
The sperm-oocyte switch in the C. elegans hermaphrodite is controlled through steady-state levels of the fem-3 mRNA
- Simone Zanetti,
- Sonja Grinschgl,
- Marco Meola,
- Marco Belfiore,
- Samantha Rey,
- Pamela Bianchi and
- Alessandro Puoti1
Abstract
Post-transcriptional control regulates many aspects of germline development in the Caenorhabditis elegans hermaphrodite. This nematode switches from spermatogenesis to oogenesis and is, therefore, capable of self-fertilization. This sperm-oocyte switch requires 3′ UTR-mediated repression of the fem-3 mRNA. Loss of fem-3 repression results in continuous spermatogenesis in hermaphrodites. Although several factors regulating fem-3 have been identified, little is known about the mechanisms that control fem-3. Here, we investigate the steady-state levels of the fem-3 transcript and the expression pattern of its protein product. We show that FEM-3 is exclusively present in germ cells that are committed to spermatogenesis. We found that in fem-3(gf)/+ heterozygotes, mutant fem-3 gain-of-function transcripts are more abundant than their wild-type counterpart. Furthermore, we show that the penetrance of the fem-3(gf) allele correlates with inefficient FBF binding and extended poly(A) tail size of fem-3 mRNAs. Finally, we show that wild-type and gain-of-function mutated fem-3 mRNAs associate equally well with polyribosomes. We propose that the fem-3 mRNA is regulated through stabilization rather than through translatability.
Keywords
INTRODUCTION
The Caenorhabditis elegans hermaphrodite produces sperm during larval development and switches to oogenesis as an adult. Males make sperm throughout their adult lives. How can a hermaphrodite, which is essentially a somatic female, produce sperm? Throughout the years, the study of germ cell development in C. elegans has led to the discovery of numerous genes involved in gamete sex determination. Among these, fem-3 has a prominent role. In fact, fem-3 loss-of-function (lf) hermaphrodites produce only oocytes, while fem-3 gain-of-function (gf) animals only make sperm (Hodgkin 1986; Barton et al. 1987). It is now clear that post-transcriptional regulation of the fem-3 mRNA is crucial for the determination of germ cells in C. elegans (Barton et al. 1987; Ahringer and Kimble 1991; Zhang et al. 1997). fem-3 regulation requires a cis-acting repressor element in the fem-3 3′ UTR, which is mutated in all fem-3(gf) alleles (Ahringer and Kimble 1991). An earlier study reported that fem-3 mRNA steady-state levels were similar in wild type and gain-of-function mutants, indicating that transcriptional regulation was not responsible for increased fem-3 activity in the mutants (Ahringer and Kimble 1991). However, such analyses were done by comparing homozygous wild-type fem-3(+) with homozygous mutant fem-3(gf) animals, therefore causing possible discrepancies through unequal RNA extraction efficiencies, phenotypic differences, and culture conditions. We have solved this problem by analyzing fem-3(+) and fem-3(gf) mRNA levels within one single population of masculinized fem-3(+)/fem-3(gf) heterozygotes. The polyadenylation status of mRNAs often reflects their activity (for review, see Sachs 2000). Previous studies report that the size of fem-3 poly(A) tails is slightly increased in gf mutants if compared to wild type (Ahringer and Kimble 1991). Based on these results, translational control was suggested as a speculative model for fem-3 regulation. Since then, this model has been commonly accepted without further experimental evidence. The importance of the cis-acting element in the fem-3 3′ UTR, also known as the PME (point mutation element) was confirmed with the Puf proteins FBF-1 and FBF-2, which depend on the wild-type PME for fem-3 RNA binding (Zhang et al. 1997; Bernstein et al. 2005). However, the molecular processes that govern fem-3 remain speculative. In particular, the loading of fem-3 mRNAs on polyribosomes has never been tested. Trans-acting regulators of fem-3 have been found through molecular and genetic approaches. On one hand, FBF-1 and FBF-2 bind to the fem-3 3′ UTR (Zhang et al. 1997). Remarkably, FBF proteins are homologous to Drosophila Pumilio, which controls the hunchback mRNA for anterior-posterior patterning of the embryo (Barker et al. 1992; Macdonald 1992). While Pumilio represses hunchback mRNA through deadenylation and translational control, this question remains unanswered for fem-3 and FBF. On the other hand, screens for recessive mutations leading to masculinized germlines have led to the mog genes, which also control fem-3 through its 3′ UTR (Graham and Kimble 1993; Graham et al. 1993; Gallegos et al. 1998).
In this study, we use improved techniques to analyze fem-3 mRNA levels, its polyadenylation status, and its loading on polyribosomes. We have also revised the experimental design by using heterozygous animals that contain both gain-of-function mutated and wild-type copies of fem-3. In fact, fem-3(gf) alleles cause dominant temperature-sensitive masculinization of the germline (Barton et al. 1987). We, therefore, compared wild-type and mutant fem-3 mRNA levels within the same masculinized animals. This approach also has the advantage of masculinized hermaphrodites not making oocytes (Barton et al. 1987; Rosenquist and Kimble 1988; Ahringer et al. 1992). In fact, oocytes and embryos are loaded with maternal fem-3 mRNA, thus causing a bias in the assessment of RNA levels (Rosenquist and Kimble 1988).
Based on genetic evidence, fem-3 acts as a positive regulator of spermatogenesis (Hodgkin 1986). However, FEM-3 has no defined motifs and appears to have evolved extremely rapidly even in closely related species (Haag et al. 2002). Based on its sequence, FEM-3 is predicted to be a soluble, intracellular protein (Ahringer et al. 1992). It has been shown to function together with CUL-2, FEM-1, and FEM-2 for ubiquitin-mediated proteolysis for somatic sex determination (Starostina et al. 2007). Therefore, the localization of the FEM-3 protein is a key step toward the understanding of its molecular function in the germline. Using anti-FEM-3 antibodies, we show that FEM-3 is specific to the sperm lineage.
RESULTS
fem-3 mRNA expression is controlled post-transcriptionally
To gain insight into fem-3 mRNA regulation, we first compared its protein and mRNA expression patterns. In adult germlines, an antisense fem-3 RNA probe stained the proximal portion of wild-type gonads. fem-3 mRNA expression overlaps with the region that is undergoing oogenesis (Fig. 1A,B). fem-3 is required maternally and is, therefore, delivered as mRNA to the oocytes (Hodgkin 1986; Rosenquist and Kimble 1988; Ahringer et al. 1992). We found that early embryos stained for fem-3 mRNA and that this staining disappeared rapidly after the 24-blastomere stage (Fig. 1G–J; data not shown). This finding is in accordance with results obtained by Northern blotting showing a rapid decrease of fem-3 mRNA within the first 4 h of embryonic development (Rosenquist and Kimble 1988; Ahringer et al. 1992). On the other hand, in masculinized fem-3(gf) mutants, fem-3 mRNA localization extends distally and coincides with the region producing spermatocytes and spermatids (Fig. 1C,D). To determine the expression pattern of the FEM-3 protein, we raised polyclonal antibodies against its N terminus. In the hermaphrodite germline, FEM-3 expression surrounds nuclei that are determined to the male fate. In fact, FEM-3 was detected in the cytoplasm of primary spermatocytes at larval stage L3 and in spermatocytes and spermatids at L4 (Fig. 1K–P). Sperm precursors were identified by their nuclear morphology and counterstaining with SP56 anti-sperm antibodies (Ward et al. 1986). In adult males, FEM-3 was found in the cytoplasm of spermatocytes and spermatids (Fig. 1Q,R). In adult hermaphrodites, which have started oogenesis, FEM-3 was only present in the sperm lineage, while oocytes were consistently negative for FEM-3 staining (Figs. 1S,T, 2A–C). fem-3(lf) mutants do not produce sperm and, therefore, cannot be used as a negative control for FEM-3 antibody staining. We hence tested the specificity by competition with the antigen and found strongly reduced staining only in the presence of the competing antigen, indicating that the antibody is specific (Fig. 2D–F). Early embryos contain MOG-6 protein and fem-3 mRNA (Figs. 1G–J, 2G–I). However, we never found FEM-3 protein at this step of development, suggesting that the fem-3 mRNA is stored but not translated in embryos. We conclude that the distribution of fem-3 mRNA in the oogenic lineage and embryos differs from that of the FEM-3 protein, hence suggesting that fem-3 is controlled post-transcriptionally.
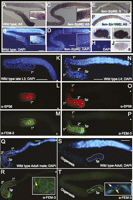
Expression of fem-3 in embryos and germlines. (A) In situ localization of fem-3 mRNA in adult germlines. (B) Nuclear DAPI staining; region with oocytes in diplotene is boxed and magnified. (C,D) In situ hybridization of fem-3 in an adult fem-3 gain-of-function mutant germline. Boxed: primary and secondary spermatocytes. The proximal portion (to the left of the box) of the gonadal arm is filled with spermatids. (E) Germline hybridized with a fem-3 sense RNA probe. (F) No signal was observed in a fem-3(e1996, null) mutant germline. (G,H) Maternal fem-3 mRNA in a two-blastomere embryo and staining with DAPI. (I) fem-3 mRNA was found in all blastomeres of a four-cell embryo: ABa (left), ABp (top), EMS (bottom), and P2 (right). (J) Staining of a four-cell embryo with a fem-3 sense RNA probe. FEM-3 protein localizes to the sperm lineage: (K–M) Wild-type L3 germline stained with DAPI, SP56 anti-sperm and anti-FEM-3 antibodies. The region outlined contains primary (1°) spermatocytes. (N–P) Staining of a wild-type germline of larval stage L4: (2°) secondary spermatocytes, (Sp) spermatids. (Q,R) Distribution of FEM-3 in an adult male germline. The region containing spermatocytes and spermatids is outlined. The inset shows high magnification of spermatids stained for DNA and FEM-3. The expression of FEM-3 is clearly cytoplasmic. (S,T) Localization of FEM-3 in a dissected adult hermaphrodite germline: FEM-3 is only present in the cytoplasm of spermatids. Inset: magnification of spermatids, merged DAPI, and anti-FEM-3 stainings. Yellow arrow, nucleus of spermatid. White arrow, nucleus of somatic cell of the spermatheca. Asterisks indicate the distal end of the germline. Bar, 10 μm.

FEM-3 is specifically expressed in the sperm lineage. (A–C) Dissected adult germlines stained with DAPI, anti-RME-2, and anti-FEM-3 antibodies. RME-2 was found in the oocyte lineage. In contrast, FEM-3 is restricted to the sperm lineage. (D–F) Anti-FEM-3 staining challenged by FEM-3 peptides. The N-terminal peptide (FEM-3N) was used to produce the antibody. Panels D–F represent a detail of the spermatogenic region of a L4 larva. All micrographs were taken under the same light conditions and time of exposure (in msec). Only samples preincubated with FEM-3N showed strongly reduced FEM-3 staining in sperm. (G–I) Eight- to 12-blastomere embryos stained only for MOG-6 but not for FEM-3. Bar, 10 μm.
Loading of fem-3 mRNAs on polyribosomes
The homology between FBF proteins and Pumilio suggest that translational control might regulate fem-3 activity (Zhang et al. 1997; Wharton et al. 1998). When efficiently translated, mRNAs associate with polyribosomes, whereas inactive mRNAs do not (Kretsinger et al. 1964; Scott and Bell 1964; Stafford et al. 1964). We, therefore, compared the distribution of wild-type and gain-of-function mutated fem-3 mRNAs along a sucrose gradient resolving free RNAs, ribosomal subunits, monosomes, and polyribosomes. To detect actively translated mRNAs, extracts were treated with cycloheximide, which blocks elongation and freezes the ribosomal subunits on the mRNA (Fig. 3A). Since fem-3(q95) harbors a 112-nt deletion, it can be distinguished from the wild-type transcript. We found wild-type and fem-3(q95) mRNAs at comparable levels in polysomal fractions, indicating that both fem-3(+) and fem-3(q95) mRNAs are loaded on polyribosomes at similar rates (Fig. 3B,C). Therefore, control of translational initiation is unlikely to cause the strong masculinization in fem-3(q95) mutants. We only detected one significant difference in mRNA distribution along the gradient: the fraction that corresponds to the 40S peak is enriched in wild-type fem-3 mRNA. Two possible explanations will be discussed later.
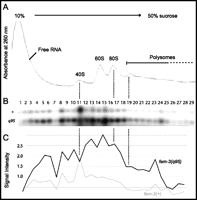
Polyribosome analysis. (A) Cytoplasmic extracts from young adult fem-3(q95)/+ heterozygotes were resolved by sucrose density sedimentation. Absorbance trace at 260 nm with peaks corresponding to free RNAs and translationally silent messenger ribonucleoprotein, ribosomal subunits (40S, 60S), monosomes (80S), and polyribosomes. (B) Analysis of fem-3 transcripts in all fractions by RT-PCR and Southern blotting. (C) Wild-type (+) and gain-of-function-mutated (q95) fem-3 cDNAs were quantified by phosphoimager analysis. We found fewer wild-type transcripts throughout all fractions, except fraction #11 that coeluted with 40S.
fem-3(gf) mRNAs are stabilized
As an alternative to translational control, we asked if fem-3(gf) alleles lead to increased stabilization of the fem-3 mRNA (Barton et al. 1987; Ahringer and Kimble 1991). As a first step, we analyzed the extent of masculinization in various mutant backgrounds at different temperatures. At 15°C, hermaphrodites are mostly fertile, producing both sperm and oocytes (Fig. 4A). At 25°C, heterozygous fem-3(q95)/+ and fem-3(q96)/+ germlines are fully masculinized (Fig. 4A; Ahringer and Kimble 1991). In contrast, even at restrictive temperature, the weak allele fem-3(q23) leads to moderate masculinization with 45% of fertile adults (Fig. 4A). In order to investigate polyadenylation of fem-3(gf) mRNAs, we tested the action of three RNA-modifying proteins. CCR-4 has been shown to have a deadenylation activity (Schmid et al. 2009), RNP-8 functions in polyadenylation and oogenesis together with GLD-2, and LARP-1 is an RNA binding protein that localizes to P bodies and is synthetically required with nos-3 for oogenesis (Nykamp et al. 2008; Kim et al. 2009; Zanin et al. 2010). In the absence of either ccr-4, rnp-8, or larp-1, fem-3(q23)/+ heterozygous germlines are increasingly masculinized (Fig. 4A). To check if strong Fem-3 (gf) phenotypes were linked to increased stabilization of the mutant fem-3 mRNA, we analyzed steady-state fem-3 mRNA levels in different mutant backgrounds, using various experimental approaches. The fem-3(q95) mRNA lacks 112 nt that span the PME (Fig. 4B,C; Ahringer and Kimble 1991). Therefore fem-3(q95) can be readily distinguished from wild-type fem-3 by RT-PCR. As a first approach for the analysis of this allele, we used two primers that span two exons, one of which includes the 3′ UTR of fem-3. PCR products were analyzed by Southern blotting with a probe that spans the last exon except the region that is deleted in allele q95. We found 77% of mutant fem-3(q95) versus 23% of wild-type fem-3 mRNA (Fig. 4B). To ensure that the primers did not preferentially amplify fem-3(q95) cDNAs, we added a small amount of genomic DNA derived from fem-3(q95)/+ heterozygotes, which was amplified at similar rates (Fig. 4B). fem-3(q95) was also quantified by real-time RT-PCR using TaqMan probes that are specific for the two RNA species. Again, in four independent experiments, we found 82% of fem-3(q95) and 18% of wild-type cDNAs. Allele fem-3(q96) corresponds to a G to U transversion: It was analyzed by sequencing of cloned PCR products. We found that among 34 clones analyzed, 32 were mutant, and two were wild type (Fig. 4C, right).
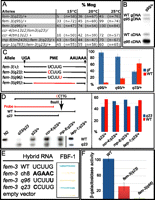
Steady-state levels of fem-3 mRNAs. (A) Masculinization of the germline (Mog) is dependent on the temperature and the genetic background. Germlines analyzed are heterozygous for fem-3(gf) and homozygous for ccr-4, rnp-8, or larp-1. (n) Number of animals tested. Mutant phenotypes increase in severity with temperature. 15°C is permissive, 20°C intermediate, and 25°C restrictive temperature. (B) Analysis of fem-3(q95)/+ extracts by semiquantitative PCR and Southern blotting. Genomic (gDNA) and reverse-transcribed (cDNA) products are visible. gDNA allows monitoring of amplification efficiency. (C) fem-3(gf) alleles. Left: Mutations in the fem-3 3′ UTR analyzed in this study. fem-3(q95) deletes the entire PME. Right: Compilation of fem-3 mRNA steady-state levels in fem-3(gf)/fem-3(+) heterozygotes as obtained by different techniques. The proportion of each transcript is indicated as a percentage of total fem-3 mRNA. The data from three to four independent experiments are shown, including standard deviations: fem-3(q95,gf) was analyzed by real-time PCR and Southern blotting; fem-3(q96,gf) was tested by sequencing; fem-3(q23,gf) was assayed by restriction fragment polymorphism analysis and by sequencing. (D) In the absence of ccr-4 and rnp-8, fem-3(q23) mRNA levels are slightly increased relative to wild type. Removal of larp-1 does not modify the amount of fem-3(q23) mRNA when compared to wild type. The probe used for the blot was directed against a fragment that is common for both alleles. Right: Quantification of the Southern blot by phosphoimager analysis. The proportion of fem-3(q23) and fem-3(+) mRNAs is indicated as a percentage of total fem-3 mRNA. Three independent experiments are compiled. (E,F) FBF-1 binds weakly to the fem-3(q23) mRNA. The results from three independent yeast three-hybrid assays on plates (E) and in solution (F) are shown. The interactions were tested on wild-type and mutant PME sequences.
Is the germline masculinization phenotype of fem-3(gf) mutants caused by inappropriate levels of the fem-3 mRNA in adults? To answer this question, we compared the ratio of fem-3(+) versus fem-3(q96) mRNAs in larvae and young adults. The rationale behind this is that fem-3 should not be regulated before larval stage 4, when spermatogenesis is initiated. In adult fem-3(q96)/+ heterozygotes, we observed highly increased steady-state levels of mutant transcript relative to wild-type transcript (Fig. 4C). However, in younger larvae, fem-3(q96) and fem-3(+) transcripts were found in comparable amounts: Out of 17 cDNAs sequenced, nine were fem-3(q96), and eight were fem-3(+). This result suggests that the fem-3(+) mRNA may lose its stability in adults, which are oogenic, while fem-3(gf) transcript levels are steady. Alternatively, the amount of fem-3(gf) mRNAs could be increased and fem-3(+) remain steady. Our interpretation is that the increase of fem-3(gf) versus fem-3(+) mRNA causes the Mog phenotype.
The weak fem-3(q23) allele leads to less severe masculinization. We have analyzed fem-3(q23) mRNA levels by sequencing of cDNA clones and by restriction fragment length polymorphism analysis. In fact, the U to C transition in fem-3(q23) generates a cleavage site for BsaXI in the corresponding cDNA (Fig. 4C,D). Unlike the two strong alleles, fem-3(q23) mRNA was found at slightly reduced levels when compared to wild type (Fig. 4C, right). Therefore increased dosage of fem-3 mRNA coincides with the extent of germline masculinization. We addressed this aspect by taking a different approach and checked fem-3 mRNA levels in heterozygous mutants lacking ccr-4, rnp-8, or larp-1, as shown in panel A. PCR-amplified cDNAs were resolved and probed with a fragment of fem-3 designed to recognize both uncut wild-type and digested mutant products with the same efficiency (Fig. 4D). When normalized to wild type and homozygous fem-3(q23), fem-3(q23) mRNA levels in heterozygotes are slightly increased in the absence of ccr-4 or rnp-8 (Fig. 4D). However, they never reached levels that were similar to those obtained with strong alleles such as q96 or q95. In the absence of larp-1, the abundance of fem-3(q23) did not change significantly (Fig. 4D). Therefore, fem-3(q23) retains a basal activity, independently of its quantity and perhaps of FBF activity. To test this possibility, we verified the binding of FBF to the fem-3(q23) 3′ UTR. An earlier study reports that FBF binding to the fem-3 3′ UTR is totally abrogated either by a fem-3(q96) mutation or by the deletion of the PME (Zhang et al. 1997). We found that FBF binds to the fem-3(q23) 3′ UTR, albeit less efficiently than to wild-type fem-3 (Fig. 4E,F). One interpretation of this result is that reduced binding of FBF leads to an incomplete penetrance of the fem-3(q23) allele. On the other hand, strong fem-3(gf) phenotypes might be caused by the complete absence of FBF binding to the fem-3 3′ UTR.
Polyadenylation analysis of fem-3 mRNAs
Stabilization of mRNAs is achieved through efficient polyadenylation (Bernstein and Ross 1989; Decker and Parker 1993). We found that, in the absence of deadenylase CCR-4 or RNA polymerase RNP-8, masculinization caused by a weak fem-3(q23) allele was increased from 45% to ∼70% (Fig. 4A). In fem-3(q23) homozygotes, penetrance increased from 59% to almost 100% in the absence of ccr-4 or rnp-8. However, mutations in poly(A) polymerase GLD-2 and in its binding partner GLD-3 did not cause masculinization (Fig. 5A). Using the PAT assay, we asked whether stabilized fem-3(q23) mRNAs had longer poly(A) tails than their wild-type counterparts (Salles and Strickland 1995). Upon Southern blot analysis and sequencing of the products, we found that wild-type fem-3 poly(A) tails contain, on average, 28 adenosines (Fig. 5B). In fem-3(q23) and fem-3(q96) gain-of-function animals, fem-3 poly(A) tails extended to 35–45 adenosines (Fig. 5B, lanes 1,7,8). Therefore, poly(A) tail length correlates with the extent of masculinization in fem-3(gf) animals. In the absence of ccr-4, poly(A) tails of wild-type and fem-3(q23) mRNAs extended to more than 50 adenosines (lanes 2,10), while removal of rnp-8 slightly increased fem-3(+) mRNA polyadenylation but had no effect on fem-3(q23) (Fig. 5B, lanes 3,9). In gld-2 and gld-3 mutant backgrounds, wild-type and gain-of-function mutated fem-3 mRNAs remained unchanged (lanes 4,5 and 11,12). FBF proteins were found to function toward deadenylation of fem-3 mRNA. In fact, the sizes of fem-3 poly(A) tails were increased in fbf-1 fbf-2 double mutants and fbf-1 fbf-2; fem-3(q96) triple mutants (lanes 6 and 13). Taken together, our experiments show that mutant fem-3(gf) poly(A) tails are longer than wild type and that the increased masculinization in the double mutants listed in panel A may be caused by augmented polyadenylation of fem-3 mRNAs.
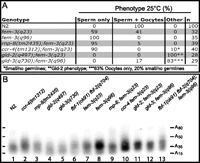
Polyadenylation analysis. (A) Phenotypes of homozygous mutants grown at restrictive temperature. (n) Numbers of germlines tested. (B) Poly(A) tail analysis performed on L4 larvae. cDNAs of fem-3 3′ UTRs including poly(A) tails were resolved and analyzed by Southern blotting and sequencing. Poly(A) tails comprise between 12 and 50 adenosines.
FEM-3 expression coincides with the extent of germline masculinization
FEM-3 protein was detected in the cytoplasm of spermatids and in developing spermatocytes of wild-type hermaphrodites (Fig. 1K–T). We analyzed FEM-3 expression in different adult mutant germlines that are predicted to have enhanced fem-3 activity. FBF-1 and FBF-2 directly bind to the fem-3 3′ UTR (Zhang et al. 1997). Due to redundancy of the FBF proteins, fbf-1 single mutants still produce oocytes. In such germlines, GLD-1 was found in the meiotic region (Fig. 6B). FEM-3 was detected in spermatids and in a few spermatocytes that had not yet been pushed to the proximal end of the germline (Fig. 6A,C). fem-3(q96) strong gain-of-function mutants are fully masculinized. In these animals, FEM-3 was found in spermatocytes and in spermatids and even extended more distally but never overlapped with GLD-1 expression (Fig. 6D–F). In hermaphrodites, GLD-1 expression is robust in the transition zone and the pachytene region, then rapidly decreases during diplotene and is absent at diakinesis (Fig. 6A–C; Jones et al. 1996). We detected FEM-3 only in cells that are located more proximally to the pachytene region. Therefore, we propose that in a masculinized fem-3(gf) germline, cells in diplotene express FEM-3. A similar expression pattern was observed in male germlines (Fig. 1Q,R), and in fbf-1fbf-2 and mog-1 mutants that are fully masculinized (Fig. 6G–J). In fbf-1fbf-2 mutants, the mitotic and pachytene regions are dramatically reduced (Crittenden et al. 2002; Lamont et al. 2004). Therefore, FEM-3 can be found in the vicinity of the distal end of the germline (Fig. 6G,H). In mog-1 mutants, the pachytene region comprises about 10 rows of cells. Here, FEM-3 is visible already at diplotene (Fig. 6I,J). We conclude that distal expression of FEM-3 in masculinized germlines correlates with the extent of germline masculinization. However, we have never found FEM-3 in cells in oogenic or spermatogenic pachytene.
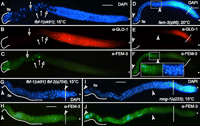
Expression of FEM-3 in mutant germlines. (A–C) fbf-1 mutants produce both spermatids and oocytes. Staining of one adult gonadal arm with DAPI (A). GLD-1 is expressed in the cytoplasm of meiotic germ cell precursors, from the transition zone throughout pachytene. GLD-1 levels decrease rapidly in the diplotene region (B). FEM-3 is found in primary spermatocytes and later steps of spermatogenesis (C); white arrows indicate secondary spermatocytes. The region containing spermatids (Sp) is outlined. Open arrows point at oocyte nuclei. (D–F) Adult fem-3(q96) germlines are masculinized at 20°C. GLD-1 is expressed throughout the early meiotic prophase in a fully masculinized germline. FEM-3 expression is found between two white arrowheads; it corresponds to the spermatogenic region. Insets (anti-Fem-3, left, and DAPI, right) show expression of FEM-3 in the diplotene region and in primary spermatocytes. (G,H) Adult fbf-1 fbf-2 germlines are fully masculinized. The distal expression of FEM-3 indicates an early onset of spermatogenesis. (I,J) mog-1 mutants produce less sperm than fbf double mutants but also show early expression of FEM-3. Asterisks indicate the distal end of the germline, dashed lines indicate the mitosis-to-meiosis transition zone. Bar, 10 μm.
FEM-3 and MOG expression are mutually exclusive
The mog genes are excellent candidates for encoding trans-acting factors that repress fem-3 through its 3′ UTR (Graham and Kimble 1993; Gallegos et al. 1998). We reasoned that if MOG proteins down-regulated fem-3 RNA, both proteins should not be found within the same germ cells. MOG proteins are present in the nuclei of many germ cells, including oocytes (Belfiore et al. 2004; Kasturi et al. 2010; Zanetti et al. 2011). We show that FEM-3 labels the spermatogenic lineage. We found that in L3 larvae, FEM-3 is present in proximal germ cells, suggesting that spermatogenesis has started, although no spermatocytes are yet visible (Figs. 7A–F, 1K–M). In such L3 larvae, MOG-6 is present in the entire germline, including cells that are committed to spermatogenesis and express FEM-3 (Fig. 7C–F). Early L4 germlines stain for FEM-3 in primary and secondary spermatocytes and in spermatids (Fig. 1N–P). However, MOG-6 is absent in secondary spermatocytes and spermatids (Fig. 7G,I). Earlier studies report that mog-6 acts as a negative regulator of the fem-3 mRNA (Graham et al. 1993; Gallegos et al. 1998). Our finding that cells that express MOG-6 are negative for FEM-3 staining supports the idea that the mog genes encode negative regulators of the fem-3 mRNA. However, our findings do not provide clues on the molecular mechanism.
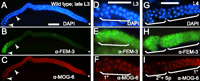
FEM-3 and MOG-6 expression in germlines. (A–C) In late L3 larvae, spermatids are not yet visible, but FEM-3 staining is present in the proximal part of the hermaphrodite germline. Sperm precursors are visible between the two arrowheads. Nuclear MOG-6 is present throughout the germline. The region between the two arrowheads is enlarged in D–F. (G–I) In L4 hermaphrodites, FEM-3 is visible throughout the sperm lineage, while MOG-6 is absent from secondary spermatocytes and spermatids. (1°) and (2°), primary and secondary spermatocytes, respectively; (Sp) spermatids. Bar, 10 μm.
DISCUSSION
Regulation of the fem-3 mRNA
fem-3 promotes sperm development in males and hermaphrodites (Hodgkin 1986). However, adult hermaphrodites produce only oocytes and thus repress fem-3. In contrast, tra-2 directs oogenesis and is repressed during larval stage 4 in hermaphrodites to allow spermatogenesis (Doniach 1986; Goodwin et al. 1993). Consequently, a fine-tuned balance between tra-2 and fem-3 activity is crucial for the sequential production of correct amounts of spermatids and oocytes. Earlier studies have shown that a strong tra-2(gf) allele can suppress the Mog phenotype of weak fem-3(gf) mutations. However, this is not true for a strong fem-3(gf) allele (Barton et al. 1987). This finding indicates that the rate of FEM-3 activity is crucial for sex determination in the hermaphrodite germline and that the balance between fem-3 and tra-2 might be achieved through a precise control of both genes. This can occur through different strategies. For instance, we hypothesize that spermatogenesis can prevail over oogenesis because high levels of fem-3 activity are reached at a critical time during germline development. Alternatively, it is also conceivable that fem-3 levels remain constant but for an extended duration.
Although several trans-acting repressors of fem-3 have been identified, the molecular mechanism of fem-3 repression remains speculative (Zhang et al. 1997; Puoti and Kimble 1999, 2000; Zanetti et al. 2011). One study reported that in the absence of larp-1, fem-3(+) mRNA levels are increased (Zanin et al. 2010). We now extend this analysis to fem-3 gain-of-function alleles. An earlier finding that the tra-2(gf) mRNA is preferentially associated with polyribosomes indicates that the tra-2 mRNA is translationally controlled (Goodwin et al. 1993). In addition, interaction studies and genetic analyses indicate that the FBF proteins are major regulators of the fem-3 mRNA (Zhang et al. 1997). The homology between FBF and Pumilio suggests a remarkable conservation of a regulatory mechanism in worms and flies. Because Pumilio represses hunchback through deadenylation and translational control, it is tempting to propose a similar mechanism for FBF-mediated repression of fem-3 (Wreden et al. 1997). However, we conclusively show that fem-3 mRNA abundance increases when its 3′ UTR is defective for the FBF binding element. In addition, we found that both wild-type and gain-of-function-mutated fem-3 mRNAs are associated with similar levels with polyribosomes, thus indicating that fem-3(gf) alleles are not translated more efficiently.
We noticed that fractions corresponding to the 40S peak are enriched with wild-type fem-3 transcripts. We propose two explanations for this peak of wild-type fem-3. On one hand, the peak corresponding to the 40S subunit could contain P-body components. P bodies contain translationally repressed mRNAs which can either be stored or directed toward decay (Brengues et al. 2005). P bodies have been found in many species, including C. elegans (Noble et al. 2008, Nykamp et al. 2008). Therefore, one hypothesis is that wild-type fem-3 mRNA could associate preferentially with P bodies and be directed toward translational silencing and/or decay. Mutant fem-3(q95) could have escaped because of its modified 3′ UTR (Noble et al. 2008). However, we do not know where P-body components elute in sucrose gradients with C. elegans extracts. Although, in yeast, P-body components are generally found within fractions that elute between free RNAs and the 40S peak (Brengues et al. 2005), their localization on a sucrose gradient with C. elegans remains speculative. An alternative explanation is that the wild-type fem-3 mRNA preferentially associates with the 40S subunit. Therefore, fem-3(+) would be recruited more efficiently for translational initiation than the mutated fem-3(gf). This does not, however, explain why fem-3(gf) alleles are expressed more efficiently. In fact, wild-type and mutant fem-3 mRNAs are loaded on polyribosomes at similar rates. However, we consistently find higher amounts of gain-of-function-mutated fem-3 mRNAs throughout most fractions of the sucrose gradient. We, therefore, suggest that, at least for the sperm-oocyte switch, fem-3 mRNA is regulated through its stability rather than through its translation.
fem-3 mRNA is repressed in embryos and larvae
Maternal fem-3 mRNA is found in embryos and oocytes (Rosenquist and Kimble 1988; Ahringer et al. 1992). This maternal mRNA is, however, rapidly degraded (Ahringer et al. 1992). FEM-3 protein was found neither in oocytes nor in early embryos, in spite of the presence of its mRNA. Therefore, maternal fem-3 mRNA must be repressed through a mechanism other than RNA decay. There are numerous examples of maternal mRNAs that are stored as a silent form at or near fertilization (for review, see Wickens et al. 2000). For example, the glp-1 mRNA is first repressed in the C. elegans zygote and then silenced only in the anterior blastomeres (Evans et al. 1994; Marin and Evans 2003). Zygotic fem-3 transcription starts later during larval development (Rosenquist and Kimble 1988). Therefore, when analyzing the sperm-oocyte switch, we most likely monitor zygotic transcripts.
fem-3 determines male germ cell fate
fem-3 mutants (Hodgkin 1986; Barton et al. 1987) and the expression pattern of the FEM-3 protein in developing spermatocytes and spermatids (this study) suggest that fem-3 directs spermatogenesis. Other studies have shown that a fem-3::gfp reporter is expressed in spermatids in adult males and hermaphrodites (Sassi et al. 2005). Therefore, we propose that fem-3 activates differentiation in sperm by activating sperm-specific factors in male gametes, rather than inactivating oogenesis. This finding is in contrast with a study that reported FEM-3 expression from the pachytene region through diplotene and diakinesis in oocytes. Moreover, in this study, FEM-3 was absent in the sperm lineage in hermaphrodites (Arur et al. 2011). Intriguingly, the results published in this other study were obtained with the same antibodies, which we made available as unpurified serum. However, we have never been able to reproduce FEM-3 staining in oocytes. Although we used identical staining procedures and tried various alternative protocols with the purified and unpurified antibody, we consistently saw staining in the sperm lineage but never in oocytes. Because meiosis is different in spermatocytes and oocytes, germ cell precursors should already be sexually determined when entering meiotic prophase (Barton and Kimble 1990). If FEM-3 was necessary for commitment to spermatogenesis, it would be expected to be present at earlier steps of gametogenesis. We, therefore, favor a model where FEM-3 promotes spermatogenesis at a later step. This model is supported by the late temperature-sensitive period of fem-3(gf) alleles (Barton et al. 1987). In fact, fem-3(gf) hermaphrodites raised at permissive temperature can revert to spermatogenesis even if they have already started oogenesis. Another example comes from fog-1, which is expressed late during spermatogenesis, yet mutant fog-1(lf) males produce oocytes (Barton and Kimble 1990). Alternatively, FEM-3 levels might be too low to be detected in the distal part of the germline.
Implications on post-transcriptional control
This study provides information on FEM-3 expression in the germline and its consequence on germ cell sex determination. We also show that fem-3 gain-of-function mutants that are impaired in FBF binding are characterized by increased steady-state fem-3 mRNA levels. Therefore, it is possible that FBF causes destabilization of the fem-3(+) mRNA. Furthermore, the polyadenylation of its mRNA correlated with the penetrance of fem-3 mutant alleles. Altogether, this study supports the following model for transient production of spermatids in hermaphrodites: At larval stage 4, tra-2 is repressed, thus tilting the balance in favor of fem-3, which is not regulated at this point (Goodwin et al. 1993). High fem-3 activity results in sperm production. In the adult, fem-3 mRNA levels decrease through degradation of the transcript. Since FBF does not bind efficiently to gain-of-function mutated fem-3 3′ UTRs, we speculate that fem-3 mRNA is controlled, at least in part, through the action of FBF. The yeast Saccharomyces cerevisiae encodes six members of the Puf protein family, including Puf3p and Puf5p, which are required for the decay of COX17 and HO mRNAs, respectively (Olivas and Parker 2000; Tadauchi et al. 2001). Furthermore, FBF represses lip-1 mRNA by controlling its abundance (Lee et al. 2006). Therefore, although Pumilio, the founding member of the Puf family, controls hunchback translation, this and previous studies indicate that other Puf proteins instead act on target mRNA decay. The future will tell if Puf action depends on a particular Puf protein or perhaps on its specific mRNA target.
MATERIALS AND METHODS
Strains and phenotype analysis
All strains were maintained at 15°C under standard conditions, unless noted otherwise. For the analysis of mutant phenotypes, hermaphrodites were allowed to lay for 6 h at the corresponding temperature (15, 20, or 25°C). Eggs were incubated at the same temperature until adulthood. Germline phenotypes were scored by nuclear morphology after DAPI staining. Analysis of heterozygous animals was done by picking fluorescent worms containing nT1g. Nonglowing animals were taken for fem-3(q23), fem-3(q95), or fem-3(q96) homozygotes. Double and triple mutants were obtained by crossing the following strains with the strains listed above: rnp-8(tm2435) I, ccr-4(tm1312) IV, gld-2(q497)/hT2 I,III, gld-3(q730)/mIn1 II, fbf-1(ok91)fbf-2(q704)/mIn1 II, fbf-1(ok91) II, mog-1(q223)/hT2 I,III, and larp-1(q783) III. mIn1(II), nT1g(IV;V), and hT2(I;III) are semidominant markers; N2 was used as a wild-type reference strain.
Quantification of fem-3 mRNA
Fertile fem-3(gf)/nT1g hermaphrodites grown at 15°C were shifted to 25°C and allowed to lay eggs for 6 h. Heterozygous F1 worms were picked under fluorescent light and scored for the Mog phenotype. RNA was extracted and reverse-transcribed as described using either random oligonucleotides or specific antisense primers (Zanetti et al. 2011). The 3′ untranslated region of fem-3 was amplified by PCR and cloned into pBluescript for the sequencing assay (alleles q23 and q96). fem-3(q95) was analyzed by Southern blotting or real-time PCR. fem-3(q23) was also detected through digestion with BsaXI; after extensive digestion, products were analyzed by Southern blotting and quantified with an Amersham Biosciences Phosphoimager (Zanetti et al. 2011). Quantitative real-time PCR was performed on cDNAs using two FAM-conjugated TaqMan probes that are specific either for the wild-type or the fem-3(q95) mutated fem-3 3′ UTR. Raw data from the TaqMan reactions were processed as described (Zhao and Fernald 2005).
In situ hybridization and antibody staining
Single-stranded Digoxigenin-labeled cDNA probes were synthesized using the entire fem-3 coding sequence (Epstein and Shakes 1995). Gonads were dissected, fixed in 0.25% glutaraldehyde, 3% formaldehyde, permeabilized with Proteinase K, and fixed again in glass tubes (Jones et al. 1996). The denatured probe was used at a dilution of 1:3. Hybridization was performed on slides for 30 h at 48°C in 50% formamide, 5× SSC, 0.1% Tween 20, 50 μg/mL heparin, and 50 μg/mL of denatured salmon sperm DNA. Probe detection was done with alkaline phosphatase-conjugated anti-DIG antibodies, 5-bromo-4-chloro-3-indolyl phosphate and Nitro blue tetrazolium chloride (Sigma). Embryos were isolated from gravid hermaphrodites, permeabilized by freeze-cracking and Proteinase K treatment, fixed for 20 min in 3.7% formaldehyde at room temperature, and hybridized for 16 h at 48°C. Nuclei were costained with 4′,6-diamidino-2-phenylindole (DAPI).
Anti-FEM-3 antibodies were generated in a rat using a KLH-conjugated synthetic peptide corresponding to 19 amino acids of the FEM-3 N terminus. Antibodies were then purified using a 900-nt portion of the fem-3 cDNA expressed in bacteria (Arur et al. 2011). Antibody staining was performed on dissected gonads as described (Jones et al. 1996). Incubations with primary antibodies were done o/n at 4°C at the following dilutions: anti-FEM-3 (1/300); anti-SP56 (1/500); anti-GLD-1 (1/150); anti-MOG-6 (1/500). FITC- or Cy-3-conjugated secondary antibodies were diluted to 1/1000. Staining with anti-FEM-3 antibodies gave similar results using alternative protocols, for instance, on whole nematodes (Bettinger et al. 1996). For competition, antibodies were incubated on a wheel at 4°C for 1.5 h in blocking solution (1× PBS Tween 0.1% + 5% BSA) with 20 μg of the corresponding peptides (1 μg/μL). Anti-FEM-3 antibodies were diluted 1:250 and secondary FITC- and Cy-3-conjugated antibodies were used at 1:1000. The expression of RME-2 was analyzed either using a transgenic line containing unc-119(ed3) III; pwIs116(rme-2::GFP + unc-119[+]) or using anti-GFP antibodies (1:200, Q-Biogene). Alternatively, we used anti-RME-2 antibodies (1:400) (Grant and Hirsh 1999). For immunocytochemical staining, embryos were transferred on a slide coated with poly-L-lysine (0.5 mg/mL) and fixed under a glass coverslip for 10 min in a humid chamber with 3 μL of 2.5% paraformaldehyde, 1× PBS, 20 mM NaOH. The preparation was frozen on dry ice for 5 min and cracked by popping off the coverslip. After 5 min in cold methanol and 5 min in cold acetone, samples were washed for 10 min in 1× PBS Tween 0.1% and blocked for 1 h in blocking solution (1× PBS Tween 0.1% + 5% BSA). Primary antibodies (α-MOG-6 [1:500] and α-FEM-3 [1:300]) were added and the sample incubated overnight at 4°C. Secondary antibodies were used at 1:1000.
Poly(A) tail assay
Homozygous L4 larvae raised at 25°C were picked. Total RNA was extracted and tested for poly(A) tail size as described (Salles and Strickland 1995). The forward primer is located in the 3′ UTR of fem-3. All products were migrated on gel and detected by Southern blotting or cloned into pBluescript and sequenced.
Polyribosome analysis
Synchronized fem-3(q95)/+ hermaphrodites were grown at 25°C until late L4 or young adults. Worms were collected in lysis buffer, frozen in liquid nitrogen, and crushed in a mortar to fine powder (Ding and Grosshans 2009). The lysate was cleared by centrifugation (10 min at 10,000g, 4°C) (Ding and Grosshans 2009). 400 μL of lysate was centrifuged on a 10-mL linear sucrose gradient (10%–50% w/v) for 3 h at 40,000 rpm at 4°C. Sucrose gradients were made by overlaying stock solutions with 10%–50% sucrose (Ding and Grosshans 2009). The gradients were collected by aspiration from the top. Absorbance profiles were recorded at 260 nm. RNA was extracted from the fractions and tested by RT-PCR. Products were analyzed by Southern blotting.
Quantitative RT-PCR specifications
Quantitative real-time PCR was performed on cDNA using two FAM-conjugated TaqMan probes, that are specific either for the wild-type (5′cttcttgtgtcattcaact) or the fem-3(q95) mutated (5′catttgaaagtctaactctct) fem-3 3′ UTR. The oligonucleotides for qRT-PCR used were as follows:
-
wt_sense (gattgctccaggaaacgatgat),
-
wt_antisense (ttcaaaatgtaaaccagaggattcg),
-
q95_sense (gattgctccaggaaacgatgat),
-
q95_antisense (atatggaaggagc/aggaaatctg).
The slash indicates the end points of the 112-nt deletion in q95.
ACKNOWLEDGMENTS
We thank Judith Kimble for comments on the manuscript and for providing mutant alleles of fem-3. Some nematode strains used in this work were supplied by the CGC, which is funded by the NIH. Other mutants came from the National Bio Resource Project in Japan. We also thank Tim Schedl for anti-GLD-1 antibodies and Barth Grant for anti-RME-2 antibodies. We thank the University of Fribourg and the Novartis Foundation (project 10B34) for financial support.
Footnotes
-
↵1 Corresponding author
E-mail alessandro.puoti{at}unifr.ch
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.031237.111.
- Received November 4, 2011.
- Accepted May 1, 2012.
- Copyright © 2012 RNA Society








