
Association of polyadenylation cleavage factor I with U1 snRNP
Abstract
Splicing and polyadenylation factors interact for the control of polyadenylation and the coupling of splicing and polyadenylation. We document an interaction between the U1 snRNP and mammalian polyadenylation cleavage factor I (CF Im), one of several polyadenylation factors needed for the cleavage of the pre-mRNA at the polyadenylation site. Sucrose density gradient centrifugation demonstrated that CF Im separated into two fractions, a light fraction which contained the known CF Im subunits (72, 68, 59, and 25 kD), and a heavy fraction, rich in snRNPs, which contained predominately the 68- and 25-kD CF Im subunits. Using specific antibodies we found that the heavy fraction contains U1 snRNP/CF Im coprecipitable complexes. These complexes were insensitive to RNase treatment, suggesting that the coprecipitation is not due to RNA tethering. In vitro binding experiments show that both the 68- and 25-kD subunits bind to and comigrate with U1 snRNP. In addition, the 25-kD CF Im subunit binds specifically to the 70K protein of U1 snRNP (U1 70K). This binding may account for the CF Im/U1 snRNP interaction. During these studies we found that mAb 2.73 (mAb 2.73), an established U1 70K antibody, efficiently precipitates the bulk of the CF Im from cellular extracts. Because mAb 2.73 has been used in a number of previous studies related to the U1 snRNP and the U1 70K protein, the precipitation of CF Im must be considered in evaluating past and future data based on the use of mAb 2.73.
Keywords
INTRODUCTION
Polyadenylation (PA) is the process by which the 3′ ends of most mammalian mRNAs are formed. In a tightly coupled set of reactions, the pre-mRNA is endonucleolytically cleaved at a specific polyadenylation site and then approximately 250 adenosine residues are polymerized to the cleaved end, forming the poly(A) tail. Nearly all mammalian cellular mRNAs are so processed in the nucleus. The polyadenylation signal in the pre-mRNA defines the site of polyadenylation through specific binding with a large complex of polyadenylation factors that orchestrate the cleavage and polyadenylation (for reviews, see Wahle and Keller 1992; Manley 1995; Wahle and Keller 1996; Colgan and Manley 1997; Wahle and Ruegsegger 1999).
Polyadenylation is not an isolated mRNA processing event; in nature it is highly coordinated, or coupled, with splicing. The coupling of the two processing reactions is also integrally linked to exon definition (Niwa et al. 1990a; Robberson et al. 1990; Niwa and Berget 1991a,b; Berget 1995), particularly the definition of the last exon of a pre-mRNA, which involves protein–protein interactions across the exon between splicing factors at the 3′ splice site and components of the polyadenylation complex at the polyadenylation signal (Niwa et al. 1990a,b; Niwa and Berget 1991a,b; Scott and Imperiale 1996; Cooke et al. 1999; Cooke and Alwine 2002). Studies from this laboratory and others have identified a number of such interactions. The U1A protein, an essential component of the U1 snRNP, can interact with the polyadenylation complex through the 160-kD subunit of the cleavage and polyadenylation specificity factor (CPSF; Lutz et al. 1996). In vitro polyadenylation reactions done with purified polyadenylation factors and purified recombinant U1A protein have shown that the U1A-CPSF interaction stabilizes CPSF binding to the AAUAAA and increases polyadenylation efficiency (Lutz et al. 1996). The U1A and U1 70K proteins have also been shown to interact with the poly(A) polymerase, causing the inhibition of polyadenylation (Boelens et al. 1993; Gunderson et al. 1994, 1998). For the U1A protein, this appears to be a specific negative feedback autoregulatory mechanism in which U1A protein binds to two sites on its own mRNA, interacts with poly(A) polymerase and inhibits polyadenylation (Gunderson et al. 1994). It has also been proposed that poly(A) polymerase contributes to the coupling of splicing and polyadenylation through interactions with the splicing factor U2AF bound to the polypyrimidine tract in the last intron (Vagner et al. 2000). Recently an RNA–protein complex associated with coupling has been identified (Cooke and Alwine 2002). This coupling complex appears to be made up of splicosomal and polyadenylation complex proteins. Thus it is likely to contain well over 70 proteins, providing the potential for many interactions between splicing and polyadenylation factors for the stabilization of the complex and the regulation of processing through coupling.
In the present study, we document an interaction between a major splicing factor, the U1 snRNP, and mammalian polyadenylation cleavage factor I (CF Im), one of several polyadenylation factors needed for the nucleolytic cleavage of the pre-mRNA at the polyadenylation site. CF Im has been highly purified and appears to consist of three major polypeptides of 68, 59, and 25 kD and a minor subunit of 72 kD (Ruegsegger et al. 1996, 1998). The 68-kD, 59-kD, and 72-kD subunits appear to be related, sharing common amino acid domains (Ruegsegger et al. 1998). These peptides may combine with the 25-kD subunit to constitute different dimer isoforms of CF Im (Takagaki et al, 1989; Ruegsegger et al. 1998); however, the complex of the 68- and 25-kD subunits can reconstitute functional CF Im activity in vitro (Ruegsegger et al. 1998). We find that three of the CF Im subunits (72, 68, and 25 kD) migrate with snRNPs on a sucrose gradient and coimmunoprecipitate with the U1 snRNP and possibly other snRNPs. The CF Im subunits also interact with U1 snRNP in vitro and GST-binding studies show that the 25-kD CF Im subunit interacts specifically with the U1 70K protein. Although a definitive link to processing has not been established, our data demonstrate the type of interaction between splicing and polyadenylation components that is expected for exon definition and for coupling of the splicing and polyadenylation reactions.
During these studies we found that monoclonal antibody 2.73 (mAb 2.73; Billings and Hoch 1984; Takeda et al. 1991), an established antibody to the U1 70K protein, strongly precipitates CF Im. Because this antibody has been used in a number of previous studies of U1 snRNP and U1 70K function, the precipitation of CF Im must be considered in evaluating past and future data based on the use of mAb 2.73.
RESULTS
Immunoprecipitation analysis using anti-U1 70k antibodies
In previous work, we have described a snRNP-free form of the U1 snRNP-specific A protein (U1A; O’Connor et al. 1997); in the present work, we initially set out to determine whether a similar snRNP-free form of the U1 snRNP-specific 70K protein (U1 70K) could be detected, and whether it was complexed with other proteins. We approached this question by separating nucleoplasmic proteins from [35S]-methionine/cysteine-labeled 293T cells on 5%–30% sucrose density gradients and immunoprecipitating the proteins in the fractions using mAb 2.73, a characterized anti-U1 70K monoclonal antibody (Billings and Hoch 1984). Thirty fractions (1 mL) were collected from the top of the gradient and the proteins from alternate fractions were immunoprecipitated and separated by 12% SDS-PAGE (Fig. 1). The U1 snRNP specific proteins U1 70K, U1A, and U1C and the Sm core proteins, B/B′, D1, D2, D3, E, F, and G, all precipitated in fractions 18 to 24 (herein called the heavy fractions and complexes). Figure 1 shows that U1 70K in the heavy fractions was less intensely labeled than U1A and the other U1snRNP proteins as has been previously observed (Hinterberger et al. 1983; Billings and Hoch 1984). This apparent low rate of synthesis suggests that U1 70K may have a long half life and that its synthesis may be autoregulated, as has been suggested previously (Ko and Gunderson 2002).
In addition to the heavy snRNP-containing fractions, a second region of mAb 2.73 immunoprecipitable 35S-labeled material was detected in fractions 10–14 (herein called the light fractions and complexes). These fractions contained strongly radioactive bands migrating at 70 kD and approximately 25 kD. The precipitation of the light complexes was specific to the mAb 2.73, as anti-U1A monoclonal antibody 1E1 (O’Connor et al. 1997) and a control antibody, anti-myc, failed to immunoprecipitate the light complexes (data not shown). Although the 70-kD band in the light region could represent an snRNP-free form of U1 70K, its intense labeling did not agree with the labeling of the expected U1 70K band in the heavy, snRNP-rich fractions. This suggested that it may be a different protein that immunoprecipitated with mAb 2.73 and migrated similarly to U1 70K.
To determine whether the 70-kD band in the light and heavy fractions was U1 70K protein, we prepared an unlabeled nucleoplasmic extract, fractionated it on a 5%–30% gradient, and immunoprecipitated every other fraction with mAb 2.73 as above. The precipitates were then separated on an SDS-PAGE gel and subjected to Western analysis probing with mAb 2.73; for better separation, a 10% gel was used (Fig. 2) instead of the 12% gel (Fig. 1). As expected, mAb 2.73 detected a 70-kD band in the heavy, snRNP-rich fractions (Fig. 2), confirming that it both precipitates U1 70K and detects it by Western analysis. However, by Western analysis, mAb 2.73 detected no 70-kD protein in the light fraction, suggesting that the 35S-met-labeled 70-kD protein in the light fraction seen in Figure 1 is not U1 70K. Thus it appears that mAb 2.73 efficiently precipitates a 70-kD protein in the light fraction that it cannot detect by Western analysis. Western analysis using mAb 2.73 did detect protein(s) in the 120–130-kD size range (X, Fig. 2) in the light fractions. Thus it is possible that mAb 2.73 cross-reacts with the large protein(s) and the 70-kD protein coprecipitates with them. Interestingly, the 120–130-kD protein(s) are not very prevalent in Figure 1, suggesting that they were not highly labeled by 35S-methionine/cysteine.
Partial purification of the light complexes and identification of CF Im subunits
To identify the 70- and 25-kD proteins in the light fraction, we prepared an immunoaffinity column by coupling mAb 2.73 to protein G agarose beads and immunopurified the proteins from concentrated light fractions. The column was washed and the light complexes eluted, concentrated, and separated on SDS PAGE. The 25-kD band on the stained gels was well separated from other proteins; therefore it was excised and analyzed by tryptic digestion and mass spectrometric analysis. The results strongly indicated that it was the 25-kD subunit of CF Im.
CF Im consists of three major polypeptides (68 kD, 59 kD, and 25 kD) and one minor polypeptide (72 kD; Ruegsegger et al. 1996, 1998). These molecular weights matched well with the sizes of the proteins in the light fractions that were immunoprecipitated with mAb 2.73 (Fig. 1). To show that mAb 2.73 immunoprecipitated CF Im, we immunoprecipitated the light fractions with (1) mAb 2.73; (2) mAb 12E12, a monoclonal antibody that immunoprecipitates snRNP-free U1A protein, which sediments in the light fractions (O’Connor et al. 1997); and (3) mAb 1E1, a U1A specific monoclonal antibody that precipitates U1A bound to snRNPs (O’Connor et al. 1997). Neither of the U1A-specific antibodies was expected to interact with CF Im subunits in the light fractions. The immunoprecipitates were analyzed by Western analysis probed with a mixture of two antibodies specific for the 68-kD and the 25-kD subunits of CF Im; because the 72-, 68-, and 59-kD subunits share sequences, they were all detected by the anti-68-kD antibody (Ruegsegger et al. 1996, 1998). Figure 3A shows that mAb 2.73 specifically and efficiently immunoprecipitated the entire CF Im complex (72, 68, 59, and 25 kD), whereas the anti-U1A antibodies did not.
In Figure 3B,C, the 68-kD and 25-kD CF Im subunits were prepared by in vitro transcription/translation and immunoprecipitated using mAb 2.73. mAb 2.73 specifically precipitated the 68-kD subunit (Fig. 3B) and not the 25-kD subunit (Fig. 3C). However, when the 68- and 25-kD subunits were mixed, the 25-kD subunit could be coprecipitated because of its binding to the 68-kD subunit to form a CF Im complex (see below). Hence the data show that mAb 2.73 immunoprecipitates CF Im, presumably through reactivity with the 68-kD subunit and the related 72- and 59-kD subunits. However, as shown above, mAb 2.73 does not recognize the CF Im subunits by Western analysis.
Cleavage factor Im subunits are present in snRNP fractions
We next determined whether there was a snRNP-free form of U1 70K protein, similar to the snRNP-free forms of U1A, which migrates in the light region of the gradient (O’Connor et al. 1997). The above data indicate that U1 70K migrates primarily in the snRNP-rich heavy fractions, suggesting that there is little, if any, snRNP-free U1 70K in these preparations. This was established more definitively by immunoprecipitating sucrose gradient fractions of unlabeled nucleoplasmic extract with mAb 2.73, similar to the experiment in Figure 2. The immunoprecipitates from every other fraction were subjected to Western analysis with sequential probing using (1) mAb 2.73, which will detect the U1 70K protein but not CF Im subunits; (2) anti-U1A (mAb 12E12), which will detect U1A that coprecipitated with U1 70K as part of the U1 snRNP; (3) anti-CF Im 68 polyclonal antibody, to detect the CF Im subunits precipitated by mAb 2.73; and (4) anti-CPSF 160, which will detect the 160-kD subunit of the cleavage and polyadenylation specificity factor (CPSF). Figure 4 shows the results; note that although the Westerns were stripped between each probing, there was some carryover. Probing with mAb 2.73 (second panel) visualized U1 70K only in the heavy fractions, all of which cosedimented with U1A (bottom panel), suggesting that it is all bound to the snRNP and that there is no snRNP-free U1 70K detectable by this analysis.
Western analysis probing with antibody to the 68-kD subunit of CF-Im (Anti-CF I 68; Fig. 4, third panel) showed that mAb 2.73 efficiently precipitated CF-Im subunits (72, 68, and 59 kD) from the light fractions as expected. In addition, a significant amount of the 72- and 68-kD CF Im subunits, but not the 59-kD subunit, were detected in the heavy, snRNP-rich fractions, suggesting that they are associated with larger complexes, possibly snRNPs. It should be noted that a band corresponding to U1 70K can be seen in the anti-CF I 68 panel; this is due to carryover from the previous probing with mAb 2.73, which was not completely removed by stripping. The anti-CF Im 68 antibody does not cross-react with U1 70K.
To determine if the heavy sedimenting CF Im was associated with other polyadenylation factors, we probed the Western with anti-CPSF 160 to identify where in the gradient the polyadenylation complex sedimented. The results (top panel) show that complexes containing CPSF 160 sediments near the very bottom of the gradient. Although some CF Im subunits sedimented with CPSF 160, it is clear that a significant portion of the heavy sedimenting CF Im is not associated with complexes containing CPSF 160. In addition, there is no CPSF 160 in the light fractions. Similar results were seen when the Western was probed for CstF 64, the 64-kD subunit of the cleavage stimulatory factor (not shown). These data suggest that a substantial amount of the CF Im in snRNP fractions is not associated with polyadenylation complexes.
In vitro synthesized CF Im 68- and 25-kD subunits associate with heavy complexes, including U1 snRNP
To further establish that CF Im subunits associate with snRNPs, we prepared [35-S]-methionine-labeled 68- and 25-kD subunits by in vitro transcription and translation. The translation mixes for the two subunits were incubated together and sedimented on a 5%–30% sucrose gradient (Fig. 5A); every other fraction was immunoprecipitated using mAb 2.73, which effectively precipitates CF Im 68-kD subunit and will coprecipitate bound 25-kD subunit. Both subunits migrated together and coprecipitated in light regions of the gradient. The bands indicated by the asterisk are premature translation termination products or breakdown products of the 68-kD subunit. The same mix of translation products was incubated with HeLa cell nuclear extract, sedimented on the sucrose gradient, and every other fraction was precipitated with mAb 2.73 (Fig. 5B). In this experiment, we see that the CF Im in the light region has shifted to fractions 11, 13, and 15 compared to fractions 5, 7, and 9 in Figure 5A. In addition, a significant amount of the CF Im sediments in the heavy fractions, suggesting that all of the CF Im has stably interacted with other proteins and protein complexes in the nuclear extract. In Figure 5C, we have taken the alternate fractions from the gradient used in Figure 5B and precipitated them with anti-U1A (mAb 1E1), which precipitates the U1 snRNP and does not cross-react with CF Im subunits (Fig. 3; data not shown). Note that the panel in Figure 5C is offset from the others because it starts with fraction 2. The results show that CF Im coprecipitates with the U1 snRNP. However, comparing Figure 5B and C, it is clear that CF Im associated with complexes sedimenting faster than U1 snRNP. This was also indicated in Figure 4, where some CF Im sedimented with large complexes containing CPSF 160, indicative of the polyadenylation complex.
CF Im subunits coimmunoprecipitate with snRNPs
To further establish the interaction between snRNPs and CF Im, the snRNP-containing heavy fractions were pooled, immunoprecipitated with anti-snRNP antibodies, and analyzed by Western analysis, probing with anti-CF-Im 68 antibody to determine whether CF Im subunits (72, 68, and 59 kD) could be coimmunoprecipitated (Fig. 6). The positive control (first lane, Fig. 6A) was a light fraction precipitated with mAb 2.73, to visualize the 72-, 68-, and 59-kD components of CF Im. The second lane shows the total CF Im subunits precipitated by mAb 2.73 from the heavy fraction. Note that we again observe that the 59-kD subunit present in the light fractions is specifically absent in the heavy fractions. In the bottom panel of Figure 6A, the Western transfer was probed for U1A protein as an indication of the precipitation of U1 snRNP.
Next the heavy fraction was precipitated with mAb 1E1, which recognizes U1A protein and precipitates U1 snRNP. The 72- and 68-kD subunits were coimmunoprecipitated, suggesting that they can associate with the U1 snRNP. The same result was seen using the Sm antigen-specific antibody Y12, which precipitates all snRNPs containing the Sm complex and does not cross-react with CF-Im subunits (data not shown). In both the 1E1 and Y12 precipitations, the bottom panel of Figure 6A shows that U1A was precipitated, confirming the precipitation of snRNPs. The final lane in Figure 6A is a negative control using mAb 12E12, which recognizes snRNP-free U1A and cannot precipitate U1 snRNP (O’Connor et al. 1997); because there is no snRNP-free U1A in the heavy fraction, no U1A is detected (bottom panel). The cumulative data suggest that the CF Im subunits can associate with U1 snRNP and possibly other snRNPs.
The data in Figure 6B provide additional proof of the association between the CF Im subunits and snRNPs. In this experiment, the in vitro synthesized [35-S]-met-labeled 68- and 25-kD subunits were incubated (for 15 min at 37°C) with nuclear extract and then the total snRNP fraction was isolated using affinity chromatography with the 3-methyl G cap antibody, which specifically recognizes the 3-methyl G cap structure unique to the U RNAs. The column was washed and eluted using cap analog (see Materials and Methods). Equivalent proportions of the depleted nuclear extract and purified snRNPs were separated by 10% SDS PAGE, and autoradiographed. Figure 6B shows that a significant proportion of [35-S]-labeled 68- and 25-kD subunits copurify with snRNPs. There appears to be a preference for binding the 25-kD subunit, consistent with the in vitro binding data presented below, which shows that U1 70K and the 25-kD CF Im subunit interact.
In sum, these data show that several different anti-snRNP and anti-U1 snRNP antibodies coimmunoprecipitate the CF-Im subunits, suggesting a specific interaction between CF Im and U1 snRNP and possibly other snRNPs.
Association between snRNP and CF Im is not due to RNA tethering
It is possible that the CF Im subunits may coprecipitate with snRNPs due to RNA tethering; for example, the snRNP may be bound at a splice site and CF Im may be bound at the polyadenylation signal of the same RNA. Thus these proteins may appear to coprecipitate. To rule out this possibility, HeLa cell nuclear extract was treated with 1 μg/mL RNase A for 15 min at 37°C in the presence of protease inhibitors. Subsequently, the RNase was not inactivated; hence, it was present and active during further steps of the experiment. This level of RNase A treatment has only a small effect on the integrity of snRNP RNAs and snRNP structure, but completely degrades mRNA precursors that may tether the proteins (data not shown).
Mock-treated (No RNase) or RNase A-treated samples were subjected to serial immunoprecipitation. The first antibody used was mAb 1E1, which is specific for U1A protein and precipitates U1 snRNPs; the supernatant of the mAb 1E1 precipitation was then treated with anti-Sm antibody Y12, which precipitates all snRNPs containing the Sm complex. In Figure 7, the serial immunoprecipitates were separated by 10% SDS-PAGE along with a nuclear extract control lane (Nuc. Ext.). Western analysis was then performed, probing with (1) anti-CF Im 68-kD antibody to visualize CF Im components; (2) mAb 2.73 to visualize U1 70K protein; and (3) mAb 1E1 to visualize U1A protein. By comparing the RNase-treated and untreated (No RNase) samples, it can be seen that both mAb 1E1 and anti-Sm antibody Y12 precipitated CF Im subunits (68 and 72 kD), U1 70 K protein, and U1A similarly from both RNase-treated and untreated samples, indicating that the association of CF Im with snRNPs is not due to RNA tethering.
Amount of CF Im associated with the heavy fraction and snRNPs
Several of the experiments shown here, as well as others (not shown) were subjected to PhosphorImager, densotometric, and other quantitative imaging analyses to determine the amount of CF Im in the heavy fraction. The analyses suggested that 20 ± 10% of the CF Im detected on the gradient sediments in complexes in the heavy region of the gradient. Within this heavy sedimenting fraction, 50%–70% appears to be associated with snRNPs in the snRNP-rich fractions and the rest is associated with larger complexes, for example, polyadenylation and coupling complexes, sedimenting near the bottom of the gradient (Fig. 4).
In vitro interaction between the 25-kD CF Im subunit and U1 70K protein
Given the interaction of CF Im with U1 snRNP, the structural similarities between U1 70 K protein and the 68-kD subunit of CF Im (see Discussion) and the cross-reactivity of mAb 2.73 with the 68-kD subunit of CF Im, we asked whether U1 70K could interact with CF Im subunits. A GST fusion with the 25-kD CF Im subunit was used to test binding to in vitro synthesized 35S-met-labeled U1 70K and 68-kD CF Im subunit (Fig. 8). The first three lanes show half of the input amounts of the 35S-met-labeled luciferase protein (nonspecific control), CF I 68-kD subunit, and U1 70K protein. As noted above, the 68-kD subunit migrates slower than U1 70K protein. The next three lanes (Bead Control) show that none of the 35S proteins bind nonspecifically to the glutathione beads. The next four lanes show the binding to GST CF I 25. As expected, the CF I 68-kD subunit binds efficiently to GST CF I 25 (Ruegsegger et al. 1996). In addition, we found that the U1 70K protein bound as efficiently to GST CF I 25 as the 68-kD subunit. When mixed, both proteins bound efficiently to the 25-kD subunit; however, this does not establish whether they can bind the 25-kD subunit simultaneously. The nonspecific luciferase control showed no binding. Additionally, the 68-kD subunit and U1 70K did not bind to GST-IκB, a nonspecific GST fusion protein (last two lanes). These data suggest that U1 70K protein binds specifically and efficiently to the CF Im 25-kD subunit in vitro. In sum, the data suggest a specific means for the interaction between CF Im with U1 snRNP.
DISCUSSION
The mAb 2.73 was spontaneously derived from the lupus-prone MRL/n strain of mice and shown to be reactive with U1 70K protein (Billings and Hoch 1984; Takeda et al. 1991); thus, the original antigen responsible for the genesis of the autoantibody is not known. In this study we have found that mAb 2.73 immunoprecipitated a complex of proteins that does not contain the U1 70K protein. We have established that this complex contains the known subunits of the mammalian polyadenylation cleavage factor Im (72, 68, 59, and 25 kD; Ruegsegger et al. 1996, 1998). In addition, mAb 2.73 precipitated the 68-kD subunit, which had been synthesized by in vitro transcription and translation in a reticulocyte lysate. The possibility of cross-reactivity is supported by structural similarities between the U1 70K protein and the 68-kD subunit of CF Im. The epitope recognized by mAb 2.73 was characterized as the arginine/aspartic acid (RD) repeats found in the carboxyl terminus of U1 70K protein (Pelsue et al. 1993). The C-terminal region of the 68-kD subunit of CF Im contains a similar alternating charged domain consisting of arginine residues alternating with glutamate or aspartate residues (Ruegsegger et al. 1998). That this region is the epitope for mAb 2.73 is supported by previous studies showing that the U1 70K monoclonal antibody detected RD repeats on proteins other than U1 70K (Staknis and Reed 1995). However, the Staknis and Reed data do not establish the strong interaction between the antibody and CF Im.
The C-terminal region of the 68-kD subunit also contains areas that match the consensus sequence for poly(A) polymerase regulatory domains (PRDs; Ko and Gunderson 2002). These domains have been characterized in a number of proteins (including U1 70K protein) that interact with and inhibit poly(A) polymerase (Ko and Gunderson 2002). The potential for such elements in CF Im suggests the interesting possibility that in the polyadenylation complex, CF Im may inhibit poly(A) polymerase function until the cleavage event has succeeded.
The most likely reason that precipitation of CF Im by mAb 2.73 was not previously observed is because the 68-kD subunit of CF Im migrates with U1 70K protein and the 25-kD subunit migrates with the Sm proteins B/B′ on SDS-PAGE. In addition, mAb 2.73 does not detect CF Im subunits by Western analysis. The ability of the antibody to precipitate the bulk of CF Im in an extract must be considered in evaluating past and future experimental results based on the use of this antibody; for example, U1 snRNP depletion of an extract using mAb 2.73 also removes most, if not all, of the CF Im (data not shown).
In addition to the CF Im in the light fraction, the sucrose density gradient analyses showed that 25%–30% of the CF Im sediment in heavier snRNP-rich fractions. Unlike the light fraction, which contains all of the CF Im subunits, the heavier fractions predominately contain the 68- and 25-kD subunits, a small amount of the 72-kD subunit, and no 59-kD subunit. Previous work (Takagaki et al. 1989; Ruegsegger et al. 1998) has suggested that CF Im subunits form stable heterodimers and may exist in three forms: 68/25, 72/25, and 59/25. It is not known why three forms exist; however, they may have different preferences for substrates or other factors as suggested by the interaction with CF Im. Specifically, our data suggest that the 68/25 dimer preferentially binds snRNP, the 72/25 dimer has less affinity for snRNP, and the 59/25 dimer has no affinity for snRNPs.
Targeted precipitation of U1 snRNP suggests that the CF Im subunits associate with U1 snRNP; however, they may associate with other snRNPs as well. The subunits also sediment with very large complexes that appear to represent polyadenylation complexes or complexes associated with the coupling of polyadenylation and splicing (Cooke and Alwine 2002).
The association of CF Im with U1 snRNP is not due to RNA tethering. A potential functional interaction was indicated by in vitro GST pull-down experiments that showed that the 25-kD subunit binds not only its normal partner, the 68-kD subunit, but also U1 70K protein. The structural similarity between the 68-kD subunit and U1 70K (discussed above) may account for this interaction. In this experiment, it cannot be determined whether the 25-kD subunit can interact with both the 68-kD subunit and U1 70K simultaneously. However, as shown in Figure 5, when in vitro synthesized 68- and 25-kD subunits are mixed, they interact and migrate as a complex in light fractions; when nuclear extract is added, these in vitro complexes migrate in the snRNP-rich heavy fractions, suggesting that the complexes can interact with another protein(s), for example, U1 70K.
The observation that the U1 snRNP–CF Im complex is not associated with CPSF 160 and CstF 64 suggests that it may be a product of the coupled processing reaction. Indeed, U1 snRNP is known to be released from the splicesome after U4, U5, and U6 entry and prior to catalysis. Thus the successful coupling of polyadenylation and splicing, resulting in the removal of the last intron (Cooke et al. 1999; Cooke and Alwine 2002), would release U1 snRNP, along with polyadenylation factors with which it interacted. Given the established interdependence of last intron removal and polyadenylation, it is interesting to consider that the release of U1snRNP may signal that spliceosome formation has been successful, providing a positive signal for polyadenylation. In addition, if the U1 snRNP–CF Im complex is a product of the reaction, it is likely to be transient, as the components of the processing complexes dissociate in order to recycle. This may explain why only a moderate amount of CF Im is found in these complexes.
The observation that only a subset of the CF Im subunits (primarily 68 and 25 kD) interact with snRNPs suggests that the complex results from a specific, functional interaction or is the product of a specific reaction. The particular exclusion of the 59-kD subunit would not be expected if this were a random, nonspecific interaction. However, experiments to demonstrate the function of the U1 snRNP–CF Im interaction in RNA processing are difficult at this point, because depletion experiments do not fully address the question. For example, depletion of CF Im would inhibit both polyadenylation and coupling, providing an obvious and uninformative result. Depletion of the snRNPs is equally problematic; in experiments not shown we have depleted extracts of snRNPs using anti-3mG cap antibody coupled to Sepharose. This removed all of the snRNP-bound CF Im. In vitro polyadenylation cleavage assays using the SV40 late polyadenylation signal showed that depleted extracts had significantly decreased cleavage activity (30% of mock-depleted extracts). Much of this activity was restored by reconstitution of the snRNP/CF Im fraction. Although this suggests a functional significance for the snRNP/CF Im complex, it is equally possible that the effects resulted from the removal of the snRNPs and/or other snRNP bound factors that normally enhance the polyadenylation reaction. In addition, if the snRNP–CF Im complex is a dissociation product of a larger processing complex, as discussed above, then its depletion may not matter and reconstruction experiments would be uninformative.
The interaction between U1 snRNP and CF Im adds to a growing number of documented interactions between splicing and polyadenylation factors that may contribute to the specific control of polyadenylation and/or the coupling of splicing and polyadenylation.
MATERIALS AND METHODS
Antibodies and plasmids
Monoclonal antibody 2.73 (anti-U1 70K) was a generous gift from S. Hoch, La Jolla Institute for Experimental Medicine, La Jolla, CA (Billings and Hoch 1984; Takeda et al. 1991). Polyclonal antibodies against the 68-kD and 25-kD components of CF Im (anti-68 and anti-25) were the kind gift of W. Keller, University of Basel, Switzerland (Ruegsegger et al. 1998). Monoclonal antibodies against U1A (1E1 and 12E12) have been previously described (O’Connor et al. 1997). Anti-myc antibodies were purchased from Santa Cruz Biotechnology, Inc. Anti-sm and anti-3-methyl G cap antibodies were purchased from Lab Vision and Oncogene Science.
Plasmids encoding U1-70K (PET-70K; Romac et al. 1994), CF Im 68 polypeptide (pBS-68; Ruegsegger et al. 1996, 1998), and CF Im 25 kD (pCF-25, gift of Jim Manley, Columbia University, New York) were used to prepare each protein by in vitro transcription/translation. The GST fusion with the 25-kD subunit of CF Im was prepared by amplifying the coding region in pCF-25 and inserting it into the XhoI site of pGEX-5x2.
Cell culture and labeling
Human 293T cells were grown in Delbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, 2 mM glutamine, and 1% penicillin-streptomycin at 37°C in 5% CO2. For in vivo labeling, cells were grown in 100-mm petri dishes to 80% confluence. Cells were washed with PBS and then labeled for 6 h at 37°C with [35S]-methionine and [35S]-cysteine (Amersham) at 300 μCi in 3 mL methionine-free DMEM supplemented with 5% dialyzed fetal calf serum, 2 mM glutamine, and 1% penicillin-streptomycin.
Extract preparation and immunoprecipitation procedures
Nucleoplasmic extracts from 293T cells were prepared as previously described (Pinol-Roma et al. 1990). For immunoprecipitation of [35S]-labeled proteins, 0.5 mL of extract were incubated with mAb 2.73 for 4 h at 4°C. After incubation, 30 μL (packed volume) of protein G-agarose beads were added to each immunoprecipitation, and incubated for 1 h at 4°C. The protein G beads had been washed and suspended in NET-2 buffer (50 mM Tris at pH 7.4, 150 mM NaCl, 0.05% NP-40, and 1 mM PMSF). After incubation, the beads were washed four times with NET-2 buffer. Proteins were eluted from the beads in 50 μL of Laemmeli sample buffer and separated by electrophoresis on 10% or 12.5% SDS-polyacrylamide gels. Gels were fixed in 40% methanol and treated with 0.5 M salicylic acid for 40 min, then dried and autoradiographed. Other immunoprecipitations of unlabeled proteins and in vitro synthesized proteins were done using similar procedures.
Sucrose density gradients and fractionation
Five percent to 30% sucrose density gradients were prepared in RSB-100 (10 mM Tris at pH 7.2, 100 mM NaCl, 1 mM MgCl2, 2 mM DTT, and 1% Triton X-100) using a BioComp Model 106 Gradient Master (New Brunswick). Nucleoplasmic extract (1 mL; approximately 10 mg unlabeled protein or 0.15 mg labeled protein) was layered on the gradient and centrifuged at 25,000 rpm at 4°C for 42 h in a Beckman SW-28 rotor. Thirty 1-mL fractions were collected from the top using a BioComp Model 150 Gradient Fractionator (New Brunswick) at 4°C.
Affinity purification of the CF Im complexes from the light fractions
Monoclonal antibody 2.73 was coupled to protein G agarose beads as described elsewhere (Harlow and Lane 1988). Sucrose density gradient fractions rich in the light complex were concentrated and incubated with antibody-coupled beads at 4°C for 4 h. Beads were washed with 50 volumes of NET-2 buffer. The light complexes were eluted with 100 mM glycine (pH 2.5). Five 1-mL fractions were collected in tubes containing 10 μL 1 M phosphate buffer (pH 8.0). Fractions were concentrated and proteins separated by SDS-PAGE as described above. For microsequencing, the gels were silver stained and the relevant band was excised.
Western analyses
Proteins separated by SDS-PAGE were electrophoretically transferred to nitrocellulose membrane as previously described (Tobin et al. 1979). Nitrocellulose membranes were blocked by treatment for 1 h with 5% nonfat dried milk dissolved in PBS. The blocked membranes were then incubated for 1 h at room temperature with primary antibodies diluted in 5% nonfat dried milk in PBS. After three 10-min washes in PBS plus 0.5% Tween-20, the membranes were incubated with HRP-conjugated secondary antibodies for 45 min. The antigen–antibody interaction was detected by chemiluminesence (ECL System, Amersham).
In vitro transcription/translation, immunoprecipitation, and GST pull-down experiments
[35S]-labeled 25- and 68-kD subunits of CF Im were prepared by coupled in vitro transcription/translation (TNT, Promega) using the manufacturer’s protocol. The expression plasmids used are described above. For coimmunoprecipitation analyses, equal amounts of translated protein (based on trichloroacetic acid precipitable counts) were incubated, either with or without nuclear extract, at 37°C for 15 min in the presence of protease inhibitors (1 mM PMSF, 1 μM leupeptin, and 0.2 μM CLTK). Density gradient centrifugation, fractionation, and immunoprecipitation were as described above.
For GST pull-down experiments, equivalent amounts of fusion proteins were bound to glutathione agarose beads and incubated with 1 × 106 counts of each in vitro labeled protein with the exception of the control luciferase protein, which was 4 × 106 counts. All reactions were performed at 4°C with constant mixing for 90 min. The beads were then washed five times with NETN+ buffer (20 mM Tris at pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40, 1 mM DTT, 1 mM PMSF, 1 mM TLCK). Proteins were eluted by boiling the beads in SDS sample buffer and separated by 8% SDS-PAGE. Gels were fixed and treated with 0.5% Salicylic acid for 40 min, dried, and autoradiographed.
Immunoaffinity purification of snRNPs on anti-m3G column
Anti-m3G cap antibody was coupled to protein A Sepharose beads at a concentration of 5 mg/ml. HeLa cell nuclear extract was incubated with beads for 2 h. The flow through nuclear extract was collected and kept at −70°C. The column was washed with 50 volumes of buffer, and snRNPs were eluted with cap analog.
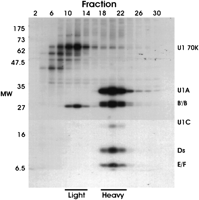
Analysis of [35S]-methionine/cysteine-labeled nuclear proteins by sucrose gradient fractionation and immunoprecipitation using anti-U1 70K antibody, mAb 2.73. Nucleoplasmic proteins from [35S]-methionine/cysteine-labeled 293T cells were separated by centrifugation on 5%–30% sucrose density gradients. Alternate fractions were immunoprecipitated using mAb 2.73 and the immunoprecipitated proteins were separated by 12% SDS-PAGE and autoradiographed. The position of the U1 snRNP specific proteins U1 70K, U1A, and U1C and the Sm core proteins, B/B′, D1, D2, D3, E, and F are shown.
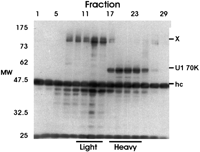
Analysis of unlabeled nuclear proteins by sucrose gradient fractionation and immunoprecipitation using anti-U1 70k antibody, mAb 2.73. Unlabeled nucleoplasmic extract was fractionated on a 5%–30% sucrose gradient. Every other fraction was immunoprecipitated with mAb 2.73 as in Figure 1. The precipitates were then separated on a 10% SDS-PAGE gel and subjected to Western analysis probing with mAb 2.73. The position of U1 70K protein, the unknown 120–130-kD proteins (X), and the immunoglobulin heavy chain (hc) are indicated.
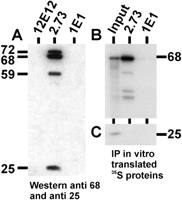
The anti-U1 70K antibody, mAb 2.73, immunoprecipitates CF Im from the light fractions. (A) The CF Im-rich light fractions were immunoprecipitated with mAb 2.73, mAb 12E12, and mAb 1E1. The immunoprecipitated proteins were separated by 10% SDS-PAGE and subjected to Western analysis probing with antibodies to both the 68-kD and 25-kD subunits of CF Im. The positions of the 72-, 68-, 59-, and 25-kD Cf Im subunits are shown. (B, C) 35S-met-labeled 68-kD (B) and 25-kD (C) subunits, prepared by in vitro transcription and translation, were immunoprecipitated with mAb 2.73 and mAb 1E1. The immunoprecipitated proteins were separated by 10% SDS-PAGE and autoradiographed. The input lane represents 1/10th of the amount of labeled proteins used for the immunoprecipitations.
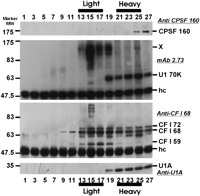
Association of CF Im subunits with snRNPs. Unlabeled nucleoplasmic extract was fractionated on a 5%–30% gradient. Every other fraction was immunoprecipitated with mAb 2.73 as in Figure 2. The precipitates were then separated by 10% SDS-PAGE and subjected to Western analysis sequentially probing with anti-CPSF 160 (top panel), mAb 2.73 (second panel), anti-CF Im 68 (third panel), and Anti-U1A (mAb 12E12; bottom panel). The Westerns were stripped between each probing; however, there was some carryover. The positions of significant proteins are indicated as well as the position of the immunoglobulin heavy chain (hc).

In vitro synthesized CF Im 68- and 25-kD subunits associate with heavy complexes, including U1 snRNP. [35-S]-methionine-labeled CF Im 68-kD and 25-kD subunits were synthesized by in vitro transcription and translation and subjected to 5%–30% sucrose density gradient centrifugation either alone (A) or after mixing with nuclear extract (B and C). In panels A and B, every other gradient fraction was immunoprecipitated using mAb 2.73. In panel C, every other fraction was precipitated with anti-U1A (mAb 1E1); note that panel C is offset from the others because it starts with fraction 2. The precipitated proteins were separated by 10% SDS-PAGE, the gels dried and autoradiographed.
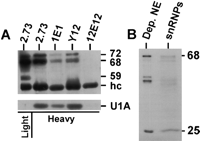
CF Im subunits coimmunoprecipitate with snRNPs. (A) The snRNP-containing heavy fractions were immunoprecipitated with mAb 2.73, mAb 1E1, Y12, and mAb 12E12. The first lane (light) is the light fraction immunoprecipitated with mAb 2.73 for visualization of the 72-, 68-, and 59-kD CF Im subunits (the amount of light fraction precipitated was one-fourth of that used for the heavy fraction precipitation). The precipitated proteins were separated by 10% SDS-PAGE and subjected to Western analysis probing with antibody against the 68-kD subunit of CF-Im (top panel) or antibody against U1A protein (bottom panel). (B) In vitro synthesized, [35-S]-met-labeled 68- and 25-kD subunits were incubated with nuclear extract. The total snRNP fraction was isolated using affinity chromatography (see text and Materials and Methods), equivalent proportions of the depleted nuclear extract and purified snRNPs were separated by 10% SDS PAGE, and the gel dried and autoradiographed. Positions of significant proteins and the immunoglobulin heavy chain (hc) are indicated.
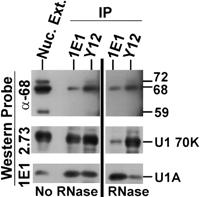
The association of CF Im subunits with U1 snRNP is not due to RNA tethering. Nuclear extracts were either mock treated (No RNase) or ribonuclease A treated (RNase) and then serially immunoprecipitated, first using anti-U1A mAb 1E1. The supernatant was then precipitated with anti-snRNP Y12. Precipitates were separated by 10% SDS-PAGE and subjected to Western analysis probing with antibody against the 68-kD subunit of CF Im (top panel) mAb 2.73 (middle panel), and 1E1 (bottom panel). The positions of significant proteins are indicated.
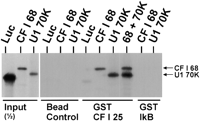
GST-25-kD CF Im subunit binds U1 70K protein. The first three lanes show the intensity of one-half of the input amounts of in vitro synthesized 35S-met-labeled luciferase, 68-kD CF Im subunit, and U1 70K protein. The next three lanes show the bead controls where the labeled proteins were incubated with uncharged glutathione beads. The next three lanes show the binding resulting from incubating the labeled proteins with glutathione beads charged with GST-25-kD CF Im subunit (GST CF I 25). The last two lanes show the binding resulting from incubating the labeled proteins with glutathione beads charged with a nonspecific GST fusion protein, GST-IκB. Bound proteins were eluted from the beads and separated by SDS-PAGE (see Materials and Methods).
Acknowledgments
The authors thank Walter Keller and Jim Manley for plasmids and antibodies related to CF Im subunits and Sally Hoch for antibody mAb 2.73. We also thank the members of the Alwine laboratory for support. Cheers to all. This work was supported by NIH Grant GM45773 provided to J.C.A. by the Public Health Service.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
-
Article and publication are at http://www.rnajournal.org/cgi/doi/10.1261/rna.5104603.
-
- Accepted August 13, 2003.
- Received June 11, 2003.
- Copyright 2003 by RNA Society








