
Terminal loop sequences in viral double-stranded RNAs modulate RIG-I signaling
- 1Department of Molecular Microbiology, Washington University School of Medicine, St. Louis, Missouri 63110, USA
- 2Center for Women's Infectious Disease Research, Washington University School of Medicine, St. Louis, Missouri 63110, USA
- Corresponding author: clopezzalaquett{at}wustl.edu
-
Handling editor: Ling-Ling Chen
Abstract
Detection of foreign RNAs is a crucial activation step for innate immunity pathways in response to viral infections. Retinoic acid–inducible gene I (RIG-I) is a cytoplasmic RNA sensor that triggers type I and III interferon (IFN) expression and activates the antiviral response during RNA virus infections. The activating ligand for RIG-I has been shown to be 5′-triphosphated, blunt-ended, double-stranded (ds)RNA, but questions remain on the impact of other RNA motifs on RIG-I activation. Here we show that immune-activating copy-back viral genomes (cbVGs) contain RNA stem–loops away from the 5′ end of the RNA that enhance RIG-I signaling and IFN expression. Importantly, the sequence of the terminal loops of the activating motifs impacts the strength of IFN expression. Additionally, we show that synthetic versions of these cbVG-derived stem–loops trigger innate immune responses in mice, demonstrating their potential as immunostimulants in vivo.
Keywords
INTRODUCTION
Retinoic acid–inducible gene I (RIG-I) and melanoma differentiation-associated protein 5 (MDA5) are important cytoplasmic sensors of viral infections that initiate the antiviral immune response upon binding of viral RNA (Kato et al. 2011; Thoresen et al. 2021). Binding of RIG-I or MDA5 by foreign RNA triggers a signaling cascade through the mitochondrial antiviral signaling (MAVS) protein that ends in the expression of antiviral proteins, including type I and III interferons (IFNs) (Tanner and Linder 2001; Yoneyama et al. 2004; Kawai et al. 2005; Seth et al. 2005; Xu et al. 2005). RIG-I and MDA5 contain a C-terminal regulatory domain that recognizes the foreign RNA and drives conformational changes in the helicase domain to wrap around the RNA (Cui et al. 2008; Lu et al. 2010, 2011; Wang et al. 2010; Kowalinski et al. 2011; Luo et al. 2011). These conformational changes release caspase activation and recruitment domains (CARDs) that promote oligomerization of the proteins and signaling through MAVS (Yoneyama et al. 2004; Peisley et al. 2013; Ramanathan et al. 2016). RIG-I and MDA5 recognize unique pathogen-associated molecular patterns (PAMPs) found in foreign RNA but not in the host cell RNAs.
The most well-studied viral RNA PAMPs are within the RNAs that activate RIG-I. The regulatory domain of RIG-I detects terminal 5′ triphosphates on RNA (Kim et al. 2004; Pichlmair et al. 2006; Schlee et al. 2009; Schmidt et al. 2009), and the detection of this motif can be inhibited by methylation of the 5′ end of the RNA (Schuberth-Wagner et al. 2015; Devarkar et al. 2016). While single-stranded (ss)RNA can activate RIG-I, the highest activation occurs when blunt-ended, double-stranded (ds)RNA (Marques et al. 2006) contains the 5′ triphosphate (Pichlmair et al. 2006). The addition of overhangs to the end of the dsRNA limits activation of RIG-I (Schlee et al. 2009; Schmidt et al. 2009). RIG-I is activated best by short dsRNA, while longer synthetic dsRNAs such as poly I:C (>500 nt in length) activate MDA5 (Kato et al. 2006, 2008; Binder et al. 2011). With these guidelines in place, minimal ligands for activating RIG-I that contain a short 10 bp dsRNA stem with a 5′ triphosphate have been synthetically generated (Kohlway et al. 2013; Linehan et al. 2018). While these studies have provided critical data to identify motifs that RIG-I recognizes and binds in the RNA, in general, they are not based on RNAs found naturally during viral infections. Further work is thus needed to determine whether other RNA structures or domains can further improve RIG-I recognition and identify the characteristics of viral RNAs that activate RIG-I signaling during actual infections.
Previously, we and others determined that the RNA that most robustly activates RIG-I signaling during Sendai virus (SeV) infection is a copy-back viral genome (cbVG) (Baum et al. 2010; Mercado-López et al. 2013; Sun et al. 2015). cbVGs contain complementary ends predicted to form a blunt-ended dsRNA PAMP with a 5′ triphosphate, characteristic of the canonical RIG-I ligand (Kolakofsky 1976; Lazzarini et al. 1981). However, detailed folding analyses of the most prominent cbVG from SeV (cbVG 546) revealed a much more complex folding of the molecule, including an internal stem–loop that is necessary for strong activation of the RIG-I pathway to stimulate IFN expression. This stem–loop is comprised of nucleotides 70–114 of cbVG 546, and when transferred to inert RNAs, it significantly enhanced the immunostimulatory capability of the RNA (Xu et al. 2015). Interestingly, the removal of stem–loop 70–114 from cbVG 546 severely decreased RIG-I activation even though the cbVG still contained the 5′ triphosphate and predicted blunt-end dsRNA (Xu et al. 2015).
It is unknown how SeV motif 70–114 enhanced RIG-I activity or whether other RIG-I stimulatory cbVGs contain stem–loops similar to SeV 70–114. We therefore sought to determine if other viral cbVGs contain RNA stem–loops that can transfer RIG-I stimulatory activity to otherwise inert RNAs and identify features of these RNA stem–loops that are necessary for robust RIG-I activation. Here, we identified multiple sequences in cbVGs from human respiratory syncytial virus (RSV) and Nipah virus (NiV) that can transfer RIG-I stimulatory activity to the inert X RNA from the hepatitis C virus. Through mutations of the terminal loop sequences of the cbVG-derived RNAs, we discovered that specific nucleotides in the terminal loop of the RNA are necessary for strong activation of RIG-I signaling in cells. Lastly, in vivo administration of the cbVG-derived RNA loops activated innate immune signaling pathways, suggesting their potential use as immunostimulants for vaccines and immunotherapies.
RESULTS
RSV and NiV cbVGs stimulate interferon expression
To identify additional natural RNA stem–loops that could activate antiviral innate immune pathways, we selected previously identified cbVGs from either RSV (RSV cbVG 236) (Sun et al. 2019) and NiV (DI15, here called NiV cbVG 378) (Welch et al. 2020). We first determined if these cbVGs can activate the IFN signaling pathway in human A549 lung cells and found that in vitro transcribed RNAs of both RSV cbVG 236 and NiV cbVG 378 could stimulate the transcription of type I (IFNB1) and type III (IFNL1) interferons at 6 h post transfection. In contrast, the hepatitis C virus X region, a well-characterized inert viral RNA (Stone et al. 2013; Fisher et al. 2018), did not stimulate IFN expression (Fig. 1A,B). IFN expression was largely RIG-I- and MAVS-dependent, similar to what we had reported for SeV cbVG 268, a shortened version of cbVG 546 (Fig. 1C,D; Xu et al. 2015). The activation of this signaling pathway was also confirmed by the presence of phosphorylated TANK-binding kinase 1 (TBK1) (Fig. 1E).
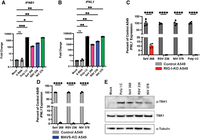
cbVG from RSV and NiV induce MAVS-dependent IFN expression. (A,B) Quantitative PCR (qPCR) for (A) type I interferon (IFNB1) and (B) type III interferon (IFNL1) expression by A549 cells at 6 h post transfection (hpt) with 5 pmol of HCV X RNA, SeV cbVG 268, RSV cbVG 236, or NiV cbVG 378 or 100 ng of poly I:C. One-way ANOVA with Tukey's HSD post hoc test was performed for statistical analysis. (ns) P > 0.05, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001. Data are represented as fold change over Mock. (C) qPCR for IFNL1 of control or RIG-I-KO A549 cells at 6 hpt with 5 pmol of SeV cbVG 268, RSV cbVG 236, or NiV cbVG 378 or 100 ng of poly I:C. One-way ANOVA with Tukey's HSD post hoc test was performed for statistical analysis. (****) P < 0.0001. (D) qPCR for IFNL1 of control or MAVS-KO A549 cells at 6 hpt with 5 pmol of SeV cbVG 268, RSV cbVG 236, or NiV cbVG 378. One-way ANOVA with Tukey's HSD post hoc test was performed for statistical analysis. (****) P < 0.0001. Data reported are biological replicates. (E) Western blots of cell lysates collected from A549 cells 6 hpt with 5 pmol of SeV cbVG 268, RSV cbVG 236, or NiV cbVG 378 or 100 ng of poly I:C. Blots were stained for phosphorylated-TBK1, TBK1, and α-tubulin.
RSV cbVGs contain immunostimulatory stem–loops
To identify which specific RSV cbVG 236 sequence confers a strong RIG-I stimulatory activity to the molecule, we first made a series of deletions in cbVG 236 that either removed internal sequences or removed a complementary end of the cbVG (Fig. 2A). We found that deleting the complementary ends of the cbVG (nucleotides 1–48 and 189–236) reduced RIG-I activation, as deletion mutants del 1–48 and 189–236 lost immunostimulatory activity (Fig. 2B). Interestingly, we also found that nucleotides 49–100 are necessary for maximum IFN expression since deletions in this range (del 49–100, del 49–187, and del 64–187) showed reduced stimulation of the IFN response, but del 101–147 and del 137–187 induced IFN expression similar to the wild-type cbVG 236.
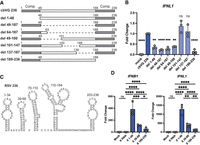
RSV cbVG sequences 1–48 and 39–68 are immunostimulatory. (A) Indicated deletion versions of RSV cbVG 236 were generated by in vitro transcription, then 1 pmol of IVT RNA was transfected into A549 cells. Gray boxes represent complementary regions (Comp.) of the cbVG. (B) Expression of IFNL1 was measured by qPCR at 6 hpt. One-way ANOVA with Tukey's HSD post hoc test was performed for statistical analysis. (ns) P > 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. Data are represented as fold change over cbVG 236. (C) Predicted structure of RNA stem–loops from RSV cbVG 236. (D) Sequences from RSV cbVG 236 were attached to X RNA and then in vitro transcribed. One picomole of RNA was transfected into A549 cells, and expression of IFNB1 and IFNL1 was measured by qPCR at 6 hpt. One-way ANOVA with Tukey's HSD post hoc test was performed for statistical analysis. (ns) P > 0.05, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. Data are represented as fold change over the X RNA control. Data reported are biological triplicates.
To determine if specific immunostimulatory stem–loops were present within the immunostimulatory sequences, we then performed in silico prediction of the RNA stem–loops using RNAfold (Zuker and Stiegler 1981). Since the complement ends of the cbVGs interfere with the in silico folding predictions based on minimal free energy and it is likely that favorable secondary structures form during RNA synthesis rather than once the entire molecule has been produced, we determined all possible RNA structures on partial cbVGs lacking either the 3′ or 5′ complementary sequences. RSV cbVG 236 was predicted to form five RNA stem–loops (Fig. 2C; Supplemental Fig. S3) in the absence of 3′-5′ complementation with two of the stem–loops (1–34 and 39–68) present within the immunostimulatory sequences. RNA folding of X region + RSV 1–48 predicted an immunostimulatory stem–loop of sequence 1–34 similar to the full-length cbVG (Supplemental Fig. S2).
To determine if the immunostimulatory sequences identified within cbVG 236 retained their activity outside the context of the original cbVG, we transferred sequences within the immunostimulatory regions containing stem–loops, or control stem–loop sequences from regions that showed no immunostimulatory activity, to the inert X RNA motif. We ensured that the overall structures of the final molecules were similar (Supplemental Fig. S2; Stone et al. 2013) and tested for their ability to induce IFN expression. We hypothesized that stem–loops that were disrupted by the deletion of nucleotides 49–100 (such as stem–loop 39–68) would induce the highest IFN expression. We found that X-region chimeras carrying RSV cbVG sequences 39–68 induced IFN expression when transfected into A549 cells, but the highest IFN signal was stimulated by transfer of nucleotides 1–48. Interestingly, the reverse complement of nucleotides 1–48 (nucleotides 189–236) induced weaker IFN expression in this context despite contributing to IFN induction in the context of the full cbVG, suggesting that the activity of this region depends on additional sequences present in the full-length cbVG (Fig. 2D). Consistent with data on full cbVGs, the X RNA constructs induced expression of IFNL1 in a RIG-I dependent manner (Supplemental Fig. S1). Overall, these data suggest that while stem nucleotides 49–100 are crucial for IFN stimulation by cbVG 236, multiple unique RNA stem–loops can confer varying strengths of IFN activation to an inert RNA.
NiV cbVGs contain immunostimulatory stem–loops
Similar to RSV, we made a series of deletions to remove specific RNA sequences from NiV cbVG 378 and determined which portions of the cbVG are necessary for strong IFN expression. We found that nucleotides within the complementary ends of the cbVG were necessary for strong expression of IFNs and removal of nucleotides 325–360 also decreased IFN expression (Fig. 3A,B) suggesting that a stem–loop in region 325–360 is necessary for a strong IFN response. NiV cbVG 378 is predicted to have eight RNA stem loops (Fig. 3C; Supplemental Fig. S4). The deletion of region 325–360 is predicted to remove stem–loop 324–361 from the cbVG, which suggests that this stem–loop enhances IFN production during NiV cbVG 378 transfection. When attached to the X RNA, we again found that while NiV 325–360 induced IFNB1 and IFNL1 expression, other NiV sequences, such as 44–78 and 85–136, induced greater IFNL1 expression (Fig. 3D). Unexpectedly, 44–78 or 85–136 sequences, which are the strongest IFN-stimulating stem–loops during X RNA attachment, were not necessary for IFN induction in the cbVG as deletion of either 44–78 or 81–140 did not reduce IFN expression. This suggests that either the RNA folds differently in the context of whole cbVG than when adding X RNA stem–loops, or other secondary structures impact the motif's activity. Further work is necessary to determine the exact structures of these RNAs in both contexts.
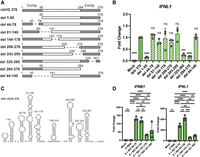
NiV cbVG sequences 44–78 and 85–136 are immunostimulatory. (A) Indicated deletion versions of NiV cbVG 378 were generated by in vitro transcription, then 1 pmol of IVT RNA was transfected into A549 cells. Gray boxes represent complementary regions (Comp.) of the cbVG. (B) Expression of IFNL1 was measured by qPCR at 6 hpt. One-way ANOVA with Tukey's HSD post hoc test was performed for statistical analysis. (ns) P > 0.05, (***) P < 0.001, (****) P < 0.0001. Data are represented as fold change over NiV cbVG 378. (C) Predicted structure of RNA stem–loops from NiV cbVG 378. (D) Sequences from NiV cbVG 378 were attached to X RNA and then in vitro transcribed. One picomole of IVT RNA was transfected into A549 cells, and expression of IFNB1 and IFNL1 was measured by qPCR at 6 hpt. One-way ANOVA with Tukey's HSD post hoc test was performed for statistical analysis. (ns) P > 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. Data are represented as fold change over the X RNA control. Data reported are biological triplicates.
Terminal loop sequences are critical for strong IFN expression
Since we observed multiple stem–loops with different strengths of RIG-I activation, we next investigated the characteristics conserved among stronger immunostimulatory RNA sequences versus weaker immunostimulatory sequences (Fig. 4A). The cbVG RNAs are predicted to form stem–loops that are diverse in their size and shape. However, two complementary sequences that are predicted to form loops with the same structure (RSV 1–34 and RSV 203–236) exhibit different immunostimulatory capabilities (Fig. 2C). We did not observe significant differences in the minimum free energy (MFE) between the stem–loops, most of which had a predicted MFE between −6 and −10 kcal/mol. In addition, we did not detect increased levels of the stronger RNA stem–loops by qPCR at 6 h posttransfection, suggesting that constructs with these stem–loops do not have better transfection efficiency or increased stability in the cells (Supplemental Fig. S5). Upon observation, we found that the terminal loops with strong immunostimulatory activity contain either a CAA motif or poly(A) stretch at the tip of the loop (Fig. 4A). In contrast, the weaker immunostimulatory loops are instead U-rich at the terminal loop residues, except for NiV 324–361, which contains an A-rich terminal loop.
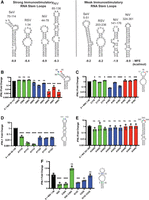
Terminal loop adenines are necessary for high IFN expression. (A) Predicted RNA structures of strong immunostimulatory and weak immunostimulatory stem–loops derived from cbVGs. (B–F) Indicated mutations were generated for cbVG sequences attached to the X RNA. The loop sequences of (B) SeV 70–114, (C) RSV 1–48, (D) NiV 85–136, and (E) NiV 324–361 were mutated. (F) RSV 1–48 stem nucleotides were mutated to alter RNA structure. RNAs were in vitro transcribed, and 1 pmol of IVT RNA was transfected into A549 cells. Expression of IFNL1 was measured by qPCR at 6 hpt. One-way ANOVAs with Tukey's HSD post hoc test were performed for statistical analysis. (ns) P > 0.05, (*) P < 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. Data are represented as fold change over the wild-type stem–loops. Data reported are biological triplicates.
We then hypothesized that the terminal loop sequences played a key role in triggering RIG-I signaling. To test this hypothesis, we mutated the terminal loops of the X-RNA attached sequences and tested their ability to induce IFN expression. Mutations of the terminal loop sequence were not predicted to alter the overall structure of the stem–loops (Supplemental Figs. S6–S9). For X + SeV 70–114, we found that mutation of the A89 or A90 terminal residues, specifically A89C, A90G, A90C, and A90U, greatly reduced IFN activation by the RNA construct; however, mutation of the C88 residue did not (Fig. 4B). Mutations in X + RSV 1–48 showed that A18G and A18U reduced IFN expression, but mutations of C17 or A19 slightly enhanced IFN expression (Fig. 4C). Additionally, mutation of X + NiV 85–136 showed that both terminal adenines, A111 and A112, were necessary for IFN signaling (Fig. 4D). However, the trend of terminal adenines was not observed for the weaker X + NiV 324–361, which did not show a reduction in IFN stimulation upon mutation of the terminal loop sequence (Fig. 4E). These data suggest that the terminal loop sequences impact the strength of the IFN response.
Our previous work showed that the stem of the RNA structures were necessary for the SeV 70–114 stem–loop to maintain strong IFN stimulation (Xu et al. 2015). We therefore then determined if the stems were also necessary for X + RSV 1–48. We found that disruption of the stems by single mutations, G5C, C30G, G13C, and C23G, resulted in reduced IFN expression (Fig. 4F), while complementing back the stem structure with double mutants, G5C C30G and G13C C23G, recovered IFN expression. Interestingly, mutation of the RSV 1–48 stem reduces the IFNL1 signal to 30%, whereas the A18G and A18U mutations in Figure 4C have a more drastic reduction (>99%) of IFNL1. These data suggest that the RNA structure as well as terminal loop sequence play a role in modulating IFN expression during RIG-I activation. Further work is necessary to determine if other features of these motifs are necessary for strong IFN induction.
RSV 1–48 activates innate immune signaling in vivo
Previously, we found that the SeV cbVG 546 stem–loop 70–114 activates a strong type I immune response in mice when administered alone or within a lipid nanoparticle (LNP) (Fisher et al. 2018, 2022; Gnazzo et al. 2025). Here, we aimed to determine whether RSV sequence 1–48 similarly activates innate sensors and induces an immune response in vivo. To ensure the RNA constructs were delivered intracellularly to trigger the RIG-I pathway, we packaged the X + RSV 1–48 and the control X + RSV 189–236 chimera constructs into LNPs (Fig. 5A) and confirmed the homogeneity of the formulations before in vivo administration (Fig. 5B). These formulations were then administered subcutaneously in the footpad of C57BL/6 mice. After 16 h, we found that LNPs containing X + RSV 1–48 induced significantly higher expression of IFNB1, MX1, IL1B, and CXCL10 compared to empty LNPs and X + RSV 189–236 (Fig. 5C). These data demonstrate that the RSV cbVG-derived 1–48 sequence retains its immunostimulatory activity in vivo when packaged into LNPs and, together with data from SeV 70–114, demonstrate that small RNA motifs with terminal loop adenines can serve as potent immunostimulators in mice.
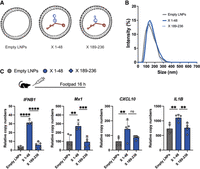
X + RSV 1–48 activates innate immune signaling in mice. (A) Schematic of lipid nanoparticles (LNPs) containing X + RSV 1–48 (X 1–48) or X + RSV 189–236 (X 189–236). (B) Dynamic light scattering measurement of LNP sizes of empty LNPs, X 1–48, and X 189–236. (C) LNPs were injected subcutaneously into the footpad of mice. Sixteen hours after injection, RNA was collected, and the RNA expression of IFNB1, MX1, CXCL10, and IL1B was quantified by qPCR. One-way ANOVAs with Tukey's HSD post hoc test were performed for statistical analysis. (ns) P > 0.05, (**) P < 0.01, (***) P < 0.001, (****) P < 0.0001. Data reported are five individual mice.
DISCUSSION
We have identified multiple RNA stem–loops derived from cbVGs of RSV and NiV that can transfer robust IFN stimulatory capabilities to an inert RNA. One unique feature of the higher immunostimulatory RNA stem–loops is a richness for adenines at the tip of the loop. When these sequences were mutated, the mutant RNAs stimulated significantly less IFN upon transfection into cells. Upon packaging into lipid nanoparticles, the RSV cbVG-derived stem–loop was able to activate innate immune signaling in mice.
While the enhanced stimulation appears to occur with adenine-rich sequences, we also observe that a stem–loop with five adenines (NiV 325–360) did not have decreased IFN activation when the adenines were mutated (Fig. 4E). Since an adenine-rich loop does not always enhance IFN expression, more work is necessary to determine the exact motif or motifs that make up an optimal terminal loop PAMP and the impact of distinct tertiary conformations. Some possibilities are that sequences in the stems of the RNA interact with the loop to alter binding or that host proteins are recognizing a specific RNA motif that is more than adenines.
One major question that remains is what step of the RIG-I pathway is impacted by these terminal adenine-rich sequences. We propose the following possibilities for how the terminal loop sequences may alter IFN expression: (1) RIG-I is heavily modulated by phosphorylation and ubiquitination by host proteins (Rehwinkel and Gack 2020), so the terminal loop may recruit or inhibit additional host factors from interacting and altering RIG-I function in cells. (2) MAVS has recently been shown to interact with host RNAs (Gokhale et al. 2024). The terminal sequence may stabilize RIG-I binding to MAVS through RNA/MAVS interactions and enhance downstream signaling to boost IFN expression. (3) The terminal loop adenines may be prone to RNA modifications such as N6 methylation to generate N6 methyladenosine (m6A) (Boulias and Greer 2023). m6A RNA modification can alter the stability of the RNA in a cell and modulate the binding of RNA binding proteins. (4) Other unknown host RNA-binding proteins support robust RIG-I signaling. Further work is necessary to parse out what interactions occur with this terminal loop sequence and why adenine-rich sequences enhance IFN expression from the RIG-I pathway. Additional mutational studies are needed to fully understand the RNA elements required for optimal activation of the RIG-I pathway.
With the advent of mRNA vaccines and the usage of synthetic RNAs as therapeutics, understanding how RNAs can activate innate immunity is crucial to optimizing RNA-based adjuvants and treatments. Synthetic RNAs can act as immune adjuvants during vaccination (Fisher et al. 2018, 2022; Linehan et al. 2018; Jiang et al. 2019; Mao et al. 2022; Gnazzo et al. 2025), and the addition of immunostimulatory stem–loops to mRNA vaccines may bypass the need for additional adjuvants such as alum. Although additional work is necessary to assess the immunostimulatory properties of RNA stem–loops in vivo, our discovery that the terminal loop sequence modulates immune activation can lead to the development of strategies to tailor the immune response not only based on dosage of stimulatory RNA but also on the sequence of attached RNA stem–loops.
MATERIALS AND METHODS
Cells and reagents
A549 human type II alveolar cells (ATCC CCL-185), RIG-I-KO A549 cells (gifted from S. Weiss), and MAVS-KO A549 cells (gifted from S. Weiss) were cultured at 37°C and 5% CO2 with Dulbecco's modified Eagle's media (Thermo Fisher 11995065) supplemented with 10% fetal bovine serum (FBS), 1 mM sodium pyruvate, 2 mM l-Glutamine, and 50 µg/mL gentamicin. All cell lines were treated with mycoplasma removal agent (MP Biomedicals 093050044) and routinely tested for mycoplasma before use. Antibodies used in this study are as follows: rabbit anti-p-TBK1 (Cell Signaling 5483, 1:2000), rabbit anti-TBK1 (Abcam AB40676, 1:2000), rabbit anti-α-tubulin (Invitrogen 13-8000, 1:2000), and goat antirabbit IgG-HRP (Cell Signaling 7074, 1:10,000).
In vitro transcription of RNA
RNAs were cloned into a pSL1180-T7 plasmid that contains T7 promoter at the 5′ end of the RNA sequence and hepatitis D virus ribozyme after the RNA sequence. Immunostimulatory RNA sequences were attached to the X RNA by PCR. In vitro transcription was performed with the MEGAscript T7 Transcription Kit (Thermo Fisher AM1334), and RNA was isolated with LiCl precipitation according to manufacturer's protocols. Purified RNA was measured by Qubit and tested for quality by Bioanalyzer (Agilent). Low molecular weight poly I:C (Invivogen tlrl-picw) was used as positive control for RIG-I activation. Full protocol is available at Protocols.io: dx.doi.org/10.17504/protocols.io.3byl49qprgo5/v2.
RT-PCR and qPCR
A549 cells were transfected with 5 pmol of indicated RNA and Lipofectamine 2000 (Thermo Fisher 11668027). After 6 h, RNA was isolated with TRIzol (Thermo Fisher 15596026). cDNA was generated with random hexamers using the High-Capacity RNA to cDNA kit (Thermo Fisher 4387406) according to the manufacturer's protocols. For qPCR quantification, cDNA was quantified with Power SYBR Green Mix (Thermo Fisher 4367660). IFNL1, IFNB1, IL1B, MX1, and CXCL10 were normalized to a housekeeping index calculated from ACTB and GAPDH genes. Sequences of primers are given in Supplemental Table S1. Full protocol is available at Protocols.io: dx.doi.org/10.17504/protocols.io.8epv5rxw6g1b/v1.
Mice
C57BL/6 mice (The Jackson Laboratory) were bred in-house. All mice were sex and age matched. Both male and female mice were included in the experiments.
Lipid nanoparticle formulation
X + RSV 1–48 and X + RSV 189–236 were encapsulated in the GenVoy Ionizable Lipid Mix (ILM) using a NanoAssemblr Ignite machine (Precision NanoSystems) following the manufacturer's instructions. Empty LNPs were performed and used as controls. Encapsulation efficiency and RNA concentration were tested by RiboGreen Assay (Thermo Fisher). Final particle size was measured by dynamic light scattering. Full protocol is available at Protocols.io: dx.doi.org/10.17504/protocols.io.36wgqnn4kgk5/v1
Mice immunization
Mice were anesthetized with isoflurane and injected subcutaneously (s.c.) into a rear footpad. For immune response studies, mice were inoculated with 0.5 µg X 1–48; 0.5 µg X 189–236; empty LNPs or PBS at a final volume of 30 µL per dose. RNA was extracted from footpad 16 h after inoculation using TRIzol (Ambion Inc.).
In silico RNA folding prediction
All RNA folding predictions were performed using the University of Vienna RNAfold website with the ViennaRNA Package (Version 2.6.3) (http://rna.tbi.univie.ac.at/cgi-bin/RNAWebSuite/RNAfold.cgi). Structures shown are the minimum free energy structures. All sequences used for folding are given in Supplemental Table S2.
Statistical analysis and reproducibility
All statistics were calculated using GraphPad Prism, version 9. Specific tests and significance values are indicated in each figure legend. Data reported are biological triplicates unless otherwise stated.
Ethics statement
All described studies adhered to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Institutional Animal Care and Use Committee, Washington University in St. Louis approved protocol 23-0083.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We acknowledge the use of the Chemical and Environmental Analysis Facility at Washington University in St. Louis for instrumentation and analytical support. This work was supported by the National Institutes of Health (NIH) T32 HL007317-44 (M.H.), NIH T32 AI007163-45 (M.H.), NIH A137062 (C.B.L.), and WashU BJC Investigator Program (C.B.L.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080913.125.
-
Freely available online through the RNA Open Access option.
- Received December 16, 2025.
- Accepted February 2, 2026.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Matthew Hackbart is the first author of this paper, “Terminal loop sequences in viral double-stranded RNAs modulate RIG-I signaling.” Matthew is a postdoctoral research fellow in Dr. Carolina B. Lopez's lab in the Department of Molecular Microbiology and Center for Women's Infectious Disease Research at Washington University in St. Louis. The main focus of his research is determining how nonstandard viral genomes alter viral replication and pathogenesis during RNA virus infections.
What are the major results described in your paper, and how do they impact this branch of the field?
In this paper, we found novel RNA sequences from viral infections that can enhance innate immune signaling through RIG-I. Uniquely, the adenine-rich sequences are located in the distal stem–loop away from the 5′ end, where most RIG-I motifs are known. These results add to the field of RIG-I signaling by providing additional RNA motifs that can be utilized for therapeutics and immune stimulants.
What led you to study RNA or this aspect of RNA science?
During my PhD, I researched how coronaviruses alter their RNA signal to escape innate immune detection. My interest in the RNA motifs and structures that are recognized by host sensors led me to work in Dr. Lopez's lab, where they studied how copy-back viral genomes interact with innate immune systems.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
We were surprised that mutating the loop of the RNA stem altered the immune stimulation. Most RIG-I work focuses on the structure of the RNA or the 5′ triphosphate, so we did not expect RNA sequences not located near these signals would alter activation. We had originally only mutated the loops of one RNA, but we changed our focus to test if other RNA loops had a similar phenotype upon getting these results.
If you were able to give one piece of advice to your younger self, what would that be?
One piece of advice I would give my younger self is to be persistent. In science, experiments will fail and hypotheses will be wrong, but keep working hard and you will find success.








