
Plant siRNAs from introns mediate DNA methylation of host genes
- Dijun Chen1,2,3,4,
- Yijun Meng1,2,4,
- Chunhui Yuan1,
- Lin Bai1,
- Donglin Huang1,
- Shaolei Lv1,
- Ping Wu2,
- Ling-ling Chen3 and
- Ming Chen1,2
- 1Department of Bioinformatics, College of Life Sciences, Zhejiang University, Hangzhou 310058, China
- 2State Key Laboratory of Plant Physiology and Biochemistry, College of Life Sciences, Zhejiang University, Hangzhou 310058, China
- 3State Key Laboratory of Crop Genetic Improvement, College of Life Science and Technology, Huazhong Agricultural University, Wuhan 430070, China
-
↵4 These authors contributed equally to this work.
Abstract
Small RNAs (sRNAs), largely known as microRNAs (miRNAs) and short interfering RNAs (siRNAs), emerged as the critical components of genetic and epigenetic regulation in eukaryotic genomes. In animals, a sizable portion of miRNAs reside within the introns of protein-coding genes, designated as mirtron genes. Recently, high-throughput sequencing (HTS) revealed a huge amount of sRNAs that derived from introns in plants, such as the monocot rice (Oryza sativa). However, the biogenesis and the biological functions of this kind of sRNAs remain elusive. Here, we performed a genome-scale survey of intron-derived sRNAs in rice based on HTS data. Several introns were found to have great potential to form internal hairpin structures, and the short hairpins could generate miRNAs while the larger ones could produce siRNAs. Furthermore, 22 introns, termed “sirtrons,” were identified from the rice protein-coding genes. The single-stranded sirtrons produced a diverse set of siRNAs from long hairpin structures. These sirtron-derived siRNAs are dominantly 21 nt, 22 nt, and 24 nt in length, whose production relied on DCL4, DCL2, and DCL3, respectively. We also observed a strong tendency for the sirtron-derived siRNAs to be coexpressed with their host genes. Finally, the 24-nt siRNAs incorporated with Argonaute 4 (AGO4) could direct DNA methylation on their host genes. In this regard, homeostatic self-regulation between intron-derived siRNAs and their host genes was proposed.
Keywords
INTRODUCTION
In eukaryotes, small RNAs (sRNAs) play key roles in diverse biological events through gene expression regulation at either transcriptional or post-transcriptional levels. Based on their biogenesis, sequence characteristics, associated effector proteins, and biological functions, the sRNAs are generally classified into two major categories: microRNAs (miRNAs) and short interfering RNAs (siRNAs) (Carthew and Sontheimer 2009).
The 21-nt to 24-nt miRNAs derived from stem–loop-structured precursors are RNA polymerase II (Pol II)–dependent (Lee et al. 2004). The miRNA genes are expressed as independent genes or are cotranscribed with their host genes and processed from certain introns (Bartel 2004; Jones-Rhoades et al. 2006). In animals, mature miRNAs are processed from pri-miRNAs (primary microRNAs) to pre-miRNAs (precursor microRNAs) by two sequential cleavages of the RNase III enzymes, Drosha and Dicer (Carthew and Sontheimer 2009). Interestingly, in invertebrates, many hairpin-structured introns, defined as mirtrons, could serve as pre-miRNAs to produce miRNAs, thus bypassing the Drosha-mediated cleavages (Carthew and Sontheimer 2009; Kim et al. 2009). In contrast, only a few mirtrons were identified in plants (Zhu et al. 2008). Instead, the generation of most plant miRNAs depends on two sequential cleavages mediated by Dicer-like1 (DCL1), converting pri-miRNAs to pre-miRNAs, then to the miRNA/miRNA* duplexes (Kurihara and Watanabe 2004).
After loading into Argonaute (AGO; usually AGO1)–associated RNA-induced silencing complexes (RISCs), miRNAs exert repressive regulatory roles on their targets through a sequence-specific manner (Voinnet 2009). Depending on the degree of sequence complementarity, interactions between miRNAs and their targets can lead to either cleavages or translational inhibition of the target transcripts (Carthew and Sontheimer 2009; Voinnet 2009). In plants, most miRNAs guide the AGO1-mediated cleavages of their targets based on the high sequence complementarity of miRNA–target duplexes (Jones-Rhoades et al. 2006; Voinnet 2009). Intriguingly, recent studies revealed that sRNAs released from the canonical miRNA (cmiRNA) precursors can direct DNA methylation in both cis and trans manner after loading into AGO4-associated RISC complexes in rice (Oryza sativa) and Arabidopsis (Arabidopsis thaliana) (Chellappan et al. 2010; Wu et al. 2010).
Compared to the miRNAs, the lengths of siRNAs are more variable, ranging from 21 to 30 nt. These siRNAs are generated from the overlapping regions of natural sense–antisense transcript pairs, from the long single-stranded hairpins formed by inverted repeats (IRs), or from the near-perfect double-stranded RNAs (dsRNAs) synthesized by RNA-dependent RNA polymerases (RDRs) (Zhang et al. 2007; Carthew and Sontheimer 2009; Ghildiyal and Zamore 2009; Dunoyer et al. 2010). According to their origins, the plant siRNAs can be divided into four major classes: heterochromatic siRNAs (hc-siRNAs), trans-acting siRNAs (ta-siRNAs), natural antisense transcript-derived siRNAs (nat-siRNAs), and long siRNAs (lsiRNAs) (Ghildiyal and Zamore 2009). The biogenesis of these siRNAs depends on specific RDRs and DCLs and Pol IV (a plant-specific DNA-dependent RNA polymerase) (Ghildiyal and Zamore 2009). The size of the siRNA is largely determined by the DCL slicer (Qi et al. 2005). Unlike DCL1 required for miRNA generation, DCL2, DCL3, and DCL4 act on perfect or near-perfect double-stranded RNA substrates (Dunoyer et al. 2010). In both Arabidopsis and rice, DCL3 is involved in the biogenesis of the 24-nt siRNAs and lmiRNAs (long microRNAs) (Xie et al. 2004; Chellappan et al. 2010; Wu et al. 2010), whereas DCL4 is responsible for the production of 21-nt siRNAs from IR genes and the ta-siRNAs (Vazquez et al. 2004; Gasciolli et al. 2005; Xie et al. 2005; Liu et al. 2007). DCL2 plays a major role in processing siRNAs from endogenous dsRNA virus in rice (Urayama et al. 2010); presumably a similar role is possessed by its homolog in Arabidopsis (Kapoor et al. 2008), which was suggested to be engaged in producing 22-nt siRNAs for viral resistance (Xie et al. 2004). Distinct kinds of siRNAs are incorporated into specific AGO-associated RISCs, guiding the complexes to the DNA or RNA targets based on sequence complementarity and triggering gene silencing transcriptionally or post-transcriptionally. Sorting siRNAs into specific AGO-associated RISCs, determined by both the lengths and the 5′-terminal nucleotide compositions of siRNAs, is essential for their normal functionalities in plants (Mi et al. 2008; Takeda et al. 2008). In this view, plants possess a sophisticated system to arrange diverse sets of endogenous sRNAs for gene regulation.
The newly developed high-throughput sequencing (HTS) technology greatly facilitated the studies on epigenomes and transcriptomes (Simon et al. 2009; Wang et al. 2009b; Hawkins et al. 2010). By using HTS, numerous conserved and non-conserved sRNAs have been identified. For some plant species, the highly integrated, whole-genome landscapes of cytosine methylomes, transcriptomes, and smRNAomes are available (Lister et al. 2008; Wang et al. 2009a; He et al. 2010). These huge and invaluable data sets provide us with a quite comprehensive view of the complex interplay among DNA methylation, gene expression, and sRNA generation. Thus, HTS opened a new era for genome- or transcriptome-wide studies, whereas the fast-growing HTS data sets remain to be fully explored.
As model organisms in the plant research area, the eudicot Arabidopsis and the monocot rice have been extensively studied, and their genomes were well annotated. Our previous study revealed that ∼7% of the sRNAs analyzed were uniquely derived from the introns in rice, while only ∼2% of the sRNAs analyzed were uniquely derived from the introns in Arabidopsis. This may be partially due to the difference of the gene structures between the two plants (Chen et al. 2010). Animal intronic canonical miRNAs and mirtronic miRNAs have been extensively described (Brown et al. 2008; Kim et al. 2009). Comparatively, comprehensive characterization of intron-derived sRNAs remains lacking in plants, although some current reports indicated that intron-derived miRNAs existed in plants (Rajagopalan et al. 2006; Axtell et al. 2007; Piriyapongsa and Jordan 2008; Zhu et al. 2008). Here, we carried out a global survey of rice intron-derived sRNAs by using publicly available HTS data sets. We discovered that some introns could produce miRNAs and siRNAs from their internal short and long hairpin structures, respectively. Besides, we identified another 22 long-hairpin-containing introns of protein-coding genes, termed “sirtrons,” that generated a diverse set of siRNAs mostly from the sense strands. The siRNAs derived from the long stem regions of the sirtrons are mainly 21 nt, 22 nt, and 24 nt in length, whose production seems to be dependent on DCL4, DCL2, and DCL3, respectively. Furthermore, based on genome-wide RNA-Seq and DNA methylation data, we demonstrated that these intron-derived sRNAs could modulate gene expression at the chromatin level. Intriguingly, the intron-derived siRNAs were indicated to be involved in DNA methylation at their own loci and seemed to be coexpressed with their host genes. It led us to propose a self-regulation model between these siRNAs and their host genes. Taken together, our study uncovered a novel intron-derived sRNA species in planta, and its biogenesis and functionalities were computationally elucidated based on HTS data.
RESULTS
Hairpin-structured introns generating both miRNAs and siRNAs in rice
While examining rice sRNAs and miRNAs residing within introns based on HTS data (Supplemental Tables S1–S3; Supplemental Data Set S1; Wu et al. 2009, 2010), we observed that, in addition to the stem–loop-structured pre-miRNAs, certain rice introns also contained long hairpin structures with both arms producing sRNA clusters (Fig. 1; Supplemental Figs. S1, S2). Moreover, mRNA HTS data (He et al. 2010) showed that the host genes containing these novel introns were actively expressed (Fig. 1B; Supplemental Fig. S2A), further indicating that these sRNAs were likely produced from the introns spliced from the host transcripts. More interestingly, the sequences of certain long hairpins were found to be highly methylated (Fig. 1B; He et al. 2010), which might be mediated by the intron-derived sRNAs. In this scenario, we will refer to these intron-derived sRNAs as siRNAs hereafter, although their functions remain to be further elucidated.
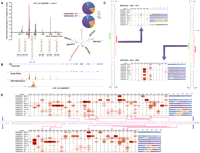
The rice introns encoding both microRNAs (miRNAs) and short interfering RNAs (siRNAs). An example of such dual-coding introns, the first intron of LOC_Os12g09290.1, is shown here. (A) Normalized abundances of the sRNAs within the intron (top left), the size distribution of sRNAs from the intron (top right), schematic presentation of the five tandem intronic miRNA precursors and a long hairpin structure (bottom left), and the predicted secondary structure of the whole intron (bottom right). The miRNAs and the miRNA*s are indicated by red and green, respectively. The complementary region of the long hairpin is highlighted in light background. The first nucleotide of the intron is set as position 1. The data are plotted at 1-base resolution. The sRNAs derived from this intron are in red on the secondary structure of the intron. The names of the miRNA genes were marked at the corresponding positions on the secondary structure of the intron with the long paired region generating siRNAs labeled with a red bar. (B) Gene and sRNA expression, and DNA methylation patterns of LOC_Os12g09290.1. The corresponding data were visualized by the UCSC Genome Browser. (C) Characterization of two newly identified MIR genes in this intron. Secondary structures of the precursor miRNAs (pre-miRNAs) (left, MIR1863d; right, MIR1863e) were predicted by RNAfold (Hofacker 2003). Configuration tables with bar charts (colored as in A) show the number and the percentage of sRNAs in each high-through sequencing library derived from these two MIR gene loci. Positions generating mature miRNAs and miRNA*s are denoted by red and green bars, respectively. (D) Number and percentage of siRNAs derived from the long hairpin, colored as in C. Only positions with more than 10 siRNA reads in at least one library are shown. For both C and D, numbers in parentheses indicate the sequence positions within the intron. (WT) Wild type.
Two miRNA genes, MIR1863b and MIR1863c, are tandemly transcribed from the sense strand of the first intron of LOC_Os12g09290.1 (Fig. 1A; Supplemental Fig. S1A,D). According to the current definition of plant miRNA genes (Meyers et al. 2008), we found that three additional sRNA-coding regions within the same intron could be annotated as bona fide miRNA gene loci (Fig. 1C; Supplemental Fig. S1B,D). All these newly identified miRNAs were 24 nt in length, and the corresponding miRNA*s could be detected in the same HTS data sets as well. Further investigation by using various sRNA HTS libraries showed that the 24-nt miRNAs were generated by DCL3 independent of RDR2 and were specifically sorted into AGO4-associated RISCs (Fig. 1C; Supplemental Fig. S1B,D). These salient features are quite similar to those of the lmiRNAs as previously described in rice (Wu et al. 2010). These three miRNA genes were designated as MIR1863d, MIR1863e, and MIR437b, respectively, based on the sequence similarities compared to the currently annotated miRNAs (miRBase, release 15). Therefore, five miRNA genes are tandemly arranged within this intron (Fig. 1A).
HTS data showed that a high abundance of siRNAs, mainly 21 nt, 22 nt, or 24 nt in length, were regularly distributed within the first intron of LOC_Os12g09290.1 (Fig. 1A). In addition to a small portion (∼6%) of siRNA reads uniquely mapped to the miRNA precursors, the remaining ones (∼94%) were mapped to a long highly complementary region (Fig. 1A,D). This ∼200-bp highly complementary region can form a pre-miRNA-like hairpin structure (Fig. 1A).
To gain deeper insights into the distribution patterns and the biogenesis of these siRNAs from the long hairpin, we used several HTS libraries prepared from different biological samples (Supplemental Table S1). The results showed that several hotspots for siRNA generation resided within the complementary stem region of this long hairpin (Fig. 1D; Supplemental Fig. S1C). Compared to the library prepared from the wild-type (WT) sample, some siRNAs were found to be down-regulated in both dcl1 and dcl3 mutants, indicating that DCL1 and DCL3 are responsible for siRNA generation in these loci. In contrast, for some loci, the siRNA read counts were increased in the dcl1 library but reduced in the dcl3 library, or vice versa, thus proposing a compensatory effect between DCL1 and DCL3 on siRNA generation. We further investigated the scene of the rdr2 library, and the result showed that nearly all the intron-derived siRNAs were unaffected compared to the WT library (Pearson's r = 0.65). From this point of view, the biogenesis of these siRNAs from the long hairpin-structured intron does not require RDR2, which is similar to the well-defined miRNAs. We named the introns containing long hairpins as “lhp-introns” for short hereafter.
Next, we investigated in detail which DCL(s) were required for the generation of these long-hairpin-derived siRNAs. At first glance, the expression of 24-nt siRNAs was severely repressed in the dcl3 library, indicating the responsibility of DCL3 for their generation. However, the accumulation of 21-nt and 22-nt siRNAs was not significantly altered in both DCL1IR (similar to the dcl1 mutant) and dcl3 libraries, compared to the WT libraries (Pearson's r = 0.68 and 0.70 for the 21-nt siRNAs; 0.60 and 0.64 for the 22-nt ones). This indicates that the generation of the 21-nt and 22-nt-long hairpin-derived siRNAs is independent of DCL1 and DCL3 activities. This observation combined with the previous findings in rice (Liu et al. 2007; Urayama et al. 2010) that DCL4 was responsible for the 21-nt siRNA generation and DCL2 for the 22-nt ones, led us to conclude that the long-hairpin-derived 21-nt, 22-nt, and 24-nt siRNAs were the products of DCL4-, DCL2-, and DCL3-mediated cleavages, respectively.
Besides, we found that another four introns—i.e., the second intron of LOC_Os06g36160.1 containing MIR1870, the second intron of LOC_Os08g15204.1 containing MIR1427, the seventh intron of LOC_Os08g25624.1 containing MIR2874, and the third intron of LOC_Os11g05562.1 containing MIR1862e and MIR2872—could also form long hairpin structures with strong siRNA-coding potential (Supplemental Fig. S2). Different from the former one, most siRNAs from the long hairpins of these introns were 24-nt ones. Interestingly, MIR1427, MIR2872, and MIR2874 originated from the corresponding long stem regions of the hairpin-structured introns (Supplemental Fig. S2B), indicating that miRNAs and miRNA-like siRNAs might share the same long hairpin precursors.
Collectively, these results indicate that the introns of the rice genes can form extremely complex hairpin structures producing both miRNAs and siRNAs. The generation of these long-hairpin-derived siRNAs depends on the activities of specific DCLs.
Genome-wide identification of lhp introns
Compared to Arabidopsis, rice possesses relatively long introns with a stronger potential for sRNA generation (Chen et al. 2010). The above results on hairpin-structured introns encoding both miRNAs and siRNAs prompted us to perform a whole-genome search for the rice introns capable of forming long hairpins. A de novo identification of rice lhp introns was carried out by using the program “einverted” embedded in the EMBOSS package (Rice et al. 2000). To distinguish the desired long-hairpin-structured introns from the short stem–loops, we used a strict criterion for this analysis (for details, see Materials and Methods). As a result, 93 lhp introns were identified in total, including nine TE (transposable element)–related introns (Supplemental Table S4).
Previous results showed that spliced hairpin introns could be retained in the nucleus (Qian et al. 1992) and naturally anneal to form long-hairpin structures that served as DCL substrates for siRNA production (Zhang et al. 2007). This is reminiscent of the short-hairpin mirtrons found in animals that are recognized by Dicer (Kim et al. 2009) and the five rice introns with dual-coding potential that we described above. Thus, it is interesting to examine whether the generation of all the lhp-intron-derived siRNAs has strand bias. To address this question, we mainly focused on the siRNAs that could be uniquely mapped to the rice genome (unique siRNAs) to unambiguously determine their origins. After mapping sRNA HTS data of rice seedlings (Wu et al. 2009, 2010) to these lhp introns, 67 lhp introns were found to possess at least one unique siRNA locus based on the WT libraries. Then, a statistical analysis was performed to examine whether siRNAs accumulated within the long-hairpin-structured regions were more enriched than random distribution (see Materials and Methods). Given a threshold that P < 0.05 and more than 10 unique siRNAs should be found within the long stem regions for further filtering, 22 lhp introns were demonstrated to produce “over-represented” siRNAs. Moreover, nearly all the lhp-intron-derived siRNAs were exclusively generated from the long complementary regions (Supplemental Data Set S2), and the vast majority of the unique siRNAs tended to be originated from the sense strands of the long hairpins (Table 1), which indicated that the 22 lhp introns were bona fide precursors encoding miRNA-like siRNAs. The siRNAs derived from the 22 lhp introns are predominantly 21 nt, 22 nt, or 24 nt in length, whose biogenesis relies on DCL4, DCL2, and DCL3, respectively (Supplemental Table S5). In the rdr2 library, the recent report showed that the production of 24-nt siRNAs from double-stranded RNAs was dramatically repressed (Wu et al. 2010). But, notably, the accumulation of all of the lhp-intron-derived siRNAs was unaffected compared to the corresponding WT library (Supplemental Table S5). In Arabidopsis, RDRs, mostly RDR2, are required to convert long single-stranded RNAs (ssRNAs) to double-stranded RNAs (dsRNAs). Based on the examinations of rdr2 mutants in both rice and Arabidopsis (Kasschau et al. 2007; Wu et al. 2010), the role of RDR2 in converting ssRNAs to dsRNAs tends to be highly conserved. Together with the previous findings in Arabidopsis (Lindow et al. 2007; Zhang et al. 2007; Dunoyer et al. 2010), we concluded that the lhp-intron-derived siRNAs were diced from the single-stranded, long-hairpin-structured precursors by specific DCLs, independent of RDRs. Here, we defined these lhp-introns as “sirtrons,” which generated highly abundant siRNAs from their long stem regions with sense-strand bias.
List of 22 sirtrons with a dominant portion of short interfering RNAs (siRNAs) generated from the sense strands
One typical example of such sirtron is the fifth intron of LOC_Os07g01240.1 (Fig. 2). We observed that the highly abundant siRNAs (4801 distinct reads with 39,969 total counts in two rice seedling libraries) were symmetrically distributed within the ∼1600-bp complementary region (Fig. 2A). Moreover, the host gene was actively expressed, and the gene body was highly methylated within this intron (Fig. 2B). The siRNAs generated from this sirtron share the common features as described above (Fig. 2C). A deeper insight into these sirtron-derived siRNAs revealed that the siRNAs in different sizes share nearly the same hot regions for generation (Fig. 2D), further supporting the idea that diverse DCLs acted cooperatively to produce different siRNA classes (Chellappan et al. 2010; Dunoyer et al. 2010; Wu et al. 2010).
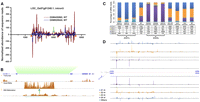
Long-hairpin-structured introns (lhp introns) producing an abundance of short interfering RNAs (siRNAs) from the sense strand. One example of this kind of intron, the fifth intron of LOC_Os07g01240.1, is shown. (A) Profiling of siRNAs derived from this intron. The normalized siRNA abundance in each library was plotted at 1-base resolution. The green arrows indicate the predicted terminal loop of the folded region, and the red triangles indicate the boundary of the hairpin. In total, 39,969 sequence reads (representing 4801 distinct siRNA sequences) were generated from the intron, with 27,060 (67.7%) and 12,909 (32.3%) mapped to the sense and the antisense strands, respectively. Of these sequences, 39,959 (>99.9%, 4793 distinct siRNA sequences) could be unambiguously mapped to the stem region of the hairpin, with 20,813 (52.1%) and 19,146 (47.9%) on the 5′ and 3′ arms, respectively. (B) Gene and siRNA expression, and DNA methylation patterns of LOC_Os07g01240.1. The long complementary stem regions are highlighted by lines at the bottom (left line indicates the 3′ arm and the right one the 5′ arm). Please note that the gene model is shown in reversed direction. (C) The size distribution of the siRNAs derived from this intron. The percentage of siRNAs in each size from the 11 data sets is shown in bar charts, and the number of sequence reads is listed in the table. Three AGO1 and three AGO4 family members are indicated below. For both A and C, note that the two wild-type (WT) libraries (GSM455965—reference library for AGO1s, and GSM520640—reference library for AGO4s, DCL1IR, dcl3, and rdr2) were from two independent experiments. (D) Deep insight into siRNA generation along the stem regions of the long hairpin. SiRNA populations in each size category (as colored by the listed key, the same as in B from the 5′ arm [top] and 3′ arm [bottom]) were plotted after alignment by the 5′ terminus. The positions of the stem regions along the predicted secondary structure (middle) are shown in parentheses.
The expression of sirtron-derived siRNAs is highly correlated with their host genes
Previous studies in animals showed that intronic miRNAs were coexpressed with their host genes (Baskerville and Bartel 2005; Berezikov et al. 2007). Since intron sequences were reported to remain in the nucleus after the splicing of mRNA precursors (Qian et al. 1992), one concern about the detected intronic siRNAs was whether their host genes were actively expressed. One would raise the hypothesis that the production of siRNAs from the introns of protein-coding genes depends on the expression of their host genes. The identified sirtrons are good candidates to address this question. Interestingly, we observed that the sirtron-derived siRNAs were coexpressed with the host genes in rice (Supplemental Table S6).
For the lhp introns generating very few siRNAs, i.e., the introns with under-represented siRNAs, no correlated expression pattern was observed between the siRNAs and their host genes. However, for the lhp introns with overrepresented siRNAs, their host genes were often highly expressed (Fig. 3A). We further probed this issue based on the 22 newly identified sirtrons in this study. These introns appeared to be the authentic precursors for siRNA generation, as a dominant portion of siRNAs were generated from the sense stands (Table 1). We found that the numbers of sirtron-derived siRNAs were positively correlated with the expression levels of the corresponding host genes based on the rice seedling libraries (Spearman's ρ = 0.32). Most of the sirtron-containing host genes were highly expressed according to the high-throughput mRNA-Seq data (Fig. 3A; Supplemental Table S6; He et al. 2010). As a comparison, there was no correlation between the numbers of siRNAs from the other lhp introns (non-sirtrons) and the expression levels of their host genes (Spearman's ρ = −0.08). Besides, for some lhp introns with over-represented siRNAs mostly derived from the antisense strands, the corresponding gene expression levels were very low or non-detectable (Fig. 3A). In this regard, we demonstrated that the siRNAs derived from the sirtrons were coexpressed with their host genes, at least to some extent.
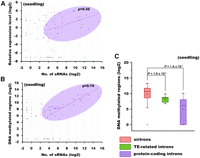
Sirtronic siRNAs are coexpressed with their host genes and direct DNA methylation in cis. (A) Correlation between the number of the intronic sRNAs and the expression levels of their host genes. (B) Correlation between the sRNA generation in lhp introns and DNA methylation at their own loci. The sRNAs and DNA methylated regions were counted for each lhp intron. For both A and B, scattered plots show log2-scaled data for each lhp intron. If the corresponding data were not detected (with “0” value unsuitable for log2-transforming), we uniformly used a constant number (smaller than lowest log2-scale detected value; “−1” for sRNA/DNA methylation data and “−7” for gene expression data) instead. The oval includes nearly all the sirtrons (red) within it, and the Spearman's rank correlation coefficient (ρ) for each category is shown. TE (transposable element)–related lhp introns (green); protein-coding lhp introns (non-sirtrons) (purple). (C) Box plots illustrating the significantly different DNA methylation pattern (log2-transformed) between the sirtronic sRNA and non-sirtronic ones, as colored in A and B. The bottom and top of the box are the 25th and 75th percentile, respectively. (Filled triangles) The smallest (upward triangles) and largest observation (downward triangles); (filled rectangles) the mean value. The significant P-values between the two groups are shown above. (TE) Transposable element.
Sirtron-derived siRNAs mediate DNA methylation of their host genes
Sorting siRNAs into the correct AGO-associated RISCs is very important for them to exert their repressive regulatory roles on the targets. In plants, the AGO1-associated siRNAs or cmiRNAs repress target genes at post-transcriptional level through cleavages, while the siRNAs or lmiRNAs recruited by AGO4-associated RISCs act at the chromatin level through RNA-directed DNA methylation (RdDM) (Hamilton et al. 2002; Llave et al. 2002; Tang et al. 2003; Vazquez et al. 2004; Borsani et al. 2005; Wu et al. 2010). In rice, there are 19 AGO homologs that can be divided into four classes: the MEL1, the AGO1, the AGO4, and the AGO7 subfamilies (Kapoor et al. 2008; Wu et al. 2009, 2010). Three members of the AGO1 subfamily, i.e., AGO1a, AGO1b, and AGO1c, preferentially associate with 21-nt siRNAs and miRNAs with 5′ U (Wu et al. 2009). AGO4a, AGO4b, and AGO16, belonging to the AGO4 subfamily, predominantly recruit 24-nt siRNAs with 5′ A, although one member, AGO4b, shows weaker bias for the siRNAs with 5′ A (Wu et al. 2010). In this analysis, we investigated into which AGO-associated RISC(s) the sirtron-derived siRNAs were sorted. The results showed that the 21-nt siRNAs, mostly with 5′ U, were highly enriched in AGO1. Comparatively, the 24-nt siRNAs, mostly beginning with A, were enriched in AGO4 (Figs. 2C, 4). Generally, the sirtron-derived 21-nt and 24-nt siRNAs are preferentially associated with AGO1- and AGO4-associated RISCs, respectively, with positive correlation with the total siRNA profiling in WT (Pearson's r = 0.38 for 21-nt siRNAs/AGO1s; r = 0.58 for 24-nt siRNAs/AGO4s). Based on the recent results in rice (Wu et al. 2009), this weak correlation between the 21-nt siRNAs and AGO1s (AGO1a, AGO1b, and AGO1c) could be partially interpreted by overlapping but nonredundant functions among the AGO1 homologs, which could also be applied to the AGO4-associated 24-nt siRNAs. Additionally, the 22-nt siRNAs were rarely detected in AGO1 and AGO4 proteins (Pearson's r = 0.19 for AGO1; r = 0.12 for AGO4), indicating that these siRNAs must be recruited by some other AGO(s).
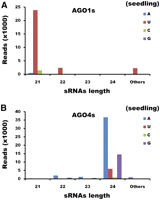
Twenty-one-nucleotide and 24-nt long-hairpin-structured intron (lhp intron)–derived siRNAs were bound to AGO1s and AGO4s, respectively. (A) lhp-intron-derived siRNA sequences counted in AGO1 (AGO1a/b/c) libraries. (B) lhp-intron-derived siRNA sequences counted in AGO4 (AGO4a/b and AGO16) libraries. For both A and B, lhp-intron-derived siRNAs associated with AGO1s or AGO4s differ in the bias of their first nucleotide and length. The 21-nt siRNAs mostly beginning with uridine were preferentially bound to AGO1s. In contrast, the 24-nt siRNAs mostly beginning with adenosine were highly enriched in AGO4 proteins. Besides, the 22-nt siRNAs were under-represented in both AGO1 and AGO4 libraries. (A) Adenosine; (U) uridine; (C) cytosine; (G) guanine.
Based on the genome-wide DNA methylation profiling data (He et al. 2010), we noticed that the long complementary regions of the sirtrons were heavily methylated when compared to the corresponding flanking sequences (Fig. 2B; Supplemental Data Set S2). The DNA methylation patterns within the hairpin structures are well correlated with the numbers of the siRNAs generated (Spearman's ρ = 0.79 depicted in Fig. 3B). This correlation became even stronger if only the 24-nt siRNAs were considered (Spearman's ρ = 0.86). Furthermore, the intensity of DNA methylation of the protein-coding sirtrons producing over-represented siRNAs is significantly stronger than that of the protein-coding non-sirtrons only generating under-represented siRNAs (P = 1.4 × 10−7) (Fig. 3C). This suggests that the sirtron-derived siRNAs are indispensable for maintaining methylation status at their own origins. Besides, the average DNA methylation status of the sirtronic loci was more than six times higher than that of the TE-related non-sirtronic loci (P = 1.9 × 10−2) (Fig. 3C). Most interestingly, nearly all of the methylated loci are restricted within the stem regions of the long hairpins. All these observations collectively demonstrated that the sirtron-derived 24-nt siRNAs were incorporated into the AGO4-associated complexes, mediating RdDM in cis.
DISCUSSION
In this study, we identified five rice introns with dual-coding potential that the miRNAs generated from the short stem–loop structures and the siRNAs from the long hairpins. Besides, the long complementary regions of 22 lhp introns belonging to the protein-coding genes, defined as sirtrons, were found to be the rich source for siRNA generation. All of the intron-derived sRNAs including siRNAs and miRNAs showed a length bias toward 21, 22, or 24 nt, whose production relied on specific DCLs (Wu et al. 2010). The 21-nt and 24-nt siRNAs were specifically loaded into AGO1- and AGO4-associated RISCs, which would further determine their manner of action. Based on the available mRNA degradome sequencing data (Wu et al. 2009; Li et al. 2010) together with target prediction algorithms, examination of the relatively highly expressed (more than 10 reads in WT libraries) and abundantly loaded (more than 100 reads in AGO1 libraries) sirtron-derived 21-nt siRNAs revealed that these 21-nt siRNAs could direct target mRNAs for cleavage (Supplemental Table 7), functionally like the cmiRNAs and 21-nt ta-siRNAs, following their incorporation into AGO1s. However, into which AGO(s) the 22-nt siRNAs were sorted remains unclear, and their biological function should be further investigated.
To determine whether this kind of sirtron exists naturally in other plant species, we applied the same strategy to the well-annotated Arabidopsis genome by using publicly available sRNA HTS data sets. Surprisingly, no such canonical sirtron was detected, although a few lhp introns were predicted (data not shown). Considering our strict criterion used for this prediction, it might be caused by the smaller introns along with fewer mapped sRNAs in Arabidopsis compared to rice (Chen et al. 2010). Or, due to the limited sRNA HTS data sets used for the analysis of Arabidopsis, also for rice, of course (Supplemental Table 5), some novel sirtrons might not be identified, which could be functional in specific tissues or under certain conditions. From another aspect, this kind of sirtron could be species-specific as indicated by the featured intronic miRNAs in rice: first, the rice intronic miRNAs tend to be less conserved, with only five conserved miRNAs (3%) from introns based on miRBase (release 15); second, so far, the 24-nt intronic lmiRNAs have only been discovered in rice, which presented a dominant portion of the intronic miRNAs (Supplemental Table 2). However, we could not rule out the possibility that such sirtrons might exist in Arabidopsis or other plant species. In fact, IR71 and IR2039 in Arabidopsis share some similar properties with the rice sirtrons, although both are not from introns (Dunoyer et al. 2010). In addition, an intronic hairpin-structured RNA (int-hpRNA) construct was reported to be capable of initiating cis- and trans-methylation by producing 24-nt siRNAs in tobacco (Dalakouras et al. 2009).
Our results also demonstrated that the sirtron-derived 24-nt siRNAs associated with AGO4 could direct RdDM in a cis manner. However, based on the observation that these 24-nt siRNAs displayed a heterogeneous distribution along sirtron loci and showed no exact correlation with the methylation density (Fig. 2B,D; Supplemental Data Set S2), the sirtron-derived 24-nt siRNAs may be involved in the amplification and maintenance of DNA methylation rather than triggering this process (Dalakouras et al. 2009). This notion could also be applied to the 24-nt siRNAs derived from the dual-coding introns (Fig. 1B,D; Supplemental Fig. S2A). Furthermore, a weak negative correlation between DNA methylation and gene expression level was observed (Pearson's r = −0.1) (Supplemental Table S6). Although body methylation could affect gene transcription in an inconsistent manner (Miura et al. 2009), i.e., either positive or negative, our data tended to support the notion that the sirtron-derived siRNA-mediated RdDM could result in transcription repression to a weak extent, which needs additional experimental validation. The results presented above together with the previous finding that intronic MIR1873 could direct RdDM at its own locus (Wu et al. 2010) led us to propose a self-regulation model (Fig. 5). In this model, the siRNAs and/or miRNAs generated from the hairpins within the spliced introns direct RdDM at their own loci, repressing the expression of their host genes at a moderate level. At the same time, the accumulation of these intron-derived siRNAs largely depends on the transcription of their host gene. Thus, the expression homeostasis between the intron-derived siRNAs and/or miRNAs and their host genes can be buffered by the self-regulation mechanism (Fig. 5). The lack of a strict correlation of the expression levels between the host genes and the intron-derived siRNAs (Fig. 3A) can be inferred from the previous reports that gene expression could be transcriptionally repressed by hypermethylation on the gene bodies, but not completely inhibited (Miura et al. 2009; Teixeira and Colot 2009), which is also reflected by the homeostatic regulation in the self-regulation model (Fig. 5). Notably, although the lmiRNAs and the 24-nt sirtronic siRNAs were both sorted into AGO4-associated RISCs, the RdDM mediated by the two small RNA species is quite different. The lmiRNAs mediated DNA methylation in trans and could significantly reduce the gene expression in WT (Chellappan et al. 2010; Wu et al. 2010), whereas the sirtron-derived siRNAs mediated methylation in cis and sometimes might not inhibit their host genes completely (Fig. 3A). Like heterochromatic siRNAs (hc-siRNAs), the 24-nt sirtronic siRNAs are DCL3-dependent. But, distinct from the hc-siRNAs, whose generation depends on Pol IV and RDR2 (Ghildiyal and Zamore 2009), the biogenesis of the 24-nt siRNAs generated from the lhp introns is Pol II–dependent and RDR2-independent (Fig. 2C; Supplemental Table S5). Besides, nearly all the unique sRNAs were generated from the sense strands (Table 1), further indicating that they were RDR-independent and/or Pol IV–independent. Supporting this notion, two discrete IR loci, IR71 and IR2039, found in Arabidopsis were shown to generate abundant sRNAs from the antisense strands independent of RDR or Pol IV (Dunoyer et al. 2010), while the vast majority of the unique sRNAs were derived from their sense strands (Zhang et al. 2007). Finally, different from the hc-siRNA-guided DNA methylation patterns that were irregularly distributed along the transposons or repetitive sequences, DNA methylation mediated by the 24-nt sirtron-derived siRNAs was limited to the long complementary regions of the hairpin-structured introns (Fig. 2B; Supplemental Data Set S2). These observations further support the proposed model that the sirtron-derived siRNAs regulate their host genes through RdDM in cis.
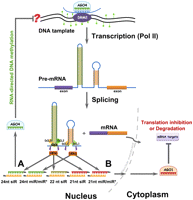
The proposed self-regulation model between the intronic microRNAs (miRNAs) and/or short interfering RNAs (siRNAs) and their host genes. The intronic miRNA/siRNA pathways initiate with the splicing of the primary mRNAs to yield the long- and/or short-hairpin-containing introns, which are the substrates of specific DCLs. Intronic miRNA precursors are processed by coordinated activities of DCL1 and DCL3 to generate canonical miRNAs (cmiRNAs, 21-nt) and long miRNAs (lmiRNAs, 24-nt), respectively. Our analysis based on deep-sequencing data from rice unraveled a strikingly symmetrical distribution of highly abundant siRNA classes, bias to 21, 22, 24 nt in length, diced by DCL4, DCL2, and DCL3, respectively, from the intronic long complementary hairpin precursors. The intronic siRNA pathway, interacting with the intronic miRNA pathway at some level, operates at both chromatin (A) and post-transcriptional levels (B). In A, the intronic 24-nt siRNAs or miRNAs bound to AGO4 proteins interact with nascent transcripts transcribed from their own loci to direct DNA methylation at the adjacent DNA (their host gene loci). Thus, intronic miRNAs/siRNAs control the transcription homeostasis of their host genes by mediating an RNA-directed DNA methylation (RdDM) pathway. Here, we proposed the potential negative correlation between host gene expression and intron-derived siRNA-mediated cis-methylation, which needs to be further validated. In B, the 21-nt siRNAs and miRNAs/miRNAs* are exported to the cytoplasm and then incorporated into AGO1 to carry out the silencing reactions (target mRNA cleavage or translation inhibition).
The identification and characterization of the intronic miRNAs and sirtronic siRNAs expanded our view of plant intron-derived sRNAs. In this study, we demonstrated that 164 introns (including 71 miRNA-containing introns and 93 lhp introns) in total contributed to the generation of nearly one-third of the intronic sRNAs in rice (Supplemental Table S8). These introns tend to be a rich source for miRNA and/or siRNA generation, suggesting their biological roles in plant development. In addition, these intron-derived siRNAs were suggested to play a regulatory role in expression control of their host genes through RdDM in cis. GO (Gene Ontology) term analysis indicated that these host genes were functionally enriched in “nucleic acid binding,” “DNA binding,” and “transcription regulator activity” (Supplemental Table S9). Interestingly, some host genes were annotated as methyltransferase genes (Supplemental Table S4).
Processing of lhp-intron-derived sRNAs by cooperative action of DCLs
Recent findings in plants showed that a diverse set of sRNAs including miRNAs and siRNAs could be generated from the same hairpin precursors (Piriyapongsa and Jordan 2008; Vazquez et al. 2008; Chellappan et al. 2010; Dunoyer et al. 2010; Wu et al. 2010), indicating cooperative processing by specialized DCLs for sRNA production. In these studies, some precursors were found to possess dual-coding potential to generate distinct sRNA classes. Chellappan et al. (2010) provided the evidence that a significant number of MIR genes in Arabidopsis could generate both miRNAs and siRNAs from the same precursors, while some MIR gene loci in rice and Arabidopsis could produce both cmiRNAs and lmiRNAs (Vazquez et al. 2008; Wu et al. 2010). All of the dual-coding MIR genes were processed by coordinated activities of DCL1 and DCL3 (Vazquez et al. 2008; Chellappan et al. 2010; Wu et al. 2010). Besides, some TE genes in rice and Arabidopsis were reported to encode both miRNAs and siRNAs (Piriyapongsa and Jordan 2008). Different from the dual-coding MIR genes and TEs, which were isolated transcription units, some introns of rice protein-coding genes identified in the study could produce miRNAs and siRNAs from their internal hairpin structures (Fig. 1; Supplemental Fig. S2), which also required precise cooperation among different DCLs (Fig. 5). All of these findings may further provide an evolutionary perspective of siRNA-to-miRNA transition, accompanied by the activity shift of DCLs (Vazquez et al. 2008; Voinnet 2009).
In this study, we showed that the sirtron-derived siRNAs were mainly 21 nt, 22 nt, and 24 nt in length, which were processed by DCL4, DCL2, and DCL3, respectively. Especially, 19 sirtrons generated all three siRNA classes, and the remaining three generated both 21-nt and 24-nt siRNAs (Supplemental Table S5). Moreover, these siRNAs of different sizes shared similar expression patterns at the hotspots for their generation (Fig. 2D), further suggesting that DCL2, DCL2, and DCL4 act cooperatively to dice the long hairpins. Two IR loci identified in Arabidopsis, i.e., IR71 and IR2039 (Dunoyer et al. 2010), required the similar machineries including DCLs and AGOs for siRNA biogenesis and their functions as the rice sirtrons shown in the present study. Experimental evidence showed that siRNAs derived from the IR-loci were hierarchically processed firstly by DCL2 and DCL3, then by DCL4 (Dunoyer et al. 2010). Similar phenomena were observed in some MIR genes (Wu et al. 2010). Based on the coordinated actions of DCL1 and DCL3, cmiRNAs and lmiRNAs could be sequentially released from the dual-coding MIR precursors (Wu et al. 2010). We observed that the 24-nt siRNAs were eliminated in dcl3, whereas the composition of 21-nt and 22-nt siRNAs was unchanged in the same library (Fig. 2C; Supplemental Table S5). This led us to propose that these sirtron-derived siRNAs were also processed in a hierarchical manner, although the detailed sequential actions of different DCLs remain elusive.
Our results together with the previous findings (Piriyapongsa and Jordan 2008; Vazquez et al. 2008; Zhu et al. 2008; Chellappan et al. 2010; Dunoyer et al. 2010; Wu et al. 2010) present complicated pathways of sRNA biogenesis and their functional implementation in plants. Advances in the plant sRNA research area indicate the existence of widespread cross talk between the miRNA and the siRNA pathways, further blurring the distinction between the two small RNA species (Voinnet 2009).
MATERIALS AND METHODS
Data sources
Rice sRNA HTS data sets of different tissues were retrieved from GEO (http://www.ncbi.nlm.nih.gov/geo/) via the accession numbers indicated in recent publications (Wu et al. 2009, 2010). DNA methylation and gene expression (RNA-Seq) data were retrieved from the UCSC Genome Browser (http://159.226.118.31:9311/cgi-bin/hgGateway?org=Nipponbare&db=nipp) (He et al. 2010). Please see Supplemental Table S1 for a summary of all the data sets used in this analysis.
The rice genomic sequences and gene annotation information were retrieved from the TIGR Rice Annotation Database (http://rice.plantbiology.msu.edu/; release 6.1), and the rice miRNAs from miRBase (http://www.mirbase.org/; release 15) and a recently published work (Wu et al. 2010).
Bioinformatics analysis of intronic sRNAs
We used relatively stringent criteria to uncover sRNAs derived from the rice introns. For details, first, the short reads in each data set were mapped to the rice genome, and those with more than five loci on the rice genome were excluded for further analyses. Filtered reads were then mapped to the repeat sequences from the Rfam database (http://rfam.janelia.org/; release 9.1) and the TIGR rice Repeat Database (ftp://ftp.plantbiology.msu.edu/pub/data/TIGR_Plant_Repeats/; release 3.3), and to the pre-miRNAs from miRBase (http://www.mirbase.org/; release 15). After removing the ones mapped to the repeats or the miRNAs, the remaining short reads were finally mapped to the rice introns. We performed these alignments by using the Bowtie algorithm (Langmead et al. 2009) with no mismatch allowed. Besides, to enable cross-library comparison, the abundance of each short read in each library was normalized in RPM (reads per million). It was calculated by dividing the count of each read by the total counts in each library, and then multiplied by 106.
Identification of long-hairpin-structured introns
To identify the long-hairpin-structured introns (lhp introns), we performed a de novo screening by using “einverted” included in the EMBOSS package (Rice et al. 2000) with default parameters. The long complementary regions within the introns are naturally formed as long hairpins, and we thus named these long-hairpin-structured introns “lhp introns.” In this study, lhp introns were defined as follows: (1) The intron should be longer than 400 nt. (2) The identity between the two complementary arms forming the stem region of the long hairpin should be >80%. (3) The highly complementary stem regions should be >500 bp for the “absolute” long lhp introns or should be more than half of the length of the intron for the “relative” long lhp introns. The secondary structures of these lhp introns were predicted by using RNAfold provided by the Vienna RNA package (Hofacker 2003).
One interesting future of such lhp introns is their “strandedness,” that is, whether most siRNAs could only be mapped to the
sense strands of the lhp introns. To address this question, the sRNA HTS data of two WT seedling libraries, representing a
quite comprehensive rice sRNA pool (GSM455965 and GSM520640) (Supplemental Table S1), were used for further studies. After
filtering, all the retained short reads were mapped to the lhp introns. To unambiguously determine the origins of the siRNAs,
the siRNAs with only one genomic locus (unique siRNAs) were mapped to the lhp introns. Then, a statistical analysis was performed
for each lhp intron to determine whether the lhp-intron-derived siRNAs were statistically enriched in its stem region compared
to the random distributions. The statistical significance was identified by hypergeometric distribution. The probability P for each lhp intron is calculated as follows: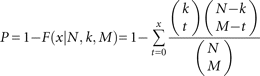 where N is the total length of the lhp intron, M is the length of the paired stem regions, k is the number of unique siRNA sequences from this intron, and x is the number of unique siRNAs from the paired stem regions.
where N is the total length of the lhp intron, M is the length of the paired stem regions, k is the number of unique siRNA sequences from this intron, and x is the number of unique siRNAs from the paired stem regions.
Statistical analysis
All statistical analyses were performed by using the R language (http://www.r-project.org/). For the GO term enrichment analysis of the introns containing miRNA genes and lhp introns, GO::TermFinder (Boyle et al. 2004) was used to detect the significantly enriched GO terms of the host gene sets (164 genes in total) compared to the genome-wide background (P < 0.05).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to Dr. Christian Klukas for comments on an earlier draft of our manuscript. We also thank two anonymous referees for their constructive and helpful suggestions. This work was supported by the National Natural Sciences Foundation of China (30971743, 31050110121); the MoST International Cooperation Project (2009DFA32030); the Key GM Project of China (2008ZX08003-005) and the Program for New Century Excellent Talents in University of China (NCET-07-0740).
Footnotes
-
Reprint requests to: Ming Chen, Department of Bioinformatics, College of Life Sciences, Zhejiang University, Zijingang Campus, Yu Hang Tang Road 388#, Hangzhou 310058, China; e-mail: mchen{at}zju.edu.cn; fax: 86-571-88206612.
-
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.2589011.
- Received December 11, 2010.
- Accepted March 15, 2011.
- Copyright © 2011 RNA Society








