
Plasticity and conditional essentiality of modification enzymes for domain V of Escherichia coli 23S ribosomal RNA
- Josefine Liljeruhm1,
- Margus Leppik2,
- Letian Bao1,
- Triin Truu2,
- Maria Calvo-Noriega1,
- Nicola S. Freyer1,
- Aivar Liiv2,
- Jinfan Wang1,
- Rubén Crespo Blanco1,
- Rya Ero2,
- Jaanus Remme2 and
- Anthony C. Forster1
- 1Department of Cell and Molecular Biology, Uppsala University, Uppsala 75124, Sweden
- 2Department of Molecular Biology, University of Tartu, 51010 Tartu, Estonia
- Corresponding authors: jaanus.remme{at}ut.ee, a.forster{at}icm.uu.se
Abstract
Escherichia coli rRNAs are post-transcriptionally modified at 36 positions but their modification enzymes are dispensable individually for growth, bringing into question their significance. However, a major growth defect was reported for deletion of the RlmE enzyme, which abolished a 2′O methylation near the peptidyl transferase center (PTC) of the 23S rRNA. Additionally, an adjacent 80-nt “critical region” around the PTC had to be modified to yield significant peptidyl transferase activity in vitro. Surprisingly, we discovered that an absence of just two rRNA modification enzymes is conditionally lethal (at 20°C): RlmE and RluC. At a permissive temperature (37°C), this double knockout was shown to abolish four modifications and be defective in ribosome assembly, though not more so than the RlmE single knockout. However, the double knockout exhibited an even lower rate of tripeptide synthesis than did the single knockout, suggesting an even more defective ribosomal translocation. A combination knockout of the five critical-region-modifying enzymes RluC, RlmKL, RlmN, RlmM, and RluE (not RlmE), which synthesize five of the seven critical-region modifications and 14 rRNA and tRNA modifications altogether, was viable (minor growth defect at 37°C, major at 20°C). This was surprising based on prior in vitro studies. This five-knockout combination had minimal effects on ribosome assembly and frameshifting at 37°C, but greater effects on ribosome assembly and in vitro peptidyl transferase activity at cooler temperatures. These results establish the conditional essentiality of bacterial rRNA modification enzymes and also reveal unexpected plasticity of modification of the PTC region in vivo.
Keywords
INTRODUCTION
Protein synthesis by ribosomes (translation) is central to all life and constitutes the major target of antibiotics. Ribosome assembly and function is exceedingly complex as ribosomes contain more than 50 protein and RNA components and they interact with numerous translation factors, tRNAs, and mRNAs. Yet, considerable strides in understanding have been made, most commonly using Escherichia coli (e.g., Davis et al. 2016) as a model system for translation and antibiotics affecting translation. The large ribosomal RNAs of all organisms are modified post-transcriptionally, with E. coli 16S and 23S rRNAs being modified at 36 nts (Purta et al. 2009; Sergiev et al. 2018), but the functions of these modifications remain largely enigmatic.
Most rRNA modifications cluster around functionally active regions, such as the peptidyl transferase center (PTC) loop (Fig. 1A), the A, E, and P sites, and the polypeptide exit tunnel, suggesting functional importance (Brimacombe et al. 1993; Decatur and Fournier 2002). On the other hand, three-dimensional rRNA modification maps (Decatur and Fournier 2002) lacked complete phylogenetic conservation (e.g., between E. coli and Saccharomyces cerevisiae) and all E. coli rRNA modification enzymes can be knocked out (KO'ed) individually (Baba et al. 2006). In these single KO strains, decreases in growth rate were minimal, if any (e.g., Purta et al. 2009; Golovina et al. 2012; Kimura et al. 2017; Pletnev et al. 2020), even for single-enzyme KOs that prevent modification at three sites in 23S rRNA (e.g., ΔrluC; Conrad et al. 1998; Huang et al. 1998). The most significant decrease in growth rate was two- to fourfold in ΔrlmE, which abolishes the 2′O methylation of U2552 in the A loop near the PTC (Bügl et al. 2000; Caldas et al. 2000; Arai et al. 2015). This KO decreases translational frameshifting in vivo (Widerak et al. 2005), but the U2552G mutation can exhibit wild-type (WT) growth if combined with U2555C (Sato et al. 2006). Even a strain combining KOs of all 11 pseudouridine (Ψ and m3Ψ) modifications of E. coli rRNAs had only a modest reduction in growth rate (O'Connor et al. 2018), despite that an rRNA pseudouridine synthase is essential in mitochondria (Zaganelli et al. 2017). Some modifications impart antibiotic resistance (Toh and Mankin 2008). Indirect effects on translation have also been implicated, such as aiding ribosomal subunit assembly by avoiding misfolding (Grosjean 2005), preventing hydrolysis of internucleotide bonds and protecting the preassembled rRNA from ribonucleases (Decatur and Fournier 2002; Nierhaus and Lafontaine 2004). In addition, modifications can increase base stacking interactions and stabilize RNA helices and other structures (Hayrapetyan et al. 2009; Westhof 2019; Wang et al. 2020).
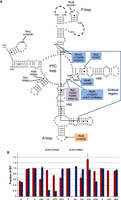
Knocking out key modifications in E. coli 23S rRNA. (A) Secondary structure of domain V including the “critical region” of the 23S rRNA (boxed) around the PTC loop (adapted from Punekar et al. 2013). Shown are all domain V modifications (ho5C is partial), modification enzymes KO'ed in this study (inside colored boxes), and known modification enzymes remaining (black). (B) Direct evidence for reductions in modified nucleotides in 50S subunits due to two different combinations of KOs (ΔrluC/ΔrlmKL/ΔrlmN/ΔrlmM/ΔrluE is abbreviated to ΔCKLNMuE). rRNAs were digested into nucleosides that were separated by HPLC (Supplemental Fig. S2), and modification peak areas were compared with WT controls normalized to 1. Standard errors of two to three biological replicates are given with absent peaks indicated by absent bars. ΔrluC/ΔrlmE should delete 3/9 Ψs (RluC) and 1/1 Um (RlmE). ΔCKLNMuE should delete 4/9 Ψs (RluC and RluE), 1/1 m7G (RlmKL), 1/2 m2G (RlmKL), 1/1 m2A (RlmN), and 1/1 Cm (RlmM). The bars decreased by amounts close to those expected.
Given the redundancy of bacterial rRNA modification enzymes, combination KOs are needed to determine their significance in cell viability. Such studies are also needed to provide a realistic pathway toward in vitro synthesis of E. coli ribosomes (and eventually self-replication; Forster and Church 2006), where the complex, partially sequential, and slow modification enzymology is a daunting bottleneck. In this vein, we have reconstituted specific and efficient post-transcriptional modification of unmodified 23S rRNA with two enzymes separately (Punekar et al. 2012, 2013) and four enzymes simultaneously (J Liljeruhm and AC Forster, unpubl.). But introduction of all 36 rRNA modifications in vitro is impractical, so we would like to know which are essential in combination. A landmark in vitro study produced noncovalently linked full-length hybrids between various 5′ and 3′ portions of the native (modified) 23S rRNA and in vitro–transcribed (unmodified) portions (Green and Noller 1996). Upon subsequent reconstitution of 50S subunits, only an 80-nt “critical natural element” was required to be modified in all cases for catalysis of the fMet-puromycin “fragment” reaction. This critical region lay around the PTC in domain V of the 2904-nt RNA and included seven modified nucleotides (Fig. 1A). To date, all but one of these seven modifications have a corresponding enzyme identified for their synthesis: RlmKL (m2G2445 modified by the L domain; m7G2069 near the critical region modified by the K domain; Lesnyak et al. 2006; Kimura et al. 2012), RluE (Ψ2457; Del Campo et al. 2001), RlhA (ho5C2501; only partially modified; Kimura et al. 2017), RlmM (Cm2498; Purta et al. 2009), RlmN (m2A2503; also m2A37 in the anticodon of tRNAs ArgICG, AspQUC, Glncmnm5s2UUG, GlnCUG, Glumnm5s2UUC, and HisQUG; Toh et al. 2008; Benitez-Paez et al. 2012), and RluC (Ψ2504; also modifies Ψ2580 and Ψ955; Conrad et al. 1998; Huang et al. 1998). Although the enzyme that synthesizes the dihydrouridine at U2449 is still unidentified and U2449 is highly conserved in other species, a point mutation of this U to a C had no direct change in phenotype (O'Connor et al. 2001). Functional 50S subunits using unmodified E. coli 23S rRNA could be achieved in in vitro reconstitutions by adding a special osmolyte and antibiotic, although activity was still down by two orders of magnitude (Semrad and Green 2002). On the other hand, ribosomal 50S subunits from two thermostable bacteria reconstituted from unmodified rRNA transcripts efficiently catalyzed the fMet-puromycin fragment reaction (Green and Noller 1999; Khaitovich et al. 1999).
Based on the survey above, we considered domain V to be a promising place to continue the search for (i) the first combination of rRNA modification enzymes that is essential in bacteria, and (ii) new dispensable combinations.
RESULTS AND DISCUSSION
Identification of a combination of 23S rRNA modification enzymes that is conditionally essential in E. coli
The first KO we picked to study was ΔrlmE because it is the only E. coli 23S rRNA modification enzyme KO with a substantial reduction in growth. However, picking a second KO to combine with it with the aim of further decreasing growth was less obvious. We chose the Ψ synthase RluC because (i) it isomerizes U2580, which is close to Um2552 (18 Å) in the 3D structure, (ii) ΔrluC provides the added benefit of preventing two other 23S rRNA modifications: Ψ2504 and Ψ955, and (iii) Ψ2504 lies in the overlap between the PTC loop and critical region of the 23S rRNA (Fig. 1A).
Nevertheless, obtaining a very severe phenotype of this double KO combination seemed an unlikely prospect given that a KO combination including ΔrluC, which prevented all 11 pseudouridine modifications (Ψ and m3Ψ), only modestly reduced the growth rate (O'Connor et al. 2018).
The ΔrluC/ΔrlmE double KO was constructed by phage P1 transduction in one single strain of E. coli MG1655 and verified (Fig. 1B; Supplemental Fig. S1 and Materials and Methods). The growth rate of the resulting strain was noticeably slower on plates at 37°C than either of its composite single KOs or WT (Fig. 2D, rows 1, 2, 5, and 7). Although E. coli is well adapted to growth at other temperatures (Barria et al. 2013), the double KO cells were nonviable at 20°C and only grew poorly at, or above, 25°C (Fig. 2A–E, row 2; note the contrast with ΔrlmE in row 7). This phenotype of ΔrluC/ΔrlmE is the most severe yet seen for bacterial rRNA modification enzyme KOs. Such cold-sensitive phenotypes are typical for mutants that destabilize RNA structure or affect ribosome assembly (Nierhaus and Lafontaine 2004; Barria et al. 2013; Aleksashin et al. 2019). Even at 37°C, the measured doubling time of ΔrluC/ΔrlmE in liquid culture was substantially slower than either of its composite single KOs (Fig. 2F; strains with just ΔrlmE or ΔrluC gave the expected decreased and similar growth rates, respectively, compared with WT; see Introduction). The surprising severity of the ΔrluC/ΔrlmE phenotype was not due to an inadvertent, nontargeted mutation(s) on the chromosome based on rescue experiments using WT genes. In quantitative growth-rescue experiments (Fig. 2F), ΔrluC/ΔrlmE (row 2) was rescued by RlmE plasmid (row 4) to give ΔrluC-level growth (row 5). Furthermore, ΔrluC/ΔrlmE (row 2) was rescued by RluC plasmid (row 3) to ΔrlmE-level growth (row 7). The dot growth assays of Figure 2A–E, although not quantitative, showed a similar overall trend. The ribosome sucrose gradients of the rescued strains in Supplemental Figure S4B (see below) were also consistent with the growth rescues.
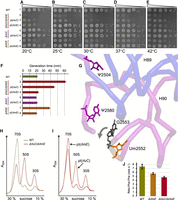
Assays of growth rate, ribosome sucrose gradient profile, and tripeptide synthesis for ΔrluC/ΔrlmE. (A–E) Representative spot growth assays at various temperatures with progressive dilutions of cells on chloramphenicol agar plates (see Materials and Methods). Rescue experiments used plasmids (pl) encoding the genes indicated (in addition to CmR). Rows 1, 2, 5, and 7 carried an empty vector to impart CmR (not indicated), and other controls are in Supplemental Figure S3. (F) Generation times in liquid cultures at 37°C. The control plasmid is not indicated (rows 1, 2, 5, and 7 had the empty vector carrying CmR). Standard errors are from at least seven biological replicates. (G) 3D proximity (Borovinskaya et al. 2007) of two of the three RluC-modified nucleotides (purple), the RlmE-modified nucleotide (orange), and the rRNA nucleotide G2553 (gray) whose relative position is affected the most (arrow) by ΔrlmE (Wang et al. 2020). Part of the nearby backbone, including that of the four nucleotides, is also shown (backbone inside the critical region is blue, outside is purple; see Fig. 1A). (H,I) Ribosome sucrose gradients of unrescued (H) and rescued (I) ΔrluC/ΔrlmE grown according to F. Sedimentation is from right to left, and other sucrose gradients according to F are in Supplemental Figure S4. Representatives of two biological replicates are shown. (J) Rates of tripeptide synthesis at 37°C. WT 7.5 ± 0.59 sec−1, ΔrlmE 5.7 ± 0.25 sec−1, ΔrluC/ΔrlmE 4.8 ± 0.24 sec−1. One-tailed P = 0.0095 for ΔrluC/ΔrlmE vs. ΔrlmE, P = 0.0045 for ΔrluC/ΔrlmE vs. WT; P = 0.015 for ΔrlmE vs. WT.
As ΔrluC exhibited no discernible growth defect, it must have potentiated the ΔrlmE defect. This potentiation may be a consequence of the close proximity of one RluC modification site (Ψ2580) to the nucleotide (G2553) which was recently reported to move the most due to the lack of modification of U2552 (Fig. 2G; Wang et al. 2020). However, it should be noted that the KO defects might not be due to the lack of modifications per se because moonlighting functions of the modification enzymes are possible.
Given the known role of RlmE in 50S assembly, which reduces the 70S to subunit ratio (Bügl et al. 2000; Caldas et al. 2000; Tan et al. 2002), we obtained ribosome sucrose gradients of the mutants. Indeed, ribosome assembly was severely impaired in ΔrluC/ΔrlmE (Fig. 2H), similar to ΔrlmE (Supplemental Fig. S4A,C). However, additional peaks due to accumulated assembly intermediates were not seen. As expected, the assembly defects were rescued by the plasmids to extents (Fig. 2I; Supplemental Fig. S4B,D) analogous to the extents of rescue of the growth rates (Fig. 2F). In vivo frameshifting assays did not explain the potentiation of the two KOs either (see below), so we turned to in vitro translation kinetics.
70S ribosomes purified from sucrose density gradients from ΔrluC/ΔrlmE, ΔrlmE, and WT strains grown at 37°C showed similar activities (generally from ∼50% to 75% active), as determined by single turnover dipeptide synthesis yields with all other components in excess of ribosomes. At equivalent active fractions of 70S ribosomes, a marked deficit was seen in tripeptide, not dipeptide, synthesis for ΔrlmE compared with WT, matching the previously published translocation deficit (see Fig. 5B and C of Wang et al. 2020). Interestingly, we found that ΔrluC/ΔrlmE had a significantly greater deficit in tripeptide synthesis than ΔrlmE (Fig. 2J). As dipeptide formations in these same reactions occurred equally fast for all three types of ribosomes, this suggested a deficit in translocation. This greater deficit, in combination with the aforementioned assembly defect, may thus be the reason for the conditional lethality of ΔrluC/ΔrlmE cells.
Feasibility of combined knockouts of “critical region” modification enzymes
Having established one conditionally essential combination of rRNA modification enzymes around the PTC, we next tested for essentiality of other combinations of domain V modification enzymes. We aimed to KO as many of the known modifying enzymes for the critical region (Fig. 1A) as possible in a single strain by successive P1 transductions. We did not KO RlhA because the gene was only identified recently (Kimura et al. 2017) and C2501 is only partially modified. Furthermore, the RlhA KO slightly increases translation activity (Fasnacht et al. 2022). We did not combine these KOs with an RlmE KO because it does not modify within the critical region.
To our surprise, MG1655 survived through five KOs of all the known enzymes that modify fully the critical region to yield ΔrluC/ΔrlmKL/ΔrlmN/ΔrlmM/ΔrluE (here abbreviated to ΔCKLNMuE, where the order in which the KOs were created is listed from left to right and uE differentiates ΔrluE from ΔrlmE). The five KOs in the strain were verified as above (Fig. 1B; Supplemental Fig. S2 and Materials and Methods). Despite lacking eight rRNA and six tRNA modifications (totaling 14), the colonies at all temperatures were only slightly smaller than WT, with the growth defect being stronger at 20°C (Fig. 3A–E). Liquid culture of ΔCKLNMuE at 37°C showed only a very modest increase in doubling time (Fig. 3F).
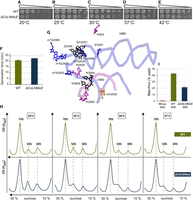
Assays of growth rate, ribosome sucrose gradient profile and peptidyl transferase with ΔCKLNMuE. (A–E) Representative spot growth assays on chloramphenicol agar plates (see Materials and Methods). Both strains carried a control plasmid (empty vector carrying CmR). (F) Generation times of liquid cultures at 37°C (standard errors of eight biological replicates). (G) 3D proximity of all modified nucleotides of domain V (Borovinskaya et al. 2007) colored according to their respective modification enzymes in Figure 1A. ΔCKLNMuE lacks the modifications on the 8 nts colored blue and purple (and also lacks m2A37 modifications in six tRNAs, which are not shown). The entire backbone of the critical region is given (blue), as is the backbone that base pairs with it in H90 (purple; see Fig. 1A). (H) Ribosome sucrose gradients of cells grown at the temperatures indicated, with representatives of two biological replicates shown. (I) Representative plots of peptidyl transferase puromycin “fragment” reactions in vitro (standard errors of three technological replicates).
These eight rRNA modifications cluster in the ribosome around the functionally important PTC (Fig. 3G). Yet, major potentiation effects of the combined KOs were not seen at 37°C, in contrast with ΔrluC/ΔrlmE. Consistent with the very modest growth effects of ΔCKLNMuE at 37°C, there was little difference in ribosome assembly seen on ribosome sucrose gradients at 37°C (Fig. 3H, right). However, with decreasing growth temperature, the heights of the 50S peaks relative to 70S peaks increased for the mutant more than the WT (Fig. 3H). This shows that ribosome assembly in the ΔCKLNMuE strain is cold-sensitive, although the effects are less pronounced than in ΔrlmE at 37°C (compare with Supplemental Fig. S4A).
Thus, our combination of modifications which, together with D2449 and partially modified ho5C2501 constitute the critical modifications in vitro (Green and Noller 1996; Semrad and Green 2002), is not critical in vivo nor even critical for fast growth at 37°C. Given the many differences between the in vivo growth assay and the in vitro reconstitution/fragment reaction assay, there are many possible explanations for this apparent discrepancy. For example:
-
The in vitro assay removed, in addition to all the critical region modifications, either all modifications upstream of the critical region (=12, not counting the RluC or RlmKL modifications) or all modifications downstream (=3, not counting the RluC modification). It is possible that the absence of these extra modifications potentiated the absence of the five modifications in the critical region.
-
The in vitro assay required hybridization of noncovalently-linked 23S rRNA pieces.
-
The in vitro defect may be in the unphysiological, two-step, rRNA folding/subunit reconstitution reaction rather than in peptidyl transferase per se.
-
The fragment reaction uses unnatural substrates and 33% methanol on ice, is very slow, and evidence indicates it is not wholly indicative of in vivo peptidyl transferase (Youngman et al. 2004; Wohlgemuth et al. 2008; Johansson et al. 2011).
-
It is possible that the ΔCKLNMuE strain is bolstered by pseudoreversions.
Given that our observed growth defect with ΔCKLNMuE became severe at the lowest growth temperature of 20°C (Fig. 3A), we wondered whether the standard 0°C incubation of the fragment reaction might have been responsible for the four orders of magnitude inhibition of peptidyl transferase measured for the unmodified critical region (Green and Noller 1996). However, compared with WT, ΔCKLNMuE 50S subunits had only about a twofold decrease in our in vitro puromycin “fragment” reaction (Fig. 3I; Supplemental Fig. S5). Nevertheless, this is still a major ribosomal catalytic defecit that may cause the cold sensitivity in cells.
Effects of combined modification enzyme KOs on translational frameshifting
Interestingly, ΔrlmE decreased translational frameshifting in vivo (Widerak et al. 2005). The explanation favored by the authors was that the unmodified A loop binds less strongly to the CCA end of the A-site tRNA, thereby increasing the relative contribution of anticodon–codon binding. In addition, several other molecular mechanisms were considered to explain the effect of this mutation (and peri-PTC mutations in general) on frameshifting. In contrast, ΔrlmN increased readthrough (Benitez-Paez et al. 2012). This effect was attributed to the single rRNA modification KO rather than the concurrent six tRNA m2A37 KOs because the tRNAs with undermodified anticodons were expected to be hyper-accurate and insufficiently near-cognate with the single UAG codon tested (Benitez-Paez et al. 2012). We therefore wondered what occurs for ΔrluC/ΔrlmE and ΔCKLNMuE.
Given the small growth defect observed at 37°C for ΔCKLNMuE, we desired improved translational assays over the original β-galactosidase plasmid assays (Widerak et al. 2005; Benitez-Paez et al. 2012). We thus used a fusion of Renilla luciferase with Firefly luciferase (Devaraj et al. 2009) because the second luciferase acts as an internal reference to control for the effects of individual sample variables such as exact plasmid copy number, efficiency of cell lysis, etc., thus reporting more specifically and precisely on translation per se. We constructed six plasmids (Fig. 4A; see Materials and Methods) differing in the linker region between the genes such that the two luciferases were either in frame or frameshifted. Five different types of frameshifting were assayed to help control for potential effects of different codons at the frameshifting sites, mindful that ΔCKLNMuE lacks modifications in six different tRNAs in addition to lacking eight rRNA modifications.
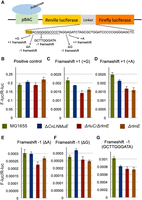
Effects on translational frameshifting in vivo of various KO strains. (A) Drawing of the dual luciferase fusion gene (top; positive control) and the sequences of five different frameshifting mutants (bottom; see Materials and Methods). (B–G) Ratio of Firefly and Renilla luciferase luminescence signals for the indicated strains carrying a plasmid from A. B used the top sequence in A while G used the 11-nt mutated region. Standard errors of at least five biological replicates are given. P < 0.05 for ΔCKLNMuE vs. WT in C, D, F, and G; for ΔrlmE vs. WT in B, C, D, F, and G; for ΔrluC/ΔrlmE vs. WT in C, D, E, and G; for ΔrluC/ΔrlmE v ΔrlmE in B.
In ΔrlmE, there was lower frameshifting for all five frameshifting constructs compared with MG1655 (Fig. 4C–G), consistent with the original report (Widerak et al. 2005), although our effects were smaller. ΔrluC/ΔrlmE behaved quite similarly to ΔrlmE, also frameshifting less than WT. ΔCKLNMuE was generally less affected in frameshifting and showed a less-consistent trend across the five constructs. This was also true at 20°C (Supplemental Fig. S6), despite the stronger growth defect at that temperature (Fig. 3A). The ΔCKLNMuE results may be indicative of codon-specific contexts where the tRNA undermodifications may decrease frameshifting in opposition to increased frameshifting due to rRNA undermodifications. Whatever the reason, the effects for ΔCKLNMuE were overall not as strong as seen with the more severe mutants ΔrluC/ΔrlmE and ΔrlmE.
Conclusions
We began this study by hypothesizing (i) that a combination KO of several rRNA modification enzymes would be required to finally prove their essentiality in bacteria, and (ii) that such a demonstration would likely involve critical-region-modifying enzymes as their modifications were reported to be required for peptidyl transferase in vitro. While we did prove conditional essentiality of the modification enzymes (at 20°C), associated with a combined defect in ribosomal assembly and in vitro tripeptide synthesis, to our surprise this only required two KOs involving only one critical-region enzyme and four modifications altogether. Also unexpected was that five of the seven critical-region enzymes, responsible for 14 rRNA and tRNA modifications altogether, were simultaneously dispensable. These KOs neither inactivated the peptidyl transferase nor even caused a major growth defect at 37°C. However, there was a strong growth defect at 20°C, again associated with reduced ribosomal catalysis measured in vitro. Hence, there is unexpected plasticity of rRNA modifications in vivo in the critical region. This clarifies the relative significance of most of the modifications in domain V and should simplify efforts to synthesize in vitro the E. coli ribosome toward the synthesis of self-replication.
Although our experiments focused on tRNA and 23S rRNA modifications in E. coli, there are implications for other organisms in which they are conserved. Um2552 (RlmE) and Ψ2457 (RluE) modifications are well conserved at homologous positions and are nearly universal. Both of them are present in S. cerevisae and Homo sapiens (Boccaletto et al. 2018). Um2552 is made by two independent enzyme systems in yeast, the stand-alone enzyme Spb1 and the small nucleolar RNA snR52-directed system, emphasizing the importance of this modification (Bonnerot et al. 2003). In spite of the doubled synthesis pathway, a lack of methylation at this position is not lethal in yeast but is accompanied by a severe growth disadvantage (Bonnerot et al. 2003). Interestingly, Spb1-directed methylation occurs at the late stages of 60S ribosomal subunit assembly (Lapeyre and Purushothaman 2004) similar to E. coli enzyme RlmE (Siibak and Remme 2010). No specific phenotype associated with Ψ2457 is known. In contrast, pseudouridines installed by RluC are found only in bacteria; the homologous uridines are not isomerized in S. cerevisae and H. sapiens (Boccaletto et al. 2018). Guanosines methylated by RlmKL (m7G2069 and m2G2445) and methylations directed by RlmM and RlmN are not conserved. Thus, the functions of most of these modifications cannot be general.
MATERIALS AND METHODS
Materials
Tritium-labeled Met was purchased from PerkinElmer. All other chemicals and reagents were purchased from Sigma-Aldrich or Merck. E. coli 70S ribosomes, translation factors, and fMet-tRNAfMet were prepared as described (Pavlov et al. 1997). Note that MRE600 was used as “WT” for these in vitro preparations because it contains low RNase I activity when compared with MG1655 WT. Synthetic XR7 fMet-Phe-Phe mRNA was prepared as described (Wang et al. 2014) with the sequence:
-
5′GGGAAUUCGGGCCCUUGUUAACAAUUAAGGAGGU
-
AUUAAAUGUUCUUCUAAUUGCAGAAAAAAAAAAA
-
AAAAAAAAAA3′
The Shine–Dalgarno sequence is underlined and the coding sequences are in bold (fMet: AUG, Phe: UUC). All kinetics experiments were conducted in HEPES-polymix buffer (pH 7.5) containing: 5 mM Mg(OAc)2, 95 mM KCl, 5 mM NH4Cl, 0.5 mM CaCl2, 1 mM spermidine, 8 mM putrescine, 1 mM dithioerythritol, and 30 mM HEPES-KOH pH 7.5.
Construction of deletion strains
The deletions were introduced sequentially into the MG1655 (F- lambda- ilvG- rfb-50 rph-1) E. coli K-12 strain by P1-mediated transduction. The order of transduction occurred as annotated in the names from left to right, initiating with MG1655 ΔrluC. The precursor single KO strains were from the KEIO collection (Baba et al. 2006) except for ΔrlmE (Caldas et al. 2000) and ΔrluE, constructed by the method of Martinez-Garcia and de Lorenzo (2011). In between each subsequent deletion step, the kanamycin resistance cassette was removed by transient expression of the flippase (FLP) recombinase (Datsenko and Wanner 2000).
Confirmations of KOs
Oligodeoxyribonucleotides that primed ∼100 bp upstream and downstream from each modification enzyme gene position were used to amplify either the genes, kanamycin cassette, or the flippase recognition target (frt) site from the WT (MG1655), ΔrluC/ΔrlmE (MG1655 ΔrluC::frt ΔrlmE::Km), ΔCKLNM (MG1655 ΔrluC::frt ΔrlmKL::frt ΔrlmN::frt ΔrlmM::frt), and ΔCKLNMuE (MG1655 ΔrluC::frt ΔrlmKL::frt ΔrlmN::frt ΔrlmM::frt ΔrluE::Km) strains. The KO strains generated PCR products with expected sizes of either the smaller fragment for each deleted gene (an frt site) or the size of the kanamycin resistance cassette, while the WT strain generated fragments of sizes expected for intact genes (e.g., Supplemental Fig. S1).
50S ribosomal subunits from the MG1655, ΔrluC/ΔrlmE, and ΔCKLNMuE strains were obtained by purifying the 70S ribosome peak on a sucrose gradient, dissociating the subunits, and then purifying the 50S peak on a second sucrose gradient (to avoid contamination by tRNAs with their associated modifications that remained bound to 70S; see below). The nucleoside composition of the combined 23S and 5S rRNAs was analyzed by HPLC (Fig. 1B; Supplemental Fig. S2) as described (Gehrke and Kuo 1989). In short, rRNAs extracted from 50S subunits were degraded by nuclease P1 (Sigma) and treated with bacterial alkaline phosphatase (Thermo Fisher Scientific). The samples were then run on a Supelcosil LC-18-S reverse-phase HPLC column, the nucleoside peaks were identified based on relative mobilities, and peak areas were integrated.
In further control experiments, severe phenotypes were rescued to rule out an inadvertent, nontargeted mutation(s) on the chromosome. Thus, ΔrlmE and ΔrluC/ΔrlmE were transformed by a low-copy plasmid (pHB, a pSC101 derivative encoding CmR) encoding RlmE downstream from a noninduced tac promoter for leaky expression (Lilleorg et al. 2020). Additional related controls were performed with plasmid-borne RluC.
Growth assays
Spot growth assays were done with 4.5 µL aliquots from six dilutions of three biological replicates corresponding to optical density measurements at 600 nm (OD600) = 10−3, 5 × 10−4, 10−4, 10−5, 2 × 10−6, and 10−6 spotted on LB agar plates, containing 25 µg/mL chloramphenicol, and incubated at 20°C (2.5–3 d), 25°C (40 h), 30°C (21 h), 37°C (16 h), or 42°C (16 h). Growth rates in liquid culture at 37°C were measured in Bioscreen C Analyzer (Oy Growth Curves Ab Ltd.) or Infinite M200 Pro plate reader (Tecan). Each well was inoculated with a 1000-fold dilution in LB media from an overnight culture grown in LB media supplemented with 25 µg/mL chloramphenicol. OD600 measurements were taken every 5 min for 24 h. The calculations were based on OD600 values between 0.03 and 0.07.
Ribosome sucrose gradients
These were done as described (O'Connor et al. 2018). Essentially, cultures were grown at 37°C in 100 mL 2xYT media until late logarithmic phase and pellets from 50 mL aliquots collected by low-speed centrifugation at 4°C. Each pellet was resuspended in 1 mL buffer (20 mM Tris-HCl pH 7.5, 100 mM KCl, 10 mM Mg(OAc)2, 6 mM 2-mercaptoethanol) and lysed with glass beads in a Precellys 24 homogenizer (Bertin Instruments) according to the manufacturer's protocol. Up to 700 µL of each lysate was layered onto a 10% to 30% (w/v) sucrose gradient prepared in the above buffer and centrifuged in a Beckman SW28 rotor at 22,000 rpm for 16 h 44 min (ω2t = 3.2 × 1011) at 4°C. Ribosome sucrose gradients were visualized by continuously monitoring at 260 nm, the peaks were quantitated, and 50S subunits were purified (see above) for modification analysis and puromycin “fragment” reactions. For tripeptide assays, ΔrlmE and ΔrluC/ΔrlmE cells were grown to ∼0.8 OD600 at 37°C in LB medium and collected by centrifugation, then the 70S peak was purified from a sucrose gradient as published (Johansson et al. 2008).
In vitro peptidyl transferase reactions
Puromycin “fragment” reactions were performed and stopped exactly as described (Green and Noller 1996), except that the 5′-truncated fMet-tRNAfMet fragment was substituted by full-length fMet-tRNAfMet prepared from overexpressed tRNA (Meinnel and Blanquet 1995). Ribosomal 50S subunits (0.1 µM final reaction conc.) were preincubated on ice for 5 min with-f[3H]Met-tRNAfMet (0.05 µM final reaction conc.) in 0.4 M KOAc, 50 mM Tris-HCl pH 8.3, 60 mM MgCl2 (65.4 µL, final reaction concs.). Reactions were initiated with aqueous, neutralized puromycin (1.6 µL; 1 mM final conc.) and ice-cold methanol to 33% (final reaction volume of 100 µL) and incubated on ice for 20 min. Reactions were stopped with 2 M KOH (0.4 M final conc.) and incubated at 37°C for 15 min. For our analytical method, the mixture was neutralized and tRNAs precipitated with formic acid (17% final conc.) and incubated on ice for 10 min. After centrifugation (14,000 rpm, 4°C, 15 min), supernatants were analyzed by C18 reverse phase HPLC coupled with a β-RAM model 4 radioactivity detector (LabLogic Systems). Separation of f[3H]Met-puromycin from f[3H]Met was achieved by elution with 42% methanol/58% H2O/0.1% trifluoroacetic acid at 0.45 mL/min for 25 min (Supplemental Fig. S5).
Tripeptide formation assay
Active concentrations of ribosomes (typically ∼50%–75% of the total within the 70S peak on a sucrose gradient for all types of ribosomes here), fMet-tRNAfMet, EF-Tu, and elongator tRNAPhe were determined through the yield of dipeptide formed when the assayed component was limiting. 70S initiation complex (IC) and elongation mixture (EM) were formed by preincubating at 37°C for 20 min separately (Wang et al. 2014). IC contained 2 μM 70S ribosomes, 5 μM XR7 fMFF mRNA, 2.4 μM [3H]fMet-tRNAfMet, 2 μM IF1, 2 μM IF2, and 2 μM IF3. EM contained 20 μM EF-Tu, 5 μM EF-Ts, 5 μM EF-G, 10 μM tRNAPhe, 0.4 mM phenylalanine, and 0.4 μM Phe-tRNA synthetase. Both IC and EM were prepared in HEPES-polymix buffer (pH 7.5) supplemented with 1 mM ATP, 1 mM GTP, 10 mM phosphoenolpyruvate, 0.05 mg/mL pyruvate kinase, and 0.002 mg/mL myokinase for energy regeneration. Fast in vitro kinetics were done at 37°C in a temperature-controlled quench-flow apparatus (RQF-3, KinTeck Corp.). Equal volumes of IC and EM were rapidly mixed, and the reactions were quenched with formic acid (17% final) at different time points. The samples were then centrifuged at 20,000g at 4°C for 30 min and the supernatant removed. Next, 120 μL of 0.5 M KOH was added to the pellet to hydrolyze all the peptides and unreacted [3H]fMet from the tRNAs. Formic acid was added (17% final) to precipitate the deacylated tRNAs. After centrifugation at 20,000g at 4°C for 15 min, the supernatant was analyzed by C18 RP-HPLC coupled with a β-RAM model 3 radioactivity detector (IN/US Systems). Separation of [3H]fMet-Phe-Phe, [3H]fMet-Phe, and [3H]fMet was achieved by elution with 50% methanol/50% H2O/0.1% trifluoroacetic acid for 20 min at 0.45 mL/min. The fraction of [3H] peptide out of the total [3H] signal at each time point was calculated, and the data were fitted to a single exponential function with Origin 7.5 (OriginLab Corp.).
In vivo frameshift assays
These used Renilla luciferase and Firefly luciferase constructs as a fusion protein under the BAD promoter on a chloramphenicol-resistant plasmid. The six plasmids (Fig. 4A) were constructed as recently published in Lilleorg et al. (2020) based on Devaraj et al. (2009). They differed in the linker region between the genes, being either in frame (pRFluc) or frameshifted, with two +1 frameshifting plasmids (pAD3, +G; pAD5, +A) and three −1 frameshifting plasmids (pAD2, ΔA; pAD4, ΔG; pAD7, CGGGGGCCCCT to GCTTGGGATA). Strains MG1655, ΔCKLNMuE, ΔRluC/ΔRlmE, and ΔrlmE, each carrying or lacking a dual luciferase plasmid (in six biological replicates), were grown in LB chloramphenicol overnight at 37°C (20°C in Supplemental Fig. S6), diluted and further grown to OD600 = 0.4–0.6 in the absence of antibiotic (to avoid its interference in the ribosome assay). They were then induced with 0.2% arabinose for 1 h, 0.25 mL of each culture was centrifuged at low speed, and the cell pellets were stored at −80°C. Lysis and addition of luminescence reagents were according to the kit manufacturer's protocol (Dual-Luciferase Reporter Assay System E1910, Promega). Briefly, pellets were dissolved in 62.5 µL Passive Lysis Buffer and 7 µL of each lysis mixture were added to 384-well black plates. Nonadjacent wells were necessary to avoid sample cross talk. The reading of relative light units (RLUs) was conducted using a SpectraMax iD5 Multimode Microplate reader (Molecular Devices) according to their software protocol. Briefly, the preprogrammed protocol dispenses 7 µL Luciferase Assay Reagent II from injector 1, waits 2 sec, reads (Firefly) luminescence for 10 sec, dispenses 7 µL Stop & Glo Reagent from injector 2, waits 2 sec, and reads (Renilla) luminescence for 10 sec. Background signals from strains lacking plasmids were subtracted from signals from the same strains bearing plasmids, and Firefly luciferase RLU was divided by Renilla luciferase RLU to obtain normalized ratios (see Supplemental Tables S1, S2 for source data).
3D images
Images were created using PDB 2QAM (Borovinskaya et al. 2007) and the PyMOL Molecular Graphics System (Schrödinger).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are very grateful to Gilbert Richarme for the ΔrlmE strain and Helena Danielsson for access to the SpectraMax. This work was supported by the Estonian Research Council (PSG295 to M.L.), an ERASMUS+ award (to M.C.-N.), the Estonian Ministry of Education and Research (PUT PRG1179 to J.R.), and the Swedish Research Council (NT project grants 2011-5787, 2016-1, and 2017-04148 to A.C.F.).
Author contributions: J.R. and M.L. designed the research; T.T. did the initial research; J.L., M.L., L.B., M.C.-N., N.S.F., J.W., R.C.B., and R.E. performed the remaining research; A.L. constructed new reagents; all authors analyzed data; A.C.F. and J.L. wrote the manuscript and all authors edited it.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079096.121.
- Received December 31, 2021.
- Accepted February 18, 2022.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Josefine Liljeruhm is the first author of this paper, “Plasticity and conditional essentiality of modification enzymes for domain V of Escherichia coli 23S ribosomal RNA.” Josefine recently completed her PhD in Professor Anthony Forster's laboratory at the Department of Cell and Molecular Biology, Uppsala University. Josefine is currently working at the company Cytiva in research and development.
What are the major results described in your paper and how do they impact this branch of the field?
After discovering ribosomal RNA modifications and identifying and characterizing their associated enzymes individually, their significance still remained largely unclear. Thus, the field is taking the next step of characterizing their importance in combination using gene knockout technology. In this manner, we established the conditional essentiality of bacterial rRNA modification enzymes and also found unexpected plasticity of modification in the active site of the ribosome.
What led you to study RNA or this aspect of RNA science?
The ribosome is a fascinating research object considering how it carries out various enzymatic tasks central to translation and evolution. In addition, the bacterial ribosome is the major target of antibiotics, and the functions of its numerous post-transcriptional RNA modifications remain enigmatic.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
As a teenager, I had a biology teacher who showed us images of an arctic fox, a red fox, and a desert fox to highlight environmental adaptations of species. I asked the obvious question (in my opinion): “How do we know the desert fox is a ‘fox’?” Right there, I first stepped into phylogenetics and DNA. My curiosity of what you could do with nucleic acid and proteins then motivated me through undergraduate and graduate school.
What are your subsequent near- or long-term career plans?
To utilize my understanding of laboratory and molecular biology techniques to further enhance the tools used in the field to drive the science forward.








