
Three critical regions of the erythromycin resistance methyltransferase, ErmE, are required for function supporting a model for the interaction of Erm family enzymes with substrate rRNA
- Rory E. Sharkey1,
- Johnny B. Herbert1,
- Danielle A. McGaha1,
- Vy Nguyen2,
- Allyn J. Schoeffler2 and
- Jack A. Dunkle1
- 1Department of Chemistry and Biochemistry, University of Alabama, Tuscaloosa, Alabama 35487, USA
- 2Department of Chemistry and Biochemistry, Loyola University New Orleans, New Orleans, Louisiana 70118, USA
- Corresponding authors: ajschoef{at}loyno.edu, jadunkle{at}ua.edu
Abstract
6-Methyladenosine modification of DNA and RNA is widespread throughout the three domains of life and often accomplished by a Rossmann-fold methyltransferase domain which contains conserved sequence elements directing S-adenosylmethionine cofactor binding and placement of the target adenosine residue into the active site. Elaborations to the conserved Rossman-fold and appended domains direct methylation to diverse DNA and RNA sequences and structures. Recently, the first atomic-resolution structure of a ribosomal RNA adenine dimethylase (RRAD) family member bound to rRNA was solved, TFB1M bound to helix 45 of 12S rRNA. Since erythromycin resistance methyltransferases are also members of the RRAD family, and understanding how these enzymes recognize rRNA could be used to combat their role in antibiotic resistance, we constructed a model of ErmE bound to a 23S rRNA fragment based on the TFB1M–rRNA structure. We designed site-directed mutants of ErmE based on this model and assayed the mutants by in vivo phenotypic assays and in vitro assays with purified protein. Our results and additional bioinformatic analyses suggest our structural model captures key ErmE–rRNA interactions and indicate three regions of Erm proteins play a critical role in methylation: the target adenosine binding pocket, the basic ridge, and the α4-cleft.
Keywords
INTRODUCTION
Methylation of adenosine residues in RNA occurs in all three domains of life (Grosjean 2009). The 6-methyladenosine (m6A) modification appears in eukaryotic mRNAs, playing a prominent role in gene regulation (Meyer et al. 2012; Schwartz et al. 2013; Pendleton et al. 2017; Frye et al. 2018). A related dimethylated adenosine, m62A, is installed in the 23S rRNA of some bacteria by an erythromycin resistance methyltransferase (Erm) and provides resistance to multiple antibiotics targeting the large ribosomal subunit (Fyfe et al. 2016).
Two protein folds account for the majority of RNA and DNA methyltransferases with the Rossmann-fold methyltransferase (RFM) fold being the most abundant and the SPOUT (SpoU-TrmD) fold the second most abundant (Czerwoniec et al. 2009). The SPOUT fold is distinguished by five parallel beta strands with the topology ↑2-↑1-↑4-↑3-↑5 and a carboxy-terminal α-helix that threads between a loop linking β-strands three and four to form a trefoil knot (Elkins et al. 2003). The RFM fold consists of seven β-strands with the topology ↑3-↑2-↑1-↑4-↑5-↓7-↑6. Key functional elements of this fold include a GxGxG motif following β-strand one that forms a binding surface for the S-adenosyl methionine cofactor, an acidic residue following β-strand two whose sidechain forms two hydrogen bonds, one with each of the hydroxyls of the S-adenosyl methionine ribose (Schubert et al. 2003). In enzymes that catalyze m6A formation in DNA or RNA, an additional motif follows β-strand four, originally denoted as [D/N/S]PP[Y/F], which is associated with positioning the substrate nucleotide (Schubert et al. 2003). Recent studies of eukaryotic, mRNA m6A methyltransferases, which consist of the RFM fold, confirmed the importance of this motif (Sledz and Jinek 2016; Wang et al. 2016; Doxtader et al. 2018). Erm proteins utilize the RFM fold and contain a slightly altered sequence following β-strand four, for example, AIPY in ErmE, that adopts the same sharply kinked structure, poised to interact with substrate adenosine, seen in other RFM fold adenosine methyltransferases (Stsiapanava and Selmer 2019).
Sequence conservation analysis indicates that Erm belongs to a protein family named the ribosomal RNA adenine dimethylase (RRAD) family (Fig. 1A; Mistry et al. 2021). In addition to Erm, this family consists of the proteins that dimethylate two conserved adenosine residues that appear in the conserved helix 45 of the small ribosomal subunit RNA of nearly all organisms (Fig. 1A,B; Cannone et al. 2002; Xu et al. 2008). Dimethylation of the two adenosine residues in helix 45 is strongly conserved across the three domains of life, due to the critical role these methylations (m62A1518 and m62A1519 in E. coli numbering) play in the maturation of the small ribosomal subunit (O'Farrell et al. 2006). Interestingly, the substrates for Erm, helix 73 of 23S rRNA, and the substrate for the other RRAD family members, helix 45 of small ribosomal subunit RNA, diverge in sequence and secondary structure (Fig. 1B). Erm proteins dimethylate an adenosine (underlined) that lies in an unpaired region between two rRNA helices within the sequence context 5′ CGGAAA 3′ (Villsen et al. 1999). The 5′ C residue is unpaired while the following two G residues participate in Watson–Crick pairs. Other RRAD family members dimethylate two adenosines located in the sequence context 5′NGAA 3′ (Xu et al. 2008). This nucleotide sequence is part of a tetraloop in helix 45 and is bordered by three Watson–Crick pairs. Understanding how Erm and other RRAD family members possess substantial structural similarity yet methylate RNA substrates differing in secondary structure and sequence will help to address the outstanding question of how the conserved RFM fold adapts to different RNA targets and whether discrete, identifiable elements of sequence or structure underlie target selectivity.
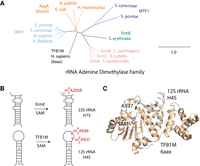
The TFB1M–rRNA crystal structure suggests how other members of the rRNA adenine dimethylase family of proteins may bind substrate RNA. (A) A phylogram of rRNA adenine dimethylase (RRAD) family proteins from model organisms. (B) A schematic for the methylation reaction performed by two RRAD family members, ErmE, and TFB1M, is shown. Both proteins dimethylate an adenosine residue adjacent to a base-paired stem region. (C) The crystal structure of TFB1M bound to substrate was recently solved, revealing details of its interactions with rRNA.
Erythromycin resistance methyltransferases, as indicated by their name, provide bacterial cells resistance to the macrolide antibiotic erythromycin and many other macrolides. These compounds consist of a 14–16 membered macrolactone ring, were among the first antibiotics to enter widespread use with the introduction of erythromycin in 1952, and include azithromycin, one of the most prescribed antibiotics in the United States (Schroeder and Stephens 2016). Macrolides inhibit bacterial growth by binding to 23S rRNA adjacent to the peptidyl transferase center of the ribosome disrupting protein synthesis (Bulkley et al. 2010; Dunkle et al. 2010; Vazquez-Laslop and Mankin 2018). Bacteria can become resistant to macrolides by expressing the mef efflux pump, by dislodging the drug from its binding site via the ABC-F protein MsrE, by modifying the drug with enzymes such as macrolide phosphotransferases (Mph), by incurring a mutation at or near the drug binding site or by dimethylation of A2058 at the drug binding site as Erm does (Fyfe et al. 2016; Wilson et al. 2020; Svetlov et al. 2021). A survey of greater than 4500 macrolide-resistant S. pneumoniae clinical isolates found that 48% contained the ermB gene alone or in conjunction with mef, underscoring the clinical importance of Erm-mediated resistance (Hawkins et al. 2015). Global surveys of the prevalence of macrolide resistance genes have also indicated a prominent role in Erm-mediated resistance (Schroeder and Stephens 2016). Additionally, Erm provides resistance to lincosamide (clindamycin), streptogramin B, and ketolide antibiotics, a phenotype known as MLSBK (McCusker and Fujimori 2012). This multidrug resistance phenotype emerges because each of these classes of antibiotics also binds 23S rRNA adjacent to the peptidyl transferase center of the ribosome in semi-overlapping sites (Bulkley et al. 2010; Dunkle et al. 2010; Noeske et al. 2014). Understanding the detailed mechanism of RNA recognition by Erm could aid in the development of selective Erm inhibitors with the potential to restore antibiotic efficacy when co-administered with relevant antibiotics, a strategy used for decades to evade β-lactamase resistance to penicillin-family antibiotics (Neu 1985).
In order to better understand how Erm enzymes interact with their RNA substrate, we generated a structural model of ErmE bound to a fragment of 23S rRNA guided by the recent RNA-bound structure of TFB1M, an rRNA methyltransferase with homology to Erm (Liu et al. 2019). We performed site-directed mutagenesis of ermE guided by the model and characterized the erythromycin resistance phenotype of cells transformed with the ermE variants. For selected ermE variants, the protein was purified and subjected to methylation kinetics assays and RNA affinity binding assays. Our data suggest that three regions of Erm proteins are crucial for recognition and methylation of RNA substrate: the adenosine pocket, the basic ridge, and the α4-cleft. Sequence analysis of a large database of pathogen-derived Erm sequences from clinical samples was used to determine to what degree our findings utilizing the ErmE model protein generalize to Erm enzymes in antibiotic-resistant pathogens.
RESULTS
A theoretical model of ErmE bound to substrate rRNA
Since no structure of an Erm protein bound to RNA exists, we searched the Protein Data Bank for structures of RRAD family members bound to substrate RNA that could provide clues to how ErmE interacts with RNA. Two structures are available that reveal an RRAD family member bound to its substrate RNA on the pathway for methylation: the recent cryo-EM structure of KsgA bound to the 30S ribosome and an X-ray crystal structure of TFB1M bound to a fragment of helix 45 of 12S rRNA (numbering of 12S rRNA is from Homo sapiens while numbering of 23S rRNA is from E. coli) (Liu et al. 2019; Stephan et al. 2021). The X-ray structure of TFB1M reveals the helix 45 tetraloop, nucleotides G934–A937, has abandoned the stacked structure, typical of a GNRA tetraloop, in which it is normally observed and instead formed intimate interactions with TFB1M (Fig. 1C). Additionally, the X-ray structure reveals that A937 has undergone base-flipping—it is dramatically rotated from its normal position stacking with adjacent tetraloop nucleotides so that it is positioned in the active site adjacent to the labile methyl group of the S-adenosylmethionine (SAM) cofactor (Fig. 1C). The KsgA-30S structure possesses an extremely similar orientation of KsgA and helix 45 of rRNA (Supplemental Fig. S1). This analysis, along with the strong sequence and structural conservation throughout the RRAD family, led us to hypothesize that all members of the RRAD family, including Erm enzymes, interact with their substrate rRNA in a similar manner.
To test the hypothesis that Erm enzymes interact with rRNA similarly to TFB1M, we superpositioned the structure of ErmE, given by pdb code 6nvm, onto the coordinates for TFB1M bound to rRNA given by 6aax. The superposition reveals a strong overlap of secondary structure elements in the Rossmann-fold catalytic domain along with conserved positioning of some elements in the noncatalytic carboxy-terminal domain (Fig. 2A). The coordinates for a 6 nt region of the TFB1M substrate RNA (5′ CUGGAA 3′) were altered in silico to the sequence of the corresponding ErmE substrate RNA (5′ GACGGA 3′). The nts that are dimethylated are underlined—TFB1M dimethylates A936 and A937 while ErmE dimethylates only A2058. Inspection of the interaction of ErmE with the in silico modeled RNA substrate revealed several clashes which was expected since induced fit structural changes are expected upon interaction of the two macromolecules. However, these clashes could be relieved with modest alterations of the ErmE coordinates. Specifically, P133 and Y134 were remodeled to conform with the position of the homologous residues (P143 and F144) in TFB1M and the rotamers of K164 (R183 in TFB1M) and R171 were altered (Fig. 2B). The active site Pro-Tyr/Phe motif is conserved across Rossmann-fold 6-methyladenosine methyltransferases and it has been previously demonstrated that the Y134A mutant of ErmE lacks activity lending support to the choice to remodel these two residues (Schubert et al. 2003; Rowe et al. 2020). K164 resides at the same position in α5 as R183 in TFB1M and the rotamer change allows K164 to form an electrostatic interaction with the RNA backbone as R183 does (Fig. 2B). The fact that only minimal changes to the ErmE coordinates were required to model its interaction with the fragment 5′ GACGGA 3′ lends credence to the hypothesis that the RRAD family members ErmE and TFB1M interact with RNA in a similar manner.

ErmE superpositioned onto TFB1M bound to rRNA suggests ErmE regions in contact with rRNA. (A) A superposition of ErmE (pdb code 6nvm) onto the structure of TFB1M bound to its RNA substrate is shown. A high degree of structural similarity is observed between ErmE and TFB1M, particularly in the Rossman-fold (RF) catalytic domain. The carboxy-terminal domain (CTD) varies between the two structures. Helices α4, α5, and α6 are poised to play a central role in RNA binding. (B) A detailed view of the ErmE–TFB1M superposition reveals that a key aromatic residue (TFB1M F144, ErmE Y134) and a basic residue (TFB1M R183, ErmE K164) are positioned in the same vicinity between the two structures but must undergo a conformational change for ErmE to bind RNA as in the TFB1M–RNA crystal structure.
To refine our theoretical model, we performed energy minimization on ErmE bound to rRNA using Rosetta Relax generating 1000 candidate structures (Tyka et al. 2011). A plot of the Rosetta score versus root mean squared deviation versus the best scoring structure, Model 0425, revealed that a handful of structures had only marginally different scores (Fig. 3A,B). Therefore, we inspected multiple structures focusing on differences in the residues predicted to interact with rRNA. Model 0515 possessed hydrogen-bonding interactions between E160 and G2057, a nucleotide whose identity was previously shown to be critical for rRNA methylation by ErmE (Fig. 3A; Villsen et al. 1999). Model 0515 possessed an electrostatic interaction between R171 and the rRNA backbone, but the distance between K164 and rRNA (4.5 Å) was too long to accommodate an electrostatic interaction. The K164A and R171A mutants of ErmE were previously shown to be associated with an erythromycin-sensitive phenotype so we expected to see interactions between both residues and rRNA in our models (Rowe et al. 2020). We nevertheless decided to use Model 0515 while designing site-directed mutants of ErmE because it sits in the nadir of a cluster of well-scoring models, a feature expected for near-native models (Tyka et al. 2011). Inspection of the interactions between ErmE and rRNA in the well-scoring models, along with sequence conservation among Erm proteins, led us to hypothesize that three regions of Erm proteins mediate the primary interactions driving RNA binding and positioning (Fig. 3C).
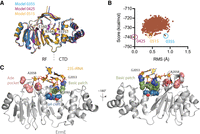
A computational model of ErmE bound to its 23S rRNA substrate suggests three regions are critical for RNA recognition. (A) A superposition of three models derived from the Rosetta Relax calculation is shown with the residues E160, K164, and R171 shown as sticks. The Rossmann-fold (RF) catalytic domain and the carboxy-terminal domain (CTD) are indicated. (B) A plot is shown of the Rosetta score versus root mean squared (RMS) deviation of Cα atoms for all models obtained versus model 0425. (C) Rosetta model 0515 is shown highlighting interactions of three regions of the protein with rRNA: the adenosine pocket, the basic patch, and the α4 cleft.
Erythromycin resistance phenotypes associated with site-directed mutants from three ErmE regions
We tested the hypothesis that the adenosine pocket, α4 cleft, and basic ridge drive the ErmE–rRNA interaction, by constructing several site-directed mutants belonging to each site and assaying the erythromycin resistance phenotype of cells transformed with the ermE variants. Within the Ade pocket, the structural model suggests π-stacking of Y134 with A2058 and Van der Waals interactions between F196 and A2058 with P198 introducing a sharp turn in the peptide backbone important for positioning of F196 (Fig. 4A). Sequence conservation analysis reveals position 134 to be Tyr or Phe in nearly all Erm proteins with F196 and P198 also nearly invariant (Fig. 4B). We assayed the erythromycin resistance phenotype of E. coli cells harboring site-directed mutants of each of these residues in two assays. An agar dilution minimal inhibitory concentration (MIC) assay was used for continuity with previous research and a microdilution MIC assay was performed in which phenotypes emerge over a much larger dynamic range (Fig. 4C; Maravic et al. 2003a; Pawlowski et al. 2018; Rowe et al. 2020). Planktonic E. coli cells are semi-resistant to erythromycin due to the efflux pump AcrAB-TolC but this phenotype can be altered by the addition of the antibiotic adjuvant phenylalanine-arginine β-naphthylamide (PAβN) that sensitizes Gram-negative bacteria to many xenobiotics through inhibition of AcrAB-TolC and potentially membrane permeabilization (Yu et al. 2005; Lamers et al. 2013; Gomes et al. 2019). Cells carrying the Y134A, F196A, and P198A mutations possessed similar phenotypes showing no erythromycin resistance in the microdilution assay and no or modest resistance (F196A) in the agar dilution assay. Cells harboring F196Y had MIC values of 512 µg/mL and 128 µg/mL in the agar dilution and microdilution assay, respectively, a phenotype of slightly less erythromycin resistance than that provided by wt ermE (Fig. 4C). Since the site-directed mutations may alter protein stability, we qualitatively assayed the levels of each variant by western blotting and observed a signal similar to wt indicating protein stability was not drastically altered in the mutants. Collectively, these data are consistent with the Ade pocket playing an important role in ErmE function. The comparison between the phenotype of cells harboring F196A versus F196Y is consistent with a van der Waals interaction between F196 and A2058, and the phenotype of P198A harboring cells indicates a role for P198 in structuring the Ade pocket. The MIC results for Y134A harboring cells are consistent with a previous report that Y134 is essential for ErmE function and our structural model that Y134 π-stacks with A2058 (Rowe et al. 2020).
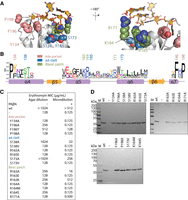
Erythromycin resistance phenotypes of site-directed mutants of the adenosine pocket, the α4 cleft, and the basic ridge. (A) A detailed view of the theoretical model of ErmE bound to Helix 73 of rRNA is shown. The model predicts residues involved in noncovalent interactions driving RNA recognition. 23S rRNA is shown in gold and selected amino acids are colored based on which region of ErmE they reside in: the adenosine pocket (pink), the α4-cleft (blue), and the basic patch (green). (B) One representative from each class of Erm proteins was used to model the sequence conservation at the three sites of interest. (C) Minimal inhibitory concentrations (MIC) for erythromycin were measured for E. coli cells expressing site-directed mutants of ErmE. Experiments were conducted as agar dilution without the antibiotic adjuvant phenyl-arginyl-beta-napthylamide (PAβΝ) or as liquid culture microdilution experiments in the presence of PAβΝ. (D) When cells displayed an erythromycin-sensitive phenotype, western blotting was used to verify that ErmE site-directed mutants were expressed and stable.
The theoretical ErmE–rRNA model places the C2055 nt of rRNA in the vicinity of the α4 cleft. The rRNA is positioned too far from the first layer of α4 cleft residues, including S138, R165, and S173, for hydrogen bonds or electrostatic interactions to form; however, we hypothesized that additional induced fit rearrangements of the rRNA and protein, not captured by the model, may occur. We assayed the erythromycin resistance phenotype of cells harboring various mutations at S138, R165, or S173 and found that S138A and S173A were associated with only modest defects in erythromycin resistance (Fig. 4C). The S138V or S173V mutants, however, were associated with a complete sensitivity to erythromycin (Fig. 4C). The phenotypes associated with the alanine substitutions suggest that residues S138 and S173 do not mediate interactions critical for RNA binding and positioning. However, the phenotypes of the valine mutants, a substitution that may alter the position of nearby residues, are consistent with the α4 cleft as a whole, playing an important role in ErmE–RNA binding and positioning. Both R165A and R165E are associated with a complete loss of erythromycin resistance suggesting R165 forms interactions critical for ErmE function (Fig. 4C). The α4 cleft mutants were also subjected to western blotting and again the signal suggested each mutant was stable and easily detectable in E. coli lysates (Fig. 4D). In sum, these data suggest the α4 cleft is essential for ErmE function in some manner but are ambiguous as to whether this involves essential contacts to the rRNA substrate.
Previous research on rRNA methyltransferases has identified electrostatic interactions with or adjacent to the methylated nucleotide that are critical for enzyme function (Dunkle et al. 2014). In model 0515, K164 and C2055 of 23S rRNA are oriented toward each other but slightly too far apart (4.5 Å) for an electrostatic interaction (Figs. 3A, 4A). However, in other models, K164 does form an electrostatic interaction with C2055. K164 is adjacent to two additional basic residues, R163 and R171, which we refer to collectively as the basic ridge. R171 forms an electrostatic interaction with the phosphate of G2053, while R163 forms an electrostatic interaction with neighboring E168, in our model, which may be important for stabilizing or positioning α5. We assayed site-directed mutants of each residue in the basic ridge. To get a detailed picture of how K164 may contribute to ErmE function, site-saturation mutagenesis was performed at this position (Supplemental Fig. S2). This technique identified that K164R is associated with an erythromycin-resistant phenotype and also identified several K164 mutations with an erythromycin-sensitive phenotype in the erythromycin selection step of our saturation mutagenesis assay (Supplemental Fig. S2). We performed detailed MIC assays for two of these mutants, K164M and K164S. These mutants were chosen because Met is sterically similar to Lys and Ser could model the effect of a polar residue at position 164. K164A, K164M, and K164S are each associated with an erythromycin-sensitive phenotype in MIC assays but western blotting indicates the protein is expressed and soluble (Fig. 4C,D). These data together with the phenotypic data indicate an Arg or Lys residue at position 164 is crucial for ErmE function. We assayed three variants of R163: R163A, R163E, and R163K. The Ala substitution was associated with a mild erythromycin resistance defect while the Lys substitution was associated with a resistance defect in the agar dilution assay but an erythromycin-resistant phenotype in the microdilution assay. The source of the discrepancy between the two MIC assays for R163K is unclear. The R163E mutant was associated with an erythromycin-sensitive phenotype in both MIC assays. In sum, the phenotypic and sequence conservation data for R163 support the Rosetta model in two ways. Firstly, the model does not include an R163 interaction with rRNA consistent with the substantial level of erythromycin resistance in cells harboring R163A. Secondly, the R163E associated phenotypes are consistent with the basic residues adjacent to R163, the basic ridge, being a crucial functional region of the protein. Presumably R163E, due to the charge alteration of the sidechain, interferes with the normal positioning of the adjacent residues. We previously determined the functional effect of variants of R171 (Rowe et al. 2020). Here we validated that R171A harboring cells are erythromycin sensitive in both the agar dilution and microdilution MIC assays. In sum, our phenotypic analyses of basic ridge mutants confirm this is a crucial region of ErmE and are consistent with a role for an electrostatic interaction between K164 and rRNA. The Rosetta models are ambiguous concerning an electrostatic interaction of R171 with rRNA, but previously published data and the phenotypes reported above indicate the interaction likely occurs (Rowe et al. 2020).
In vitro analyses of the role of the three ErmE regions in rRNA methylation
Next we sought to use in vitro assays with purified ErmE site-directed mutants to investigate, mechanistically, how each of the three regions contributes to rRNA methylation. Selected ErmE mutants were purified by two column chromatography steps and subjected to circular dichroism spectroscopy to verify normal folding of the proteins (Fig. 5A,B). 6-Methyladenosine formation by Erm requires binding of RNA and the SAM cofactor and the correct positioning of these two molecules for nucleophilic attack of the labile methyl group by the amine group of Ade. In Rossmann-fold methyltransferases, SAM is bound rigidly, in a deep cleft in the active site (Schubert et al. 2003). Substrate RNA, however, normally undergoes substantial conformational changes to achieve the conformation required for methylation (Dunkle et al. 2014; Schwalm et al. 2016). Because each of the three ErmE regions under investigation is hypothesized to contact RNA, functional defects in the site-directed mutants could arise, due to a loss of affinity for RNA or due to a loss in the ability to correctly position RNA for methylation.
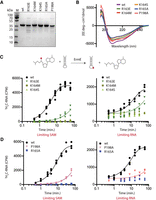
ErmE site-directed mutants associated with an erythromycin-sensitive phenotype display RNA methylation defects in vitro. (A) SDS-PAGE analysis of wild-type ErmE and variants indicates that the proteins are reasonably pure and display the expected molecular weight. (B) Wild-type ErmE and variants produce similar circular dichroism spectra indicating there is no major change in protein structure in the variants. (C) Single-turnover kinetics assays were used to characterize RNA methylation by selected site-directed mutants from the basic patch region. 3H-S-adenosylmethionine (SAM) was used as the methyl donor to follow product formation, resulting in the transfer of a radio-isotopically labeled methyl group (red circle) from SAM to RNA. (D) Single-turnover kinetics assays were performed on site-directed mutants from the adenosine pocket and α4-cleft. A scatter plot of three replicates is shown along with a best-fit line derived by nonlinear regression.
Our approach utilized methylation assays containing a 48-nt RNA that models 23S rRNA helix 73 as substrate and the assays were performed in two ways (Vester et al. 1998). Single-turnover kinetics were performed with either SAM limiting or with RNA limiting (Fig. 5C,D). This is because a kinetic defect due to a lowered affinity for RNA, for example, would be expected to appear in the limiting RNA assay but be alleviated in the limiting SAM assay where RNA is now in excess. As a direct test of RNA affinity binding, the same 48-nt substrate was 5′-labeled with fluorescein, and fluorescence polarization upon binding of the RNA to the ErmE mutants was measured (Fig. 6A). We selected site-directed mutants from each of the three ErmE regions that were associated with an erythromycin-sensitive phenotype to report on the overall contribution of that region to ErmE function. Our goal was to use the in vitro assays to assign defects in the ErmE mutants to either a loss of RNA affinity or an inability to correctly position RNA.
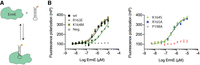
RNA affinity binding was measured for selected site-directed ErmE mutants. (A) Affinity binding of ErmE variants to an RNA oligonucleotide mimicking 23S rRNA helix 73 was measured by fluorescence polarization utilizing a 5′ fluorescein label on the RNA. (B) A scatter plot of three replicates of each ErmE variant under analysis is shown along with nonlinear regression fits. A single replicate of a titration of pepsin is shown as a negative control (Neg.) for RNA binding.
Methylation assays with basic ridge mutants, R163E, K164M, and K164S, each displayed substantial defects in methylation (Fig. 5C). Methylation by K164M and K164S was too slow for curve fitting and extraction of a kobs in our assay under both limiting SAM and limiting RNA conditions. However, the R163E defect could be quantitated and was found to have a kobs ∼10-fold slower than wild type. Similar kobs values for R163E (1.1 × 10−2 min−1 versus 1.0 × 10−2 min−1) were obtained under limiting SAM and limiting RNA conditions (Table 1). Since increasing RNA (limiting SAM conditions) did not increase kobs, the R163E kinetics results are consistent with a defect in RNA positioning as the primary explanation for the altered kinetics of R163E.
RNA affinity binding and kobs values for wild-type ErmE and variants
Methylation assays with P198A of the Ade pocket and R165A or the α4-cleft were performed and substantial defects were observed (Table 1; Fig. 5D). The R165A defects were too severe to allow curve fitting and extraction of kobs; however, kobs could be extracted from the P198A data. Under limiting SAM conditions, P198A was found to produce a kobs = 6.2 × 10−3 min−1 but kobs = 4 × 10−3 min−1 under limiting RNA conditions. These data, showing that the rate of reaction increased slightly when RNA was not the limiting reagent, are consistent with an RNA binding defect for P198A.
Each of the mutants characterized in methylation assays was also subjected to RNA affinity binding assays to aid in dissecting the mechanistic basis of methylation defects. Basic ridge mutants K164M and K164S bound RNA tightly, while R163E displayed a 3.7-fold increase in Kd for RNA (Table 1; Fig. 6B). The fact that K164M and K164S display no defect in RNA binding suggests that an electrostatic interaction between K164 and rRNA is crucial for positioning RNA for methylation, and a defect in positioning explains the lack of methyltransferase activity by these mutants. The altered Kd for RNA binding by R163E may contribute to the methylation defect but is unlikely to be the main cause. This is because in the limiting SAM methylation experiments, RNA was included at 20 µM so ErmE would remain saturated with RNA despite the 3.7 µM Kd value. The α4-cleft variant, R165A bound RNA tightly (Kd = 0.49 ± 0.05 µM for R165A vs. Kd = 0.98 ± 0.07 µM for wt) strongly suggesting that R165 plays a role in RNA positioning, explaining the methylation defects for this mutant (Table 1; Fig. 5D). The Ade pocket variant, P198A, in contrast, displayed barely detectable levels of RNA binding and we were not able to perform curve fitting on this data (Table 1; Fig. 5D). The methylation data provided a hint that RNA binding might be compromised in P198A (discussed above) and this is strongly confirmed by the affinity binding assay. The theoretical structural model suggested that P198 played a role in facilitating the Van der Waals interactions between F196 and the A2058. The in vitro data arising from the P198A mutant suggest the interaction of F196 with A2058, or this interaction and other interactions nearby that are similarly affected by the P198A mutation, are important for tight binding between ErmE and rRNA. The in vitro assays taken together suggest that the basic ridge and at least the R165 residue of the α4-cleft are required to position RNA for productive methylation, while P198 promotes an Ade pocket structure that drives RNA affinity binding.
Assaying the contribution of E160 to rRNA methylation
Our structural model of ErmE bound to rRNA predicts that when A2058 is positioned in the active site, a hydrogen-bonding interaction between E160 of ErmE and G2057 of 23S rRNA occurs (Fig. 7A). This interaction is not observed in the TFB1M–rRNA crystal structure despite the enzyme also possessing a Glu at the corresponding position in α5 (Liu et al. 2019). This may be explained by the fact that when A937 occupies the active site of TFB1M, the 5′ residue is A936 which is unable to form two hydrogen bonds with Glu. An important hydrogen-bonding interaction between ErmE E160 and G2057 is plausible because, across all Erm proteins, Glu is a common residue at the 160 position in α5 and the identity of G2057 has been shown to be crucial for rRNA methylation by ErmE (Fig. 4B; Villsen et al. 1999).
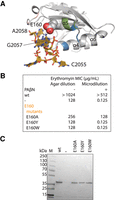
Erythromycin resistance phenotypes of E160 site-directed mutants. (Α) The ErmE–RNA structural model suggests hydrogen bonding between E160 and G2057 of rRNA. The basic ridge is colored green, the α4 cleft blue, and the Ade pocket pink. (B) Minimal inhibitory concentrations (MIC) for erythromycin were measured for E. coli cells expressing E160 site-directed mutants. Experiments were conducted as agar dilution without the antibiotic adjuvant phenyl-arginyl-beta-napthylamide (PAβΝ) or as liquid culture microdilution experiments in the presence of PAβΝ. (C) When cells displayed an erythromycin-sensitive phenotype, western blotting was used to verify that ErmE site-directed mutants were expressed and stable.
We used erythromycin resistance phenotypic assays to investigate the importance of the E160 interaction with rRNA using a two-pronged approach. First, we constructed E160A to investigate what effect the loss of the hydrogen bonds would have. Secondly, we constructed E160Y and E160W because the structural model predicts that these mutants would not only abrogate hydrogen bonding but also sterically clash with rRNA. The E160W and E160Y test whether rRNA, in fact, makes a close approach to ErmE at the E160 position of α5. We performed the erythromycin resistance assays reported earlier, this time with E160A, E160Y, and E160W (Fig. 7B). We found that cells transformed with E160A displayed a mild reduction in resistance: MIC levels fell to 256 µg/mL in the agar dilution assay and 128 µg/mL in the microdilution assay (Fig. 7B). In the case of E160Y and E160W transformed cells, the MIC values were the same as in cells lacking ermE; a completely sensitive phenotype was observed. Western blotting of lysates from each E160 variant revealed ErmE was stable and soluble (Fig. 7C). A range of erythromycin resistance phenotypes was observed in cells transformed with E160 variants likely indicating an alteration in the interaction of the protein with rRNA.
To understand mechanistically how the E160 variants might alter activity, we again assayed single-turnover methylation kinetics and RNA binding, this time on ErmE E160A and ErmE E160W (Fig. 8). Under limiting SAM conditions, E160A showed an 11-fold lower Kobs than wt, and a similar result under limiting RNA conditions (Fig. 8A). Under both limiting SAM or limiting RNA conditions, E160W did not produce measurable methylated RNA. When RNA affinity binding was investigated, E160W displayed a fourfold increase in Kd,RNA versus wt, while E160A bound RNA somewhat tighter than wt. Taken together with the phenotypic results from erythromycin resistance assays, the data indicate that E160 is positioned close to substrate rRNA and likely engages G2057 in hydrogen bonding. The hydrogen bonds are not a significant driver of ErmE's binding affinity for RNA but contribute to positioning RNA for efficient methylation.
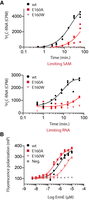
Methylation kinetics and RNA affinity binding measurements are consistent with the position of E160 in the ErmE–RNA model. (A) Single-turnover kinetics assays of RNA methylation by ErmE variants were performed using 3H-S-adenosylmethionine (SAM) as the methyl donor. Assays were performed under conditions of limiting SAM or limiting RNA. An oligonucleotide mimicking 23S rRNA helix 73 was used as the substrate. A scatter plot of three replicates is shown along with a best-fit line determined by nonlinear regression. (B) Measurements of binding affinity of E160 variants for the helix 73 analogs were performed by fluorescence polarization using a fluorescein-labeled RNA oligonucleotide. A scatter plot of three replicates is shown for wt, E160A, and E160W. A single replicate of a titration of pepsin is shown as a negative control (Neg.) for RNA binding. E160W possesses reduced affinity for RNA, consistent with a steric clash with the RNA, but E160A does not.
Sequence conservation of the three Erm regions in pathogenic bacteria
Next, we performed a bioinformatic analysis to understand how our model of the ErmE–rRNA interaction may generalize to ErmE orthologs from pathogenic bacteria. Recently, next-generation DNA sequencing of pathogenic bacteria from clinical samples and foodborne disease outbreaks has been deployed to identify the antibiotic susceptibility of the specimens and trace the origins of disease outbreaks. The NCBI database, MicroBIGG-E, catalogs antibiotic resistance gene sequences from these samples including greater than 50,000 erm gene sequences. We used this data to investigate the conservation of the three regions we believe are critical for the interaction of Erm proteins with rRNA. We focused our analysis on the erm genes closely associated with Gram-positive pathogens for which erythromycin is used as an antimicrobial, ermA, ermB, and ermC. There are over 7600, 25000, and 2400 discoverable sequences in the Micro-BIGG-E database for these erm genes, respectively, at the time of our analysis. Sequence logos show that the key residues of the Ade pocket, Y134, F196, and P198 are nearly invariant across the Erm sequences in the database (Fig. 9). Additionally, nearly all sequences appear to contain a basic residue at the basic ridge positions 163 and 164, but R171 is specific to ErmE and its most closely related homologs (Fig. 9).

Sequence conservation analysis of Εrm from clinical samples. Sequence logos were generated from pathogen-derived Erm sequences to analyze sequence variation across the adenosine pocket, α4 cleft, and basic ridge. Sequences were obtained from the NCBI MicroBIGG-E, a database of antimicrobial resistance genes derived from next-generation sequencing data of clinical samples and samples associated with foodborne disease outbreaks. Subtypes are indicated at left; MBE refers to all MicroBIGG-E sequences.
The structural convergence of the Ade pocket and basic ridge contrasts with what is seen at E160 and in the α4-cleft. E160 is observed in many Erm proteins, but Erm proteins from key pathogens contain a Gly at this position (Figs. 4A, 9). Superposition of the structure of ErmC, given by the pdb code 1qao, with our model of ErmE bound to rRNA suggests the Glu at positions 158 and 159 is unable to interact with the G2057 nucleobase despite its proximity in the primary sequence. The α4-cleft residue S138 is conserved as Thr in Erm proteins from pathogens which may form the same interactions with rRNA as Ser, but positions 165 and 173 diverge between ErmE and the Erm proteins under analysis (Fig. 9). Taken together the conservation data suggest the overall pose of rRNA on an Erm family protein is likely the same as indicated by the sequence convergence of the Ade pocket and the crucial basic residue at position 164. However, the details in how various Erm proteins interact with rRNA at E160 and the α4-cleft differ between Erm proteins from antibiotic producers, such as ErmE and Erm proteins from pathogens.
DISCUSSION
We used the crystal structure of the RRAD family member TFB1M bound to rRNA as a starting point to devise a theoretical model of ErmE bound to rRNA (Figs. 1, 2). We then used phenotypic assays and in vitro assays to characterize site-directed mutants of ErmE designed to test the model of ErmE bound to rRNA. We utilized sequence conservation data, chosen to model the sequence diversity in all Erm proteins, to inform our interpretation of the phenotypic and in vitro assays of the mutants and we used a sequence conservation analysis of Erm proteins from pathogens to gauge how well our structural model reveals the details of the interaction of ErmA, ErmB, and ErmC with rRNA. Many previous studies have been performed that attempted to explain how features of rRNA or features of the Erm protein give rise to specific methylation of A2058. Below we discuss how these data provide additional support to our structural model. Additionally, we consider how our results may guide efforts to discover specific inhibitors of Erm which, if co-administered with a macrolide antibiotic, could thwart the resistance mechanism restoring efficacy of the antibiotic in resistant bacteria.
While our studies focused on assaying site-directed mutants of ErmE, previous studies have utilized site-directed mutants of rRNA to understand how rRNA sequence and structure contribute to specific methylation of A2058 by ErmE. Studies showing that ErmE methylates A2058 of 23S rRNA in vivo and in vitro, as ErmC does, established ErmE as a useful model system, allowing for comparison of results among Erm proteins to better understand structure and function relationships in the enzyme family (Skinner et al. 1983; Vester and Douthwaite 1994; Zhong et al. 1995). Studies designed to identify the minimal RNA unit that supports efficient methylation by ErmE identified helix 73 of 23S rRNA and the single-stranded adjacent region encompassing A2058 as the minimal substrate (Vester et al. 1998). A variety of complementary techniques were then deployed to understand the role of helix 73's sequence and secondary structure in directing methylation reaching similar results (Hansen et al. 1999, 2011; Villsen et al. 1999). These studies found that the bulge formed by C2055 and the identity of G2057 are critical for rRNA methylation by ErmE but that the 3′ region of helix 73, nt 2611 to 2625, was amenable to substitution and modification indicating this region is not critical for directing methylation.
Villsen et al. (1999) quantitated the methylated product from reactions of ErmE with rRNA oligonucleotides encoding specific changes in helix 73. They found that substitutions at G2053 and A2054 led to an approximately two- to fourfold reduction in methylation consistent with the lack of sequence-specific contacts of ErmE to these nucleotides in our structural model (Fig. 4A). The C2055A substitution led to an approximately twofold reduction in methylation while removal of this nucleotide led to a fivefold reduction in methylation (Villsen et al. 1999). Our structural model does not possess any interactions between C2055 and ErmE but we speculate that if induced-fit changes in rRNA were better modeled the ribose may hydrogen bond to S138, cation-π interactions could occur between either R165 or R174 and C2055, and Van der Waals interactions could occur between various residues of the α4 cleft and C2055 (Fig 4A). Critically none of these interactions is sequence dependent. This scenario explains how the existence of a bulge formed by unpaired C2055 could be important for methylation without the identity of the nucleotide being critical. Substitution of G2057 by a pyrimidine led to a 10-fold reduction in methylation while the G2057A substitution led to a fivefold reduction (Villsen et al. 1999). This is consistent with the placement of G2057 in our model wherein G2057 is engaged in base stacking with G2053 and A2054 and E160 hydrogen bonds to the Watson–Crick face of G2057 (Fig. 7A). Presumably, the fivefold reduction in methylation observed by Villsen for G2057A, the modest changes in the erythromycin resistance levels for E160A, and the modest decreases of in vitro methylation for E160A that we observed all reflect a loss of the hydrogen bonding (Figs. 7B, 8A). The profound defects in erythromycin resistance, in vitro methylation, and the fourfold weaker binding to rRNA exhibited by E160W presumably reflect the combined loss of hydrogen bonding and the interruption of the base stacking interaction caused by the steric bulk of the Trp substitution displacing G2057 from its normal position (Figs. 7B, 8A). Therefore, our E160W ErmE mutant may produce the same rearrangements as the G2057C or G2057U rRNA substitutions. Additional investigations on the role of the 3′ side of helix 73 (nts C2611 to G2625) in rRNA methylation by ErmE found that this region of rRNA accommodated substantial modification of the nucleotides to deoxy species or locked nucleic acids without a substantial effect on methylation (Hansen et al. 2011). This is compatible with our model since we do not posit any critical interactions between the 3′ region of helix 73 and ErmE. One finding by Villsen et al. (1999) not easily explained by our model is an 11-fold reduction of methylation by the G2056C substitution. Our model does not rationalize this observation since G2056 is pointing away from ErmE toward solvent, but the majority of the findings on the effect of rRNA substitutions on ErmE methylation are rationalized by our structural model.
An important question is whether existing data support a common mode of interaction of Erm proteins with rRNA. Two types of experiments address this question: studies of the effect of rRNA substitutions on the activity of various Erm family members and studies of the effect of site-directed mutants of the Erm proteins themselves on activity. Helix 73 of 23S rRNA followed by a short single-stranded region containing A2058 has been demonstrated to be a suitable substrate for methylation by ErmC and ErmS, as well as ErmE (Kovalic et al. 1995; Schluckebier et al. 1999). The features of rRNA that contribute to methylation have not been thoroughly investigated for Erm proteins other than ErmE, however, it has been shown that for both ErmC and ErmS that the bulge created by C2055 is important (Fig. 1B; Kovalic et al. 1995; Schluckebier et al. 1999). ErmS possesses a Glu residue at the position corresponding to E160 in ErmE; however, inverting the orientation of the G2057–C2611 base pair slightly increased the activity of ErmS on this substrate suggesting the hydrogen bonding to the face of G2057 is not important for ErmS recognition of rRNA (Figs. 1B, 7A). The available data on rRNA mutants are consistent with our structural model in which a flipped C2055 is prominent and E160 hydrogen bonding to the Watson–Crick face of G2057 can occur but is not required for rRNA recognition.
Extensive site-directed mutagenesis of ErmC has been performed including mutations that belong to the three regions of interest in our structural model. In the Ade pocket, mutation of ErmC Y104 showed profound phenotypic defects in erythromycin resistance assays and in vitro methylation assays similar to results previously reported for ErmE Y134 mutants (Fig. 4A; Supplemental Fig. S3; Maravic et al. 2003b; Rowe et al. 2020). Mutations of ErmC F163 and P165 had modest phenotypic defects in erythromycin resistance assays but P165 had a substantial in vitro defect; kcat for methylation was reduced by 25-fold, consistent with our observations (Table 1; Fig. 4A; Maravic et al. 2003b). Studies of ErmC show that mutation of basic patch residues K133 and R134 (R163 and K164 in ErmE) is associated with defects similar to when these residues are altered in ErmE (Fig. 4A; Supplemental Fig. S3). K133 is associated with a modest defect in erythromycin resistance as we observed in the ErmE R163A mutant (Fig. 4C). The R134A ErmC mutant is associated with a lack of resistance to erythromycin and a 30-fold reduction in kcat similar to the assays reported herein for ErmE K164 mutants, although the defects in ErmE K164 variants rendered the proteins too defective to quantitate activity (Table 1; Fig. 4C; Maravic et al. 2003a). The R171 residue is specific to the ErmE clade so there is no corresponding basic residue in ErmC. The low sequence conservation between ErmC and ErmE in the α4 cleft is consistent with the type of interactions we believe form between protein and rRNA here: hydrogen bonds to the ribose, Van der Waals interactions with the ribose and nucleobase, and cation-π interactions. One residue that demonstrates similarity is T108 in ErmC which corresponds to S138 in ErmE (Supplemental Fig. S3). The ErmC T108A mutant is associated with a two- to fourfold reduction in erythromycin resistance and a 58-fold kcat defect (Maravic et al. 2003a). We observed no phenotypic defect for ErmE S138A in agar dilution assays and a modest defect in the microdilution assay (Fig. 4C). ErmE S138V, however, was associated with a complete loss of erythromycin resistance in phenotypic assays, and our phenotypic and in vitro assays with ErmE α4 cleft variant R165A suggest this region plays an important role in rRNA recognition by ErmE. Taken together, a comparison of assays with site-directed mutants of ErmC and ErmE suggests that the Ade pocket functions in a similar manner across Erm proteins as does the basic ridge residues corresponding to R163 and K164 in ErmE. Our structural model posits that the Ade pocket is structured to position A2058 for methylation by π-stacking and Van der Waals interactions and that these interactions, made possible by P198's contribution to structuring the Ade pocket, also contribute to the Kd for rRNA (Fig. 6B). The basic ridge residue R163 does not directly contact rRNA but may promote correct protein structure in some manner. Our model posits that K164 forms an electrostatic interaction important for correctly positioning the rRNA substrate for methylation but this interaction does not contribute to Kd for rRNA (Table 1; Fig. 4A). Assays of site-directed mutants of rRNA, ErmC, and ErmE are consistent with the α4 cleft interaction with the C2055 bulged nucleotide of rRNA, although the exact details of this interaction are not conserved across Erm proteins.
In this study, we sought to determine which structural sites were critical for Erm function and rationalize the contribution of the sites to function. If specific inhibitors of Erm proteins were known, therapies that combined macrolide antibiotics with an Erm inhibitor could potentially be developed to block the resistance mechanism. Coadministration of β-lactam antibiotics with β-lactamase inhibitors is a strategy that has extended the efficacy of this class of antibiotics successfully for decades (Leemans et al. 2014). Structure-based drug design will benefit from our data demonstrating that the Ade pocket, basic ridge, and α4-cleft are each essential for Erm function. In silico screening against each of these sites can be performed. We hypothesize that molecules which bind to one or more of these sites will interfere with enzyme function. Inhibitors of human protein arginine methyltransferases (PRMTs) and protein lysine methyltransferases (PKMTs) have been developed as therapeutics (Schapira 2016a). Small molecule inhibitors of these proteins target the SAM binding site but also target the substrate-binding site (Schapira 2016a). Recently, a therapeutic inhibitor of an RNA methyltransferase was reported confirming the suspicion that RNA methyltransferases could be targeted as well as protein methyltransferases (Schapira 2016b; Yankova et al. 2021).
One challenge in Erm inhibitor development is the potential for off-target interactions with the human m6A methyltransferases and RRAD family members DIM1T and TFB1M. METTL3 also possesses some active site homology to Erm proteins (Sledz and Jinek 2016; Wang et al. 2016). Sequence and structural conservation in the Ade pocket and a basic ridge is high among all RRAD family members, which is expected since this site must recognize an Ade residue in all cases (Liu et al. 2019; Shen et al. 2020). A structural superposition of TFB1M or DIMT1 with ErmE reveals the α4-cleft does deviate among these proteins (Fig. 10). The α5-α6 linker, which is known to affect substrate rRNA specificity, exhibits substantial structural variation which appears to impact the position of α6 (Fig. 10; Bhujbalrao and Anand 2019). Future work should use structure-guided discovery techniques to identify inhibitors of Erm proteins focusing on both the SAM binding site and the three regions we have identified as crucial for rRNA recognition. We hypothesize that molecules engineered to include α4-cleft binding may demonstrate better specificity than molecules that do not.
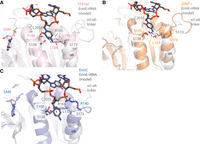
Superpositions of human rRNA adenine dimethylase family members and ErmC onto ErmE highlighting structural variation in the α4 cleft. (A) A superposition of human TFB1M given by pdb code 6aax, onto our ErmE–rRNA model. The view emphasizes the α4 cleft region. (B) A superposition of human DIMT1, given by pdb code 6w6c, onto our ErmE–rRNA model. (C) A superposition of bacterial ErmC, given by pdb code 1qao, onto our ErmE–rRNA model. In all cases only the catalytic domains of the enzymes are shown for simplicity.
MATERIALS AND METHODS
Construction of a computational model of ErmE bound to 23S rRNA
Coordinates for ErmE (pdb code 6nvm) were superpositioned onto TFB1M bound to RNA (pdb code 6aax) using the align command in Pymol. The Rossmann-fold methyltransferase domain of each protein (ErmE residues 46–211 and TFB1M residues 35–232) was exclusively used in the alignment because the proteins diverge substantially in the carboxy-terminal domain. A 6-nt mimic of the ErmE 23S rRNA substrate, ending in the methylation target A2058, was generated in the following manner. The 6aax coordinates for the five RNA residues preceding the methylation target, 932–936, and the methylation target residue, 937, were altered in Coot to the sequence of the 23S rRNA residues 2053–2058 (5′ GACGGA 3′). The methylation target of ErmE, A2058 is underlined in the sequence. Inspection of the interaction of these RNA coordinates with 6nvm revealed several steric clashes. Since current protein–RNA docking software is not capable of modeling large allosteric changes (Kappel 2019), the steric clashes were resolved manually in Coot (Emsley et al. 2010). The coordinates given by 6nvm for P133 and Y134 were altered to match the corresponding residues of 6aax, P143, and F144. This required a rotamer change in Y134 and a movement of the peptide backbone for both P133 and Y134. Additionally, clashes between K164 and R171 in 6nvm and RNA were resolved by rotamer changes at each residue. The theoretical model of ErmE–RNA derived from 6aax was refined by the Relax application in Rosetta 3.12. 1000 decoys were generated using the flags:
-
-s 6nvm_rRNA.pdb -nstruct=1000 -in:file:movemap movemap -out:file:silent 6nvm_rRNA.out
-
-out:file:silent_struct_type binary -out:file:fullatom -out:file:scorefile scorefile.s
-
and the movemap file:
-
RESIDUE * BBCHI
-
RESIDUE 245 250 NO #rRNA
-
JUMP * YES
The resulting scores were plotted versus root mean squared deviation, calculated using Cα atoms, with the lowest scoring model, 0425 as the reference. Three representative models are shown in Figure 3A and model 0515, which was used to generate all other figures, is available at https://dunklelab.ua.edu/structures.
Construction of ErmE site-directed mutants
Site-directed mutagenesis of ermE was previously described (Rowe et al. 2020). Briefly, ermE codon-optimized for E. coli was inserted into pBAD/Myc His A (Invitrogen) resulting in an open reading frame encoding the sequence given by Uniprot ID P07287 except that the 82 carboxy-terminal residues, which encode a low complexity Gly-rich segment, are removed and the MYC and hexahistidine tags are added to the carboxyl terminus. Site-directed mutants were generated in one of three ways as indicated in Supplemental Table S1 along with the oligonucleotide sequences used.
Erythromycin MIC assays
Agar dilution minimum inhibitory concentration assays were performed similarly to a previous protocol with minor modifications (Rowe et al. 2020). LB-ampicillin plates were coated with 250 µL of 0.02% w/v arabinose and dried. Cells from frozen stock cultures (E. coli TOP10 carrying a site-directed mutant of ermE on pBAD) were streaked onto the LB-Amp plates and incubated overnight at 37°C. Cells from these plates were streaked onto several 0.02% w/v arabinose coated LB plates containing a serial dilution of erythromycin ranging from 0 µg/mL up to 1024 µg/mL. The plates were incubated overnight at 37°C and the lowest concentration of erythromycin that was able to prevent confluent cell growth was recorded.
Microdilution MIC assays were also performed on the ErmE mutants. LB-ampicillin plates were coated with 250 µL of 0.02% w/v arabinose and allowed to dry. Cells from frozen permanent cultures were streaked onto the plates and incubated overnight at 37°C. Several colonies were harvested and resuspended in a sterile 300 mM NaCl solution, and the A600 was measured. Cell suspensions were diluted with the NaCl solution until the A600 measured was 0.1. Media for the microplate dilution were then prepared and consisted of media including erythromycin (Mueller-Hinton broth, 2% w/v L-arabinose, 20 µg/mL PAßN, 512 mg/mL erythromycin) and media lacking erythromycin (Mueller-Hinton broth, 2% w/v L-arabinose, 20 µg/mL PAßN). The media were added to a sterile, 96-well assay block, and a serial dilution of erythromycin was created ranging from 0 mg/mL up to 512 mg/mL with 150 µL final volume. A volume of 1.5 µL of the cell suspension was transferred to the assay block at each of the concentrations, and the block was incubated for 16 h at 37°C covered with a Breath-easy membrane. A plate reader was used to assess growth, with an A600 value of 0.01 greater than the background signal indicating a positive test for growth. The lowest concentration of erythromycin able to prevent cell growth was recorded.
Western blotting
Western blots were performed as described previously with slight modifications (Rowe et al. 2020). In an assay block, 10 µL of cells from overnight cultures were added to 500 µL of LB-Amp media, and the cells were grown for 2 h at 37°C while shaking. Protein expression was then induced with 2% w/v arabinose, and cells were grown for 2 more hours. The cells were harvested by centrifugation, resuspended in Buffer E (Tris HCl pH 7.5 50 mM, NaCl 250 mM, DTT 1 mM, glycerol 2% v/v) with lysozyme (0.1 mg/mL), and incubated on ice for 30 min. Cells were lysed with two freeze/thaw cycles in liquid nitrogen, and cell debris was pelleted by centrifugation. The supernatant was analyzed using SDS-PAGE on a 4%–20% gel, with wild type and pBAD as controls, and transferred to a nitrocellulose membrane. The membrane was blocked for 1 h in 3% w/v BSA in TBST, washed with TBST three times, and incubated for 1 h in 1:1250 anti-myc antibody at room temperature. The membrane was washed again with TBST and incubated in 1:2500 anti-mouse HRP conjugated antibody for 1 h. The membrane blots were developed with 1-Solution TMB and imaged using a gel imager.
Protein purification
ErmE variants used for in vitro assays were purified as follows. E. coli TOP10 cells harboring pBAD-ermE WT or site-directed mutants were grown in LB media at 37°C to ∼0.6 A600 at which point recombinant expression was induced with 0.02% w/v L-arabinose. Cell growth was continued at 37°C for 4 h, at which point the cells were harvested by centrifugation. Cells were resuspended in Buffer E (Tris HCl pH 7.5 50 mM, NaCl 250 mM, DTT 1 mM, glycerol 2% v/v) supplemented with 15 mM imidazole and 0.1% v/v Triton X-100 and subjected to sonication. The cell lysate was clarified by centrifugation for 30 min at 4800g and applied to HisPur immobilized Ni2+ resin. The resin slurry was applied to a column that was washed with 15 column volumes of Buffer E supplemented with 15 mM imidazole. Protein was eluted from the resin with a stepwise gradient of increasing imidazole (125, 250, and 500 mM) over 15 column volumes. Fractions were analyzed by SDS-PAGE for purity.
ErmE variants were further purified by size-exclusion chromatography with an S75 (sephadex) column. Fractions consistent with monomeric ErmE were pooled and concentrated on a Pall centrifugal device with a 10,000 MWCO membrane. Glycerol was added to a final concentration of 10% v/v and aliquots were flash-frozen until needed.
Methylation assays
Methylation assays under single-turnover conditions to extract kobs were performed as described previously with minor modifications (Rowe et al. 2020). An oligonucleotide mimicking the structure of helix 73 of 23S rRNA (V48) synthesized by Horizon was used as a substrate
(Vester et al. 1998; Rowe et al. 2020). Briefly, for reactions with limiting SAM, 3H-SAM was present at 0.05 µM and RNA was present at 20 µM. In reactions with limiting RNA, 3H-SAM was present at 5 µM and RNA was present at 0.2 µM. Reactions were performed in Buffer E diluted 1:1 with ultrapure water.
At the indicated timepoints, 2.5–5.0 µL of reaction volume were removed and quenched by dilution into 45.0–47.5 µL of 0.1
mg/mL salmon sperm DNA and the addition of 200 µL of 10% TCA. Precipitated RNA was collected by vacuum filtration on a Millipore
Multiscreen GF 96-well plate, washed with 10% TCA and ethanol, and dried. PerkinElmer Betaplate scintillation fluid was applied
and scintillation counting was performed on a MicroBeta 2 instrument. Counts per minute (product) versus time curves were
fit to the expression: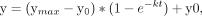 using GraphPad Prism.
using GraphPad Prism.
RNA affinity binding
RNA binding was measured by fluorescence polarization. A synthetic, PAGE purified, and 5′ fluorescein-labeled 48-nt strand
of RNA mimicking the structure of helix 73 of 23S rRNA was used (Horizon Discovery) (Vester et al. 1998). A serial dilution of protein was created in a clear bottom plate using Buffer E, with concentrations ranging from 20 µM
to 10 nM. The fluorescent RNA was diluted in water and mixed with the serially diluted protein, resulting in final protein
concentrations ranging from 10 µM to 5 nM in 0.5× Buffer E. The plate was centrifuged and incubated for 1 h at room temperature,
then polarization was measured with a plate reader. Pepsin was used as a negative control due to its lack of ability to bind
to RNA, and several background measurements were taken of the fluorescent RNA in 0.5× Buffer E with no protein. A further
measurement was taken after a 2-h incubation to ensure full binding equilibrium was reached. Data from three replicates were
fit to the equation: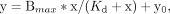 in GraphPad Prism.
in GraphPad Prism.
Sequence conservation analyses using the MicroBIGG-E database
Alignments of Erm amino acid sequences were conducted as follows. First, the structure-guided alignment function in MODELLER was used to align all Erm subtypes in the NIH Reference Gene Database (as of Jan. 23, 2021) using ErmE (PDB ID 6NVM) and ErmC (PDB ID 1QAO) structures as guides (Sali and Blundell 1993). Erm32 was excluded because it targets a different rRNA site (O'Farrell et al. 2008). Erm37, a Mycobacterium-specific methyltransferase, was excluded because it demonstrates target site slippage as well as significant sequence divergence (Madsen et al. 2005). Groups of Erm subtype sequences downloaded from the NIH MicroBIGG-E database (as of Feb. 13, 2021) were added to this reference gene profile using CLUSTAL Omega (Sievers and Higgins 2018, 2020; Sievers et al. 2011). Erm47 was excluded because it includes only two hits in the MicroBIGG-E database. Any Erm genes without subtype classifications (of which there were 98) were also excluded. Sequence logos were created using the command-line version of WebLogo (Schneider and Stephens 1990; Crooks et al. 2004).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by NIAID, National Institutes of Health grant R15AI131159 (to J.A.D. and A.J.S.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.078946.121.
- Received August 11, 2021.
- Accepted October 23, 2021.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is a new editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Rory Sharkey is the first author of this paper, “Three critical regions of the erythromycin resistance methyltransferase, ErmE, are required for function supporting a model for the interaction of Erm family enzymes with substrate rRNA.” Rory recently began graduate studies at Rice University in the Biochemistry and Cell Biology graduate program.
What are the major results described in your paper and how do they impact this branch of the field?
The major result described in the paper is the identification of three regions of the ErmE protein that are required for its function. ErmE confers resistance to macrolide and lincosamide class antibiotics by recognizing and methylating 23S rRNA. We've termed the three critical regions: the target adenosine binding pocket, the basic ridge, and the α4-cleft. Comparison to another Erm protein, ErmC, showed that previous studies of several corresponding site-directed mutants yielded similar results. Therefore, the three critical protein regions we identified may be important for the function of all Erm proteins. We believe this model may impact the design of drugs to inhibit Erm proteins. Inhibition of these antibiotic resistance enzymes could rescue the effect of antibiotics, such as erythromycin, and allow for its continued use as an effective treatment for bacterial infections. The binding of an inhibitor to one or two of the critical regions (they are structurally adjacent) would likely cause a potent inhibitory effect.
What led you to study RNA or this aspect of RNA science?
We were led to study this methylation of ribosomal RNA because it is responsible for conferring multidrug resistance to bacteria. The problem of antibiotic-resistant bacteria has become much more relevant as the use of antibiotics has increased and a better understanding of the mechanisms of such enzymes will help to better combat the issue. We were also curious as to what elements of Erm proteins cause them to target helix 73 of 23S rRNA in the large ribosomal subunit, while other RRAD family members with structural similarity target helix 45 of 16S rRNA in the small ribosomal subunit. Previous work has assayed several mutations in the RNA sequence of the protein–RNA interaction, and we aimed to better understand the contributions of the enzyme in both recognizing and methylating the rRNA substrate.
If you were able to give one piece of advice to your younger self, what would that be?
If I could give advice to my younger self, it would be to seek diverse perspectives on how to deal with issues. The importance of input and advice from those of different backgrounds or disciplines is invaluable in science. These different perspectives can lead to a breakthrough on an issue that has left one puzzled or may open one's eyes to new opportunities for investigations in the future. This can also lead to interdisciplinary collaboration, which often leads to more impactful research because of its use of multiple fields to better understand the phenomena. I would also encourage my younger self to embrace curiosity in all aspects of science. Not asking questions and pursuing rigorous explanations can lead to gaps in knowledge. Such gaps in fundamental knowledge can lead to an unstable basis for building future knowledge and will likely be a recurring problem. Embracing curiosity helps to remedy such issues and will build toward a more competent and confident scientist when conducting new research.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My philosophy and approach to science were greatly impacted during my undergraduate research with Dr. Jack Dunkle and his research laboratory group. I learned a great deal about the scientific process and how one critically evaluates a hypothesis. There was a significant focus on being detailed and thorough in all laboratory practices and experimental design. I learned the importance of appropriate positive and negative controls in every experiment as well as being able to replicate results with consistent technique. I also learned an emphasis on being critical of a hypothesis and how to continue to refine it as new information is learned from research. It is also important to be objective about a hypothesis and adapt or abandon it when results contradict the original hypothesis. Although many aspects of science can be slow and difficult, perseverance can lead to impactful research.
What are your subsequent near- or long-term career plans?
I recently began graduate school at Rice University in the Biochemistry and Cell Biology program. My short-term goal is to complete my studies here and conduct impactful research on aspects of biochemistry and structural biology related to the disease. Soon, I will be joining a thesis laboratory and hope to combine elements of what I learned from my undergraduate research with the knowledge I have gained from courses and research experiences that I have had at Rice. In the long term, I am not sure whether I would like to pursue a career in academia or industry, but I would like to continue to perform impactful science and foster habits of continual learning.








