for silencing in N. crassa, but lost in A. nidulans (Hammond and Keller 2005), whereas the ortholog encoded by rrpA is conserved, but dispensable for IRT silencing, in A. fumigatus (Fig. 2B,C). This result appears to be consistent with the described loss of the activity in A. nidulans.
We showed that a 500-bp double-stranded RNA with complementarity to the pabA mRNA is capable of silencing pabA expression to limit fungal growth when expressed from the chromosome. Surprisingly, we also observed that the pksP-IRT led to growth inhibition when heterologously expressed. We suspect that the pksP-IRT sequence either results in production of small RNAs that have off-target effects or that the overproduction of a double-stranded RNA is mistaken for a mycoviral infection, resulting in altered fungal behavior. It is possible that overexpression of double-stranded RNA, which may also mimic a mycoviral infection, sequesters RNase III proteins from their normal functions in ribosome biogenesis and limits fungal growth. It is worth noting that in the literature, heterologous overexpression of a much less structured human mRNA, RBP7, in A. fumigatus did not detrimentally alter growth (Shopova et al. 2020), suggesting that the observed growth inhibition is not due solely to resource depletion, e.g., nucleotide pool depletion. A closer inspection of the small RNAs produced from the inverted-repeat transgene constructs will be required to fully understand this observation and detail the interplay between the different functionalities of RNAi in A. fumigatus.
The RNAi pathway in fungi has been linked to diverse regulation, including in sexual differentiation, development, stress response, and growth (Ruiz-Vazquez et al. 2015; Nicolas and Garre 2016; Torres-Martinez and Ruiz-Vazquez 2017). Despite all these important functions in other organisms, A. fumigatus appears to be relatively robust in tolerating deletion of the core RNAi machinery. Minimal functional differences were observed in a wide array of stress assays, although we did observe a decrease in fitness in our serial passaging experiment for some lineages of the ΔdclA/B and ΔppdA/B strains, implying that loss of RNAi can have consequences. RNAi pathways also frequently play an important role in maintaining genome integrity, as has been described in distantly related fungi Cryptococcus neoformans (Janbon et al. 2010; Wang et al. 2012), Mucor circinelloides (Nicolas et al. 2010; Calo et al. 2017), Fusarium graminearum (Chen et al. 2015), Magnaporthe oryzae (Kadotani et al. 2003; Murata et al. 2007), Schizosaccharomyces pombe (Roche et al. 2016), and Trichoderma atroviride (Carreras-Villasenor et al. 2013). Investigation of fungal RNAi has proven a function in preserving the integrity of the host genome against invasive nucleic acids including transgenes, transposons, and invading viruses during both the vegetative and sexual cycles (Nuss 2011; Torres-Martinez and Ruiz-Vazquez 2017). Future studies into the RNAi components that contribute to genome protection are now possible using the tools we created here.
Additional work will be needed to determine the mechanism of regulation imparted by the dicer-like proteins in conidia, but some clear differences from previous reports should be noted. For example, A. fumigatus has a conserved, seemingly intact Rnt1 enzyme and the action of the dicer-like proteins seems unlikely to match that observed in C. albicans, where the only active RNase III enzyme performs the function of both dicer and Rnt1 in processing of rRNA (Bernstein et al. 2012). RNAi has also been linked to maintenance of quiescence in S. pombe, but again differences exist between the two organisms. A. fumigatus RNAi knockouts showed no defects in growth in our assays, and no change was observed when the RNAi knockouts were stressed with TBZ. The effects in conidia could also be due to regulation of heterochromatin formation as has been shown in other organisms (Martienssen and Moazed 2015), but at least preliminary studies in A. nidulans suggest that RNAi is uncoupled from regulation of heterochromatin in Aspergillus (Hammond and Keller 2005).
In the proteomics analyses, we observed only minor differences using a strict statistical analysis, whereas a more relaxed analysis led to disparate changes. As RNAi is known to play pleotropic roles in gene regulation, we can envisage that A. fumigatus may use RNAi to control gene expression in a variety of different ways. This pleotropic regulation is likely evident in the limited pool of endogenous small RNAs in A. fumigatus. The majority of reads from small RNA-seq were derived from ribosomal RNAs and tRNAs, as was observed previously (Jochl et al. 2009), with only 23 high-abundance small RNA loci. These matched poorly to observed changes in protein abundance in the knockouts, potentially suggesting alternative biogenesis pathways or regulation in the conidiophore structure. A more in-depth analysis using the inverted-repeat transgene and deletion strains produced here will be required to understand the pool of small RNAs in A. fumigatus.
In conclusion, we have shown that A. fumigatus RNAi appears to play an active role in post-transcriptional gene silencing that runs parallel to a previously unappreciated housekeeping function in regulation of conidial ribosomal biogenesis genes. The strains created and data sets collected here will likely be useful in further delineating the functions of this complex, evolutionarily diverse RNA regulatory pathway.
MATERIALS AND METHODS
Domain mapping and genomic analysis
Domain structures were created using UniProt annotations (https://www.uniprot.org; Q4WSH7, Q4WY64, Q4WEW6, Q4WWR4, Q4WUR8, Q4WC92, Q4WXX3, Q4WA22, and Q4WVE3) and created with the drawProteins (Brennan 2018) package using R version 4.1.0 (2021-05-18).
The genomic data analyzed in this study were generated in Barber et al. (2021) and are publicly available as raw reads and assembled genomes under PRJNA697844 and PRJNA595552. Isolates originated from environmental and clinical sources and displayed a global distribution. Presence/absence variation in the genomes was quantified using OrthoFinder using peptide sequences from 300 genomes. Given that no cases of gene loss or amplification were detected among the genome assemblies, analysis of sequence variation was performed by aligning raw sequencing reads to the Af293 reference genome (FungiDB release 39). PCR duplicates were marked using MarkDuplicate from Picard v.2.18.25. Variant calling to detect SNVs and short indels was performed using HaplotypeCaller and high-quality variants identified using the following filtering criteria: SNP: QD < 2.0||MQ < 40.0||FS > 60.0||MQ RankSum < −12.5||ReadPosRankSum < −8.0; indel: QD < 2.0||FS > 200.0||ReadPosRankSum < −20.0. Variant function was predicted using SnpEff v.4.3.
Strains and culture conditions
Aspergillus fumigatus strains were grown on Aspergillus minimal medium (AMM) agar plates (70 mM NaNO3, 11.2 mM KH2PO4, 7 mM KCl, 2 mM MgSO4, 1% [w/v] glucose and 1 µL/mL trace element solution at pH 6.5). The trace element solution was composed of 1 g of FeSO4 • 7 H2O, 8.8 g of ZnSO4 • 7 H2O, 0.4 g of CuSO4 • 5 H2O, 0.15 g of MnSO4 • H2O, 0.1 g of NaB4O7 • 10 H2O, 0.05 g of (NH4)6Mo7O24 • 4 H2O, and ultrafiltrated water to 1000 mL. Spores were harvested after 5 d (or 7 d for the evolution experiment) with 10 mL of sterile distilled water using a T-shaped inoculation spreader, and spore suspensions were filtered through a 30-µm cell strainer (MACS; Miltenyi Biotec GmbH) to exclude mycelium. A. fumigatus transformants were screened on AMM agar plates supplemented with 150 µg/mL hygromycin, 0.1 µg/mL pyrithiamine, or 160 µg/mL phleomycin for selection. For in vitro transcription and confocal laser scanning microscopy experiments, A. fumigatus AfS150 was grown on malt agar (Sigma-Aldrich) plates supplemented with agar to a final concentration of 2% (w/v) for 5 d. Escherichia coli strains were grown at 37°C on LB broth or agar supplemented with 100 µg/mL carbenicillin when necessary.
Genetic manipulation strategies
Knockout constructs were created using several strategies; namely, Fusion PCR or the Gibson cloning method (Szewczyk and Krappmann 2010). To make constructs by Fusion PCR, an antibiotic selection gene and ∼1 kb of DNA flanking regions (5′ and 3′) of the target gene were amplified. PCR fragments were purified using the GeneJET PCR purification kit (Thermo Fisher Scientific) and concentrations were measured by NanoDrop 2000 spectrophotometer. Next, equimolar amounts of the three fragments, two flanking regions, and selection gene, were combined using Fusion PCR with the 2× Phusion Flash High Fidelity Master Mix and 10 pmol of each forward and reverse primer for the 5′ flank and 3′ flank, respectively. Fusion PCR products were purified and validated by agarose gel electrophoresis. For Gibson assembly, a selection gene and ∼1 kb flanking regions of the target gene were amplified by PCR using primers that create overhangs (Supplemental Table S2). In addition, pUC18 vector backbone was also amplified from plasmid “pUChph Volker.” DNA fragments were assembled and the resulting plasmid was used to transform chemically competent E. coli following the NEBuilder HiFi DNA Assembly protocol (New England Biolabs). Knockouts of A. fumigatus were generated by homologous recombination following the transformation of protoplasts (Weidner et al. 1998).
Growth and viability assays
The growth capacity of different A. fumigatus strains in AMM media was investigated by determining the dry weight from shaking cultures. 1 × 107 spores were inoculated in 50 mL of AMM and incubated at 37°C and 200 rpm for 24 h. Mycelia were harvested using Miracloth (Merck KGaA), washed with deionized water, and dried at 60°C for 3 d. The susceptibility of different A. fumigatus strains to stress conditions was investigated by spotting 5 µL of spore suspension with concentrations ranging from 105 to 102/mL on AMM agar plates supplemented with different stress-inducing agents for 2 d. All plates were incubated at 37°C.
Inverted-repeat transgene silencing assays
To silence A. fumigatus pksP, sense and antisense pksP PCR products (500 bp each) were amplified with primer pairs oARK0031 & 0032 and oARK0033 & 0034, respectively. These PCR products were then cloned into the two multicloning sites in the p-Silent1 plasmid vector, which already has a hygromycin resistant cassette. The trpC promoter upstream of the inverted-repeat region originally present from the p-Silent 1 vector was replaced by a tetracycline-inducible promoter (TetON). The resulting plasmid was then digested with restriction enzymes SacI and FspI to excise the IRT-expressing region and hygromycin (hph) resistant cassette, which was then cloned into a pUC18 vector backbone flanked by 5′ and 3′ regions (∼1 kb) of a nonfunctional pyrG locus in the CEA17ΔakuBKU80 (A1163, wild type) strain to generate pSilent-pksP1. Because some of the RNAi knockout strains already have a hygromycin resistant cassette, we constructed pSilent-pksP2 by replacing the selection marker with a phleomycin (ble) resistant gene. Both plasmids were linearized by BtsI restriction enzyme before transformation of strains (Supplemental Table S3). To generate a para-aminobenzoic acid synthetase (pabA) silencing construct, pabA-IRT sequence plus a 3′ trpC terminator was synthesized. This synthesized DNA was then cloned using Gibson assembly into a PCR product amplified from pSilent-pksP2 with primer pair oARK0096 & oARK0038. The latter PCR product consist of the pUC18 backbone, pyrG flanks, phleomycin resistant cassette, and TetON promoter to drive the pabA-IRT expression. The plasmid was linearized by PstI restriction enzyme before transformation of CEA17ΔakuBKU80 wild-type strain (da Silva Ferreira et al. 2006).
Southern blot
Target gene deletion was confirmed by Southern blot, as described previously (Southern 2006). Digested genomic DNA samples were separated in a 1% (w/v) agarose gel. Separated DNA fragments on gel were transferred onto a nylon membrane (pore size: 0.45 µm, Carl Roth GmbH). The DNA was crosslinked to the membrane by UV irradiation followed by overnight hybridization with a DIG-11-dUTP-labeled DNA probe at 65°C. The membrane was washed twice for 5 min at 65°C (1× SSC, 0.1 SDS), followed by two washes with wash buffer II (0.5× SSC, 0.1 SDS) for 20 min at 65°C. The membrane was next rinsed with maleic acid wash buffer, incubated with blocking solution for 30 min to prevent unspecific antibody binding, followed by another incubation with 20 mL of blocking solution plus 0.5 µL of anti-digoxigenin-AP, Fab fragments (Roche) for 30 min. The unbound antibodies were removed by washing twice with 50 mL of maleic acid wash buffer for 15 min. Finally, the membrane was equilibrated with detection buffer, developed with several drops of the chemiluminescent substrate CDP-Star, ready-to-use (Roche) and detected with a chemiluminescence imaging Fusion FX SPECTRA instrument (Vilber Lourmat).
In vitro transcription and confocal laser scanning microscopy
For in vitro dsRNA transcription, genomic DNA was isolated from mycelium of A. fumigatus CEA17ΔakuBKU80 grown overnight in liquid cultures and used as a template for the amplification of 500 bp of the pabA sequence by PCR. The PCR reaction mix consisted of 200–300 ng of template DNA, 10 pmol of each primer, and 25 µL of 2× Phusion Flash high-fidelity PCR master mix (Thermo Fisher Scientific). Both the forward and the reverse primer were designed to contain a T7 promotor at their 5′ end for subsequent dsRNA transcription (Supplemental Table S2; T7_pabA_500_1_ for/T7_pab_A500_1_rev). PCR products were loaded onto a 1% agarose gel to check for the correct size and purified with the GeneJET PCR purification kit (Thermo Fisher Scientific). The products were then used as a template for in vitro dsRNA transcription using the MEGAscript RNAi kit (Invitrogen) according to the manufacturer's protocol with slight modifications. Briefly, the transcription reaction was assembled as specified with the addition of 0.5 µL fluorescein-labeled-UTP solution (Sigma-Aldrich). The reaction was incubated for 4 h at 37°C and the RNA products were annealed by heating the reaction for 5 min to 75°C and subsequently left to cool at room temperature. Digestion of DNA and single-stranded RNA and the purification of dsRNA was performed as described in the protocol with the exception that the purified dsRNA was eluted in two steps using a total of 100 µL of 95°C ultrapure H2O. Quantity and quality of the dsRNA was assessed by fluorometric analysis using a Qubit 4 Fluorometer (Thermo Fisher Scientific). dsRNA was stored in the dark at −80°C until usage.
For fluorescence microscopy, 1 × 104 freshly collected A. fumigatus AfS150 conidia (Lother et al. 2014) were seeded into each well of an eight-well chamber slide (µ-Slide 8 Well, Ibidi) in a total of 100 µL of RPMI medium (Sigma-Aldrich), treated with 2 µg of labeled dsRNA, and incubated for 16 h at 37°C and 5% CO2 (v/v). The following day, old media was removed from the samples and fresh media containing calcofluor white was added to the fungus. After 30 min of incubation at room temperature in the dark, hyphae were carefully washed with sterile phosphate-buffered saline (PBS). An amount of 100 µL of RPMI was added to each chamber and samples were observed by confocal laser scanning microscopy with a Zeiss LSM 780 microscope (Carl Zeiss). Images were processed using the ZEN blue edition software (version 3.4, Carl Zeiss).
RNA isolation
Fungal mycelium was collected from liquid culture using Miracloth (Millipore) and disrupted in liquid nitrogen using a precooled mortar and pestle. Roughly 0.5 g of homogenized mycelium was transferred into a 2-mL Eppendorf tube. An amount of 800 µL TRIzol was added to the ground mycelia and vortexed vigorously. Tubes were frozen briefly for 5 sec in liquid nitrogen and allowed to thaw on ice. An amount of 160 µL of chloroform was added to the thawed mixture, vortexed, and centrifugated for 5 min at 4°C at full speed. The aqueous upper phase was transferred to a fresh 2-mL tube without disturbing the interphase. RNA extraction from aqueous phase was done with one volume of phenol/chloroform/isoamyl alcohol (25:24:1, v/v). Brief vortexing preceded centrifugation for 5 min at 4°C. Extraction was repeated until no more interphase was observable, and this was followed by another extraction with 400 µL of chloroform. RNA was precipitated using 400 µL of isopropanol for 20 min, followed by pelleting by centrifugation for 20 min at 4°C. The pellet was washed with 700 µL of 70% ethanol and air dried for 5 min at 37°C prior to resuspension in RNase-free water. The RNA isolation was followed by a DNase treatment using 2 units of TURBO DNase (Thermo Fisher) per 10 µg of RNA for 30 min at 37°C in 100 µL of total volume. Total RNA was collected using the RNA Clean and Concentrator-25 kit (Zymo Research) according to the manufacturer's instructions.
Quantitative RT-PCR (qRT-PCR)
RNA was isolated from cultures of CEA17ΔakuBKU80, pabA-IRT, and pksP-IRT with and without doxycycline induction after 24 h. Complementary DNA (cDNA) was synthesized from RNA according to the iScript cDNA synthesis kit protocol (Bio-Rad Laboratories, Inc.), and the concentration was determined using a Qubit Flex Fluorometer (Thermo Fisher Scientific). A 20-µL reaction volume per well was prepared in a 96-well plate. The PCR reaction mix consisted of 10 ng of cDNA, 4 pmol of each primer pair, 1 µL of 20× Eva Green (Biotium), and 10 µL of 2× MyTaq HS Mix (Bioline). Specific primers (qRT_coxV_for / qRT_coxV_rev) for A. fumigatus cytochrome C oxidase subunit V (AFUA_5G10560) were used as internal controls to normalize the expression of target genes. The cDNAs from four biological samples were used for analysis, and all the reactions were run in triplicate. The reaction was carried out using the QuantStudio 3 Real-Time PCR system (Thermo Fisher Scientific) with the following thermal cycling profile: 20 sec at 95°C, followed by 40 cycles of amplification (5 sec at 95°C, 34 sec at 60°C). Relative expression was calculated according to the 2−ΔΔCT method (Livak and Schmittgen 2001).
mRNA seq and analysis
Total RNA was isolated from three biological replicates of CEA17ΔakuBKU80, ΔppdA/B, ΔdclA/B, and ΔrrpA/B knockout strains from conidia, 24-, and 48-h-old mycelium. RNA was isolated from mycelia as described above. For A. fumigatus conidia, the spores were first homogenized in a bead beater (0.5-mm beads; FastPrep-24, MP Biomedicals) in TRIzol before proceeding with the first chloroform extraction step as described in the RNA isolation method above. The RNA isolation was followed by a DNase treatment using two units of TURBO DNase (Thermo Fisher) per 10 µg of RNA for 30 min at 37°C in 100-µL total volume. Total RNA was then collected using the RNA Clean and Concentrator-25 kit (Zymo Research) according to the manufacturer's instructions. Total RNA collected were then sent for sequencing. Poly(A) selection and directional sequencing of mRNA was performed by Novogene using paired-end 150-bp read sequencing on a NovaSeq 6000.
Preprocessing of raw reads including quality control and gene abundance estimation was done with the GEO2RNAseq pipeline (v0.9.12) (Seelbinder et al. 2019) in R (version 3.5.1). Quality analysis was done with FastQC (v0.11.8) before and after trimming. Read-quality trimming was done with Trimmomatic (v0.36). Reads were rRNA-filtered using SortMeRNA (v2.1) with a single rRNA database combining all rRNA databases shipped with SortMeRNA. Reference annotation was created by extracting and combining exon features from corresponding annotation files. Reads were mapped against the reference genome of A. fumigatus (Af293, ASM265v1) using HiSat2 (v2.1.0, paired-end mode). Gene abundance estimation was done with featureCounts (v1.28.0) in paired-end mode with default parameters. MultiQC version 1.7 was finally used to summarize and assess the quality of the output of FastQC, Trimmomatic, HiSat, featureCounts, and SAMtools. The count matrix with gene abundance data without and with median-of-ratios normalization (MRN) (Anders and Huber 2010) were extracted. Raw files are accessible under Gene Expression Omnibus accession number GSE223618.
Differential gene expression was analyzed using GEO2RNAseq. Pairwise tests were performed using four statistical tools (DESeq v1.30.0, DESeq2 v1.18.1, limma voom v3.34.6, and edgeR v3.20.7) to report P-values and multiple testing corrected P-values using the false-discovery rate method q = FDR(p) for each tool. Gene expression differences were considered significant if they were reported significant by all four tools (q ≤ 0.05 and |log2 (MRN)| ≤ 1). GO-Slim analysis was performed on genes identified as significantly differentially expressed by all four methods described above using https://fungidb.org (Amos et al. 2022) with a P-value cutoff of 0.05 and limiting the search to only GO-Slim terms.
Proteome analysis
Spores were harvested from 5-d-old AMM agar plates incubated at 37°C. Spores were pelleted in 2-mL screw cap tubes and a 300-µL equivalent of 0.5-mm beads were added. Bead beating was done in lysis buffer (1% [w/v] SDS, 150 mM NaCl, 100 mM TEAB [triethyl ammonium bicarbonate], with the addition of each one tablet cOmplete ULTRA protease inhibitor cocktail and PhosSTOP [both Roche] per 10 mL of lysis buffer), three times for 20 sec with 2-min rest on ice in between. Harvested mycelium from 48-h cultures was disrupted by mortar and pestle with liquid nitrogen. Cell debris was then homogenized in lysis buffer. All samples were then incubated at 37°C in a water bath sonicator for 30 min with 0.5 µL of Benzonase nuclease (250 U/µL). Proteins were separated from insolubilized debris by centrifugation at 18,000g for 15 min. Each 100 µg of total protein per sample was diluted with 100 mM TEAB to gain a final volume of 100 µL. Subsequently, cysteine thiols were reduced and carbamidomethylated for 30 min at 70°C by addition of 2 µL each of 500 mM TCEP [tris(2-carboxyethyl)phosphine] and 625 mM 2-chloroacetamide (CAA). The samples were further cleaned up by methanol–chloroform–water precipitation using the protocol of Wessel and Flugge (1984). Protein precipitates were resolubilized in 5% trifluoroethanol of aqueous 100 mM TEAB and digested for 18 h with Trypsin + LysC (Promega) at a ratio of 25:1 protein:protease. Samples were evaporated in a vacuum concentrator (Eppendorf). Tryptic peptides were resolubilized in 30 µL of 0.05% TFA and 2% ACN in H2O filtered through 10-kDa MWCO PES membrane spin filters (VWR) at 14,000g for 15 min and transferred to HPLC vials.
LC-MS/MS analysis and protein database search
An improvement in technology was implemented between the two LC-MS/MS proteomics experiments described in this manuscript. Each method is described in detail below for clarity. Method 1 (M1) is related to 48-h mycelial proteomics (three biological replicates) and method 2 (M2) to conidial proteomics (four biological replicates). Each biological replicate was measured in triplicate (three analytical replicates). LC-MS/MS analysis was performed on an Ultimate 3000 nano RSLC system connected to either a QExactive Plus (M1) or an Orbitrap Exploris 480 mass spectrometer (both Thermo Fisher Scientific) equipped with a FAIMS ion mobility interface (M2). Peptide trapping for 5 min on an Acclaim Pep Map 100 column (2 cm × 75 µm, 3 µm) at 5 µL/min was followed by separation on an analytical Acclaim Pep Map RSLC nano column (50 cm × 75 µm, 2µm). Mobile phase gradient elution of eluent A (0.1% [v/v] formic acid in water) mixed with eluent B (0.1% [v/v] formic acid in 90/10 acetonitrile/water) was performed using the following gradients. M1: 0–5 min at 4% B, 30 min at 7% B, 60 min at 10% B, 100 min at 15% B, 140 min at 25% B, 180 min at 45% B, 200 min at 65% B, 210–215 min at 96% B, 215.1–240 min at 4% B. M2: 0 min at 4% B, 20 min at 6% B, 45 min at 10% B, 75 min at 16% B, 105 min at 25% B, 135 min at 45% B, 150 min at 65% B, 160–165 min at 96% B, 165.1–180 min at 4% B. Positively charged ions were generated at spray voltage of 2.2 kV using a stainless steel emitter attached to the Nanospray Flex Ion Source (Thermo Fisher Scientific). The quadrupole/orbitrap instrument was operated in Full MS/data-dependent MS2 mode to trigger fragmentation of the top 15 precursor ions (M1). For M2 the number of data-dependent scans was related to the cycle time of 1.5 sec (M2). Precursor ions were monitored at m/z 300–1500 (M1) or m/z 300–1200 (M2) at a resolution of 140,000 (M1) or 120,000 (M2) FWHM (full width at half maximum) using a maximum injection time (ITmax) of 120 msec (M1) or 50 msec (M2) and an AGC (automatic gain control) target of 3 × 106 (M1) and a normalized AGC target of 300% (M2), respectively. Precursor ions with a charge state of z = 2–5 were filtered at an isolation width of m/z 1.6 (M1) or m/z 4.0 (M2) for further fragmentation at 30% (M1) or 28% (M2) HCD collision energy. MS2 ions were scanned at 17,500 FWHM (ITmax = 120 msec, AGC = 2 × 105) for M1 and 15,000 FWHM (ITmax = 40 msec, AGC = 200%) for M2. Three different compensation voltages were applied (−48 V, −63 V, −78 V) for the FAIMS interface (M2).
Tandem mass spectra were searched against the FungiDB database (https://fungidb.org/common/downloads/Current_Release/AfumigatusAf293/fasta/data/FungiDB-57_AfumigatusAf293_AnnotatedProteins.fasta) of Aspergillus fumigatus Af293 (July 9, 2021) for M1 and against the UniProt pan proteome databases (December 21, 2022) of Neosartorya fumigata/Aspergillus fumigatus (https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/pan_proteomes/UP000002530.fasta.gz) for M2 using Thermo Proteome Discoverer (PD) 2.4 (M1) or 3.0 (M2). The following database search engines and respective scoring thresholds (in parentheses) have been applied for the different methods. M1: Mascot 2.4.1 (>30), Sequest HT (>4), MS Amanda 2.0 (>300), and MS Fragger 3.2 (>8). M2: Mascot 2.8 (>30), Comet (>3), MS Amanda 2.0 (>300), Sequest HT with and without INFERYS Rescoring (>3), and CHIMERYS (>2). Two missed cleavages were allowed for the tryptic digestion. The precursor mass tolerance was set to 10 ppm and the fragment mass tolerance was set to 0.02 kDa. Modifications were defined as dynamic Met oxidation, phosphorylation of Ser, Thr, and Tyr (M2), protein N-terminal acetylation with and without Met loss as well as static Cys carbamidomethylation. A strict false discovery rate (FDR) of <1% (peptide and protein level) and aforementioned search engine score threshold values were required for positive protein hits. The Percolator node and a reverse decoy database was used for q-value validation of spectral matches. Only rank 1 proteins and peptides of the top scored proteins were counted. Label-free protein quantification was based on the Minora algorithm using the precursor abundance based on intensity and a signal to noise ratio >5. Normalization was performed by using the total peptide amount method. Imputation of missing quan values was applied by using abundance values of 75% of the lowest abundance identified. Differential protein abundance was defined as a fold change of >2, P-value/ABS(log4ratio) ≤ 0.05 and at least identified in two of three (M1) or three of four replicates (M2) of the sample group with the highest abundance (Supplemental Data Sets S4, S5). Heat maps were created in R v. 4.2.2 using pheatmap v. 1.0.12 by first log2-transforming normalized abundance data and then calculating Z-scores for proteins that exhibited at least a log2(FC) = 2 and a P-value ≤0.05 in one of the conditions.
Library preparation for small RNA-sequencing and bioinformatic analysis
Total RNA was isolated from three separate biological experiments using A. fumigatus CEA10 conidia (5 d on AMM agar plates at 37°C) and mycelium after 24 and 48 h of growth in AMM liquid culture at 37°C using the Universal RNA Kit (Roboklon) with slight modifications. Namely, conidia and mycelia at different time points were harvested and resuspended in RL buffer followed by bead beating three times for 30 sec each using 0.5-mm glass beads (Carl Roth). The total RNA was then isolated according to the yeast protocol provided by the manufacturer (Roboklon) using an on-column DNase treatment (Lucigen Baseline-ZERO DNase). The RNA isolation was followed by an additional DNase treatment using four units of TURBO DNase (Thermo Fisher) for 30 min at 37°C in 100-µL total volume. The small RNAs were then collected using the RNA Clean and Concentrator-25 kit (Zymo Research) according to the manufacturer's instructions for small RNA enrichment.
Since the 5′ end of A. fumigatus small RNAs are not well-defined, the isolated RNA was treated with 1 µL (5 units) of RppH enzyme (New England Biolabs) in NEB buffer 2 (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM DTT at pH 7.9) for 1 h at 37°C to facilitate removal of a pyrophosphate from the 5′ end of triphosphorylated RNA. The reaction was ended with EDTA for 5 min at 65°C followed by cleanup using the RNA Clean and Concentrator-25 kit (Zymo Research) as described above. RNA was quantified using the Qubit 4 Fluorometer (Thermo Fisher). Small RNA libraries were then prepared from 10 ng of small RNA using the NEBNext Multiplex Small RNA library preparation set for Illumina (New England Biolabs #E7300S) according to the manufacturer's recommendation using NEBNext Multiplex Oligos for Illumina (Index Primer Set 1, #E7335S) (Supplemental Table S1). Size selection was performed according to the manufacturer using AMPure XP beads (Beckman Coulter). Libraries were validated for the correct size by polyacrylamide gel electrophoresis. The barcoded libraries were pooled without further adjustment, followed by 50-cycle single-end sequencing using the Illumina HiSeq 4000 platform (Eurofins Genomics). In this case, the sRNA data produced are not strictly suitable for comparison of read abundance between time points but were deemed appropriate for sRNA discovery.
Adaptors were trimmed from sRNA sequencing data using BBDuk from BBTools v38.22 (https://sourceforge.net/projects/bbmap/) and quality profiles assessed using FastQC. Candidate sRNAs were identified from merged replicate data using Manatee v1.2 (Handzlik et al. 2020) along with the Ensembl A. fumigatus A1163 genome (ASM15014v1) sequence and annotated transfer RNAs as a reference. Sequence hits between 19 and 25 nt in length and with more than 200 counts were retained for further analysis. Hits were visually inspected using sRNA- and mRNA-seq data and the complete A. fumigatus gene annotation viewed in Integrative Genomics Viewer (IGV) to validate their existence and identify antisense overlap with gene transcription (Thorvaldsdottir et al. 2013). FASTA sequences for the final candidate sRNAs were extracted using Gffread from GFF utilities (Pertea and Pertea 2020).
Data availability
The mass spectrometry proteomics data were deposited in the ProteomeXchange Consortium via the PRIDE partner repository (Perez-Riverol et al. 2022) with the data set identifiers PXD033984 (48-h mycelium) and PXD039739 (conidia). Small RNA and mRNA sequencing data sets can be found on GEO under identifiers GSE223619 and GSE223618, respectively.
Statistical analysis
The proteomics data were analyzed as described. For the remaining analyses, P-values were determined by ANOVA or two-tailed Student's t-tests where appropriate using Prism 5.0.0 software (GraphPad Software). Differences between the groups were considered significant at a P-value of <0.05. Throughout the article, significance is denoted as follows: P ≤ 0.05 (*), P ≤ 0.01 (**), P ≤ 0.001 (***), and nonsignificant (ns).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Pamela Lehenberger, Bhawana Israni, Carmen Schult, and Laura Broschat for excellent technical assistance. The work presented here was generously supported by the Federal Ministry for Education and Research (BMBF: https://www.bmbf.de/), Germany, Project FKZ 01K12012 “RFIN—RNA-Biologie von Pilzinfektionen.” Additional funding support came from the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) under Germanýs Excellence Strategy—EXC 2051—Project-ID 390713860 and the DFG-funded Collaborative Research Center/Transregio FungiNet 124 “Pathogenic fungi and their human host: Networks of Interaction” (210879364, project A1, INF, and Z2).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079350.122.
-
Freely available online through the RNA Open Access option.
- Received July 28, 2022.
- Accepted March 23, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Abdulrahman Kelani is the first author of this paper, “Disruption of the Aspergillus fumigatus RNA interference machinery alters the conidial transcriptome.” Abdulrahman is a doctoral researcher in the laboratory of Matthew Blango (Junior Research Group RNA Biology of Fungal Infections) at the Leibniz Institute for Natural Product Research and Infection Biology: Hans Knöll Institute (Leibniz-HKI), Jena, Germany. The collective aim of the laboratory is to understand the role of RNA and RNA regulatory proteins during fungal infection. His research goal as a member of this team is to characterize the small RNA species produced by conserved RNA regulatory mechanisms like the RNAi pathway and understand the importance of this pathway in the human fungal pathogen Aspergillus fumigatus.
What are the major results described in your paper and how do they impact this branch of the field?
Though there are studies that have attributed quite a lot of functions to RNAi in fungi, a lot is still unknown. Our results show a role for RNAi machinery in control of the conidial transcriptome, especially on ribosome biogenesis genes. The relevance of this role was shown later in our publication as the loss of RNAi resulted in decreased fitness over time, since knockout mutants produced less spores upon serial passaging. Our study has provided a number of tools and assays that can now be used to further dissect the role of RNAi in an important filamentous fungal pathogen.
What led you to study RNA or this aspect of RNA science?
Over the last decade a lot of impactful studies have reported several functions attributed to RNA other than the conventional knowledge of genetic message translation. A plethora of RNA species have been discovered with various functions, which is already very exciting to know. However, I decided to study RNAi in pathogenic fungi because of the urgent need to find other therapeutic options for treating fungal infection. I believe that the RNAi pathway in fungi could be exploited in this sense and therefore needs to be studied.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
As a kid in high school I was very curious about science, especially the existence of life forms, and so I picked up an interest in biology. Once in high school we had a science fair, and on this occasion I saw bacteria for the first time using a microscope. It was just like a light bulb was lit in my head. Then I knew I had to study microorganisms for my bachelor's degree. Since then, knowing that there is an endless amount of processes taking place on a cellular level of these very little life forms has always kept me curious.
If you were able to give one piece of advice to your younger self, what would that be?
Keep giving it your 100% in spite of the challenges.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My supervisor has been a very huge influence on my approach to science. I believe I have grown so much as a scientist under his mentorship. In addition, working at the Leibniz Institute for Natural Product Research and Infection Biology, which has a reputation for fungal infection research, has also contributed to my development as a scientific researcher.








