
Genetic and genomic approaches to explore roles for the conserved 3′-5′ exoribonuclease EXOSC10 in normal and malignant cells
- 1Université de Rennes, Inserm, EHESP, Irset (Institut de recherche en santé, environnement et travail)-UMR_S 1085, Rennes F-35042, France
- 2Faculté des Sciences et Ingénierie, Sorbonne Université, Paris F-75005, France
- Corresponding author: michael.primig{at}inserm.fr
Abstract
EXOSC10 is a conserved 3′-5′ exoribonuclease involved in processing ribosomal RNAs and degrading coding and noncoding transcripts as a catalytic subunit of the nuclear RNA exosome and in cooperation with cofactors. The protein is posttranslationally modified and shuttles between the nucleolus and the nucleus in response to oxygen deprivation in a process that involves sumoylation. EXOSC10 is of medical interest because its activity is inhibited by the anticancer drug 5-fluorouracil, which interferes with DNA replication and RNA-dependent processes. Moreover, high expression of EXOSC10 in certain somatic tumors is associated with patient survival. We discuss global and tissue-specific deletion experiments in the mouse, assess the protein's clinical relevance as a prognostic cancer biomarker in the context of human genomics data for normal versus malignant tissues, and explore EXOSC10’s transcriptional regulatory network.
Keywords
INTRODUCTION
Rrp6, the budding yeast ortholog of mammalian EXOSC10, was initially reported to be a nonessential 3′-5′ exoribonuclease involved in ribosomal RNA processing, and later found to be involved in degrading and processing aberrant mRNAs and a wide variety of noncoding transcripts (Briggs et al. 1998; Neil et al. 2009; Xu et al. 2009; Gudipati et al. 2012). The enzyme can act alone or in association with regulatory proteins, but it is thought to predominantly function as a catalytic subunit of the highly conserved nuclear RNA exosome; for review, see Stuparevic et al. (2021). The human protein includes an N-terminal protein interaction domain (PMC2NT), an internal catalytic domain comprising two elements (EXO1 and HRDC), and an unstructured C-terminal region (Fig. 1A). EXOSC10 is a hub protein that physically interacts with several core subunits and cofactors of the nuclear RNA exosome (Fig. 1B), associates with numerous proteins in multimeric complexes, and undergoes a variety of posttranslational modifications (PTMs), such as phosphorylation of serines, threonines, and tyrosines, and methylation, acetylation, sumoylation, and ubiquitination of lysine residues; for review, see Stuparevic et al. (2021) (Fig. 1C).
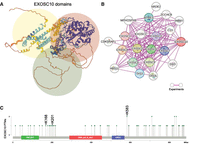
EXOSC10 structure, protein network, and sumoylation. (A) A cartoon structure from the AlphaFold database (https://alphafold.ebi.ac.uk) for human EXOSC10 (AF-Q01780-F1) is shown, and N-terminal, internal, and C-terminal regions are highlighted in yellow, red, and green, respectively. (B) An experimentally verified physical subnetwork of EXOSC10, RNA-exosome core subunits, and cofactors as provided by the String database (https://string-db.org) is shown. (C) A lollipop plot of posttranslational modifications referenced by PhosphoSitePlus (https://www.phosphosite.org) generated with cBioPortal's mutation mapper (https://www.cbioportal.org/mutation_mapper) is shown. The positions of amino acids undergoing PTMs (x-axis) are plotted against the number of PTMs for any given residue (y-axis). Three lysines (K) that are sumoylated in response to stress are indicated. The location of functional domains is indicated by colored bars. Amino acid coordinates are given at the bottom.
Human EXOSC10 is clinically relevant from several perspectives. First, it is among several RNA exosome subunits that are targeted by auto-antibodies in the polymyositis/scleroderma overlap syndrome (hence its initial name PM/Scl-100 for polymyositis/scleroderma autoantigen 100 kD) (Bluthner and Bautz 1992; Ge et al. 1992; Staals and Pruijn 2011). Second, its enzymatic activity is inhibited by the widely used anticancer drug 5-fluorouracil (5-FU) (Kammler et al. 2008; Silverstein et al. 2011). Third, EXOSC10 was reported to be a potential prognostic liver cancer biomarker (Meng et al. 2023).
Mouse gene deletion models are a major tool for functional studies of conserved mammalian genes potentially important for human development and disease (Groza et al. 2023). The global gene deletion approach disrupts a gene in all tissues from early development, while the targeted or conditional knockout (cKO) using the Cre-loxP system restricts the loss-of-function allele to specific tissues. The latter method avoids embryonic lethality and enables functional studies of so-called floxed alleles containing the loxP sites targeted by the Cre recombinase in selected organs or at defined developmental stages, depending on the promoter that drives Cre expression (Foster et al. 2024).
The increasingly rapid expansion of genome biology over the past 30 years has inspired the development of a huge variety of bioinformatics solutions for the production, management, statistical analysis, and biomedical interpretation of quantitative data. The Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org) catalogs genetic variation from high-throughput exome- and whole-genome DNA sequencing of ethnically diverse and healthy populations to facilitate interpretation of rare DNA sequence variants (Chen et al. 2024). The Catalogue Of Somatic Mutations In Cancer (COSMIC; https://cancer.sanger.ac.uk/cosmic/login) project compiles somatic mutations found in cancer, including driver mutations and gene fusions, across a wide variety of tumor types (Sondka et al. 2024). PhosphoSitePlus (https://www.phosphosite.org) compiles information about posttranslational modifications of proteins detected mostly in high-throughput studies (Hornbeck et al. 2015). The University of Alabama at Birmingham Cancer data analysis Portal (UALCAN; https://ualcan.path.uab.edu), the Gene Expression Profiling Interactive Analysis (GEPIA 2; http://gepia2.cancer-pku.cn), and Tumor Immune Estimation Resource (TIMER 2; http://timer.cistrome.org) provide a variety of cancer exploration tools, including one based on data from The Cancer Genome Atlas (TCGA) project that links gene expression and clinical outcome (Tang et al. 2019; Li et al. 2020; Chandrashekar et al. 2022).
High expression levels of tumor driver or suppressor genes in cancers and patient survival can be negatively or positively associated. Kaplan–Meier (KM) plots depict patient survival over time in the context of high biomarker expression levels in specific malignancies. Statistical significance, typically assessed by log-rank tests, indicates whether a biomarker is associated with favorable or unfavorable outcomes, which facilitates efforts to optimize therapeutic strategies (Rich et al. 2010). The outcome of this approach can depend on the type of expression data set, the parameters selected, and the methods used to analyze data. It is therefore useful to compare predictions by sources like, for example, GEPIA 2 and UALCAN to tools such as the KM Plotter (https://kmplot.com/) (Lanczky and Gyorffy 2021; Posta and Gyorffy 2025).
High-throughput in vivo protein–DNA binding data produced using, among others, the chromatin immunoprecipitation and sequencing (ChIP-seq) method to analyze protein–DNA binding activities in numerous tissue and cell samples, are available via the ChIP-Atlas database (https://chip-atlas.org). The Eukaryotic Promoter Database (EPD; https://epd.expasy.org/epd/) provides a motif search tool for promoters with experimentally verified transcription start sites (TSSs) (Meylan et al. 2020; Zou et al. 2024). Protein structure predictions are available via the European Bioinformatics Institute's AlphaFold database (https://alphafold.ebi.ac.uk), and protein network visualization and analysis tools are provided by String (https://string-db.org) (Szklarczyk et al. 2023; Varadi et al. 2024).
In this review, we summarize the outcome of genetic analyses based on global and targeted Exosc10 deletion experiments in the mouse (Fig. 2), before we focus on work on the human protein's regulation by sumoylation. We then interpret the results in the context of genomics data from gnomAD, COSMIC, ChIP-Atlas, and String databases and discuss publicly available data associating high EXOSC10 expression levels in a variety of malign somatic tumors with the survival of patients over time.
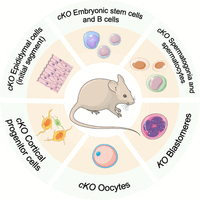
Transgenic gene deletion models for Exosc10. A schematic shows different somatic and germ cell types analyzed by conditional (cKO) or global gene knockout (KO) studies in the mouse.
Mouse Exosc10 function in embryonic, somatic, and reproductive tissues
EXOSC10 targets regulatory long noncoding RNAs in embryonic stem cells and differentiated B cells and is critical for B-cell development
Pefanis et al. (2015) gained insights into the regulatory roles of Exosc10 by RNA profiling mutant embryonic stem cells (ESCs) and B cells that they isolated from transgenic mice generated using a Cre-loxP conditional inversion (COIN) method. This approach enabled them to disrupt normal gene expression by exposing a floxed allele to 4-hydroxytamoxifen (4-OHT)–inducible Cre recombinase activity and to determine differential gene expression patterns in wild-type versus mutant cells by RNA-sequencing (Pefanis et al. 2015). RNA-profiling data for ESCs revealed a critical role for EXOSC10 in the degradation of a subset of 1506 RNA exosome-targeted long noncoding RNAs (x-lncRNAs), including 423 enhancer RNAs (x-eRNAs). Similar experiments using B cells identified 62 EXOSC10 x-eRNAs targets (Pefanis et al. 2015). The authors demonstrate that EXOSC10 targets transcripts involved in interactions of genes and so-called super-enhancer sequences, which are clusters of individual enhancers regionally enriched for active chromatin marks. Further work revealed that RNA exosome–mediated degradation of divergently transcribed eRNAs that form R-loops (cotranscriptional RNA/DNA hybrids), contributes to maintaining genomic integrity and, in B cells, immunoglobulin heavy-chain (IgH) locus class-switch recombination (Pefanis et al. 2015). Related work by Laffleur et al. (2022) provided insights into Exosc10’s peak expression in developing pro-B and large pre-B cells during V(D)J recombination, in activated B cells, during the transitional stages and in follicular B cells in the spleen. These findings were complemented by a conditional gene deletion experiment (Exosc10COIN) revealing that B cells developing in bone marrow arrest at the pro-B-cell stage and B cells fail to normally differentiate into pre-B cells in the absence of functional EXOSC10 (Laffleur et al. 2022).
In normal cells, eRNA levels are tightly controlled to keep target gene expression responsive to regulatory cues, allow for developmental-stage or tissue-specific transcription, and avoid interference with proximal protein-coding loci or formation of R-loops. Perturbed eRNA processing due to altered RNA exosome activity can lead to genomic instability and abnormal enhancer activation-mediated upregulation of oncogenes; for review, see Sartorelli and Lauberth (2020) and references therein.
Exosc10 is essential for spermatogenesis and contributes to the regulation of meiotic gene expression and rRNA processing in male germ cells
Jamin et al. (2017) observed that mouse EXOSC10 associates in late pachytene spermatocytes with the XY body (or sex body), which is a heterochromatic structure essential for transcriptionally silencing of the sex chromosomes, before the protein becomes undetectable as spermatogenesis progresses during the postmeiotic stages (Jamin et al. 2017). This points to a possible function for mouse EXOSC10 in lncRNA-dependent sex chromosome silencing in the male germline and raises the question of why the protein is downregulated during later stages of male gametogenesis. Perhaps its activity is not needed (or may even be harmful) during the development of transcriptionally and translationally inert gametes.
The authors also reported a novel critical role for the exoribonuclease in mitotic germ cells. The study was based on transgenic mice bearing floxed Exosc10 alleles in combination with Ddx4|Vasa- or Stra8-promoter (p) driven Cre recombinases expressed in early undifferentiated spermatogonia and late spermatogonia/early spermatocytes, respectively. Interestingly, mutant males were infertile or subfertile depending on the Cre line: Ddx4|Vasap-Cre caused a complete loss of germ cells, whereas Stra8p-Cre mediated deletion [STOCK Tg(Stra8-362 iCre)1Reb/J backcrossed with the C57BL/6NRj strain] yielded incomplete penetrance with residual sperm production and reduced fertility (Jamin et al. 2017). These results indicate that Exosc10 is essential for spermatogonial growth, division, and development.
A recent study by Yu et al. (2025) focusing on the same question confirmed these findings in a targeted deletion experiment employing a different Stra8p-Cre construct (Stra8-GFPCre knock-in line maintained in the C57BL/6J background) mediating Cre expression in a subset of spermatogonia and spermatocytes (Yu et al. 2025). The authors analyzed the transcriptomes in male germ cells from postnatal day 21 (P21) control and targeted deletion mice (cKO) via single-cell RNA-sequencing (scRNA-seq). Inevitably, they observed relatively enriched mitotic (spermatogonial) and strongly depleted meiotic (spermatocytes) and postmeiotic (spermatids) germ cell populations. They then—unsurprisingly—identified numerous supposedly deregulated male germline genes at successive mitotic, meiotic, and postmeiotic developmental stages in the absence of Exosc10, which in fact is easily explained by the altered germ cell composition of wild-type versus cKO testes (Yu et al. 2025). Along the same line, Fan et al. (2025) reported a comprehensive RNA/protein profiling study of testes from a wild-type (WT) control versus a targeted Exosc10 deletion mutant (Fan et al. 2025). The authors crossed Exosc10flox/flox and Ddx4-Cre strains to generate Ddx4-Cre; Exosc10flox/− (DcKO) conditional knockout mice (also in the C57BL/6J background), enabling them to analyze the exoribonuclease's function during embryonic germ cell development from embryonic day E15.5 onward. Fan et al. confirm the earlier finding by Jamin et al. (2017) that preventing spermatogonial differentiation at a prenatal stage using Ddx4-Cre disrupts postnatal spermatogenesis and therefore causes a strong male infertility phenotype (Jamin et al. 2017). Determining the transcriptome and proteome profiles of WT versus DcKO testes at postnatal day 4 (P4) identified a total of 78 cases for which mRNA/protein patterns showed coherent up- or downregulation. This result led Fan et al. to conclude that these processes are dysregulated in the absence of EXOSC10, implying that somehow this enzyme acts as a master regulator of both biological processes and the genes related to them. A simpler explanation might be that lower levels of mRNAs and proteins important for germ cell division and function are a consequence of strongly reduced spermatogonial germ cell populations in DcKO versus control testes samples. In other words, the differential expression pattern is not due to a regulatory process but might be a consequence of depleting mitotic germ cell populations expressing these genes in DcKO testis samples. In this context, it is noteworthy that the authors report induction of genes involved in inflammatory response in mutant testes. Rather than reflecting a direct link with EXOSC10, this could be a stress response of abnormal testicular tissue, reminiscent of the inflammatory-like response to human spermatogenic failure (Spiess et al. 2007).
Exosc10 is critical for early embryonic development
The embryonic lethal phenotype of homozygous Exosc10 mutant mice was first mentioned by Jamin et al. (2017) and Wu and Dean (2020) who studied the gene's function specifically in male and female gametes, respectively (Jamin et al. 2017; Wu and Dean 2020). A more detailed global gene deletion analysis of Exosc10’s phenotype during embryogenesis by Petit et al. (2022) revealed the gene's essential role at the eight-cell/morula transition prior to formation of the blastocyst. Genotyping of early embryos identified no Exosc10−/− embryos past embryonic day E4.5, indicating that the protein is essential during the first mitotic cell divisions of blastomeres (Petit et al. 2022). Maternal Exosc10 mRNA present in oocytes may allow for development through the one- to four-cell stages, but once zygotic gene expression becomes dominant, homozygous mutant embryos undergo developmental arrest. Mouse EXOSC10 localizes very early during development to the periphery of the so-called nucleolus precursor bodies in blastomeres, which suggests a role in rRNA processing and ribosome biogenesis during initial stages of embryogenesis (Petit et al. 2022).
Normal oogenesis requires functional Exosc10
Wu and Dean (2020) inactivated Exosc10 specifically in growing oocytes by combining a heterozygous floxed allele with Cre under the control of the promoter regulating Zp3 (Exosc10flox/−; Zp3-Cre designated as cKO). Zp3 encodes one of the proteins that constitute the zona pellucida, an extracellular matrix surrounding oocytes important for, among others, sperm binding. The authors report that cKO female mice displayed abnormal oocyte maturation due to defective germinal vesicle breakdown (GVBD), a phenotype likely related to impaired global degradation of both polyadenylated and nonpolyadenylated RNAs, as revealed by single oocyte RNA-profiling (Wu and Dean 2020). Interestingly, the authors observed a subfertility phenotype rather than the expected infertility phenotype. They attribute this observation to molecular heterogeneity of the cKO oocyte population that manifests itself via distinct levels of the transcriptome's perturbation (Wu and Dean 2020). This is conceptually intriguing because it raises the possibility that mammalian oocytes may possess more or less effective compensation pathways in the absence of functional EXOSC10. One possible candidate is the 3′-5′ exoribonuclease DIS3, which is the second catalytic subunit of the nuclear RNA exosome (Tomecki et al. 2010). This enzyme is typically localized in the nucleus rather than the nucleolus, but has recently been found to target cytoplasmic circular RNAs via its endoribonuclease activity independently of the RNA exosome core (Latini et al. 2025). Interestingly, Wu and Dean (2023) report that a double knockout of Exosc10 and Dis3 in oocytes indeed reveals synergistic functions of the enzymes in ensuring transcriptome integrity during oogenesis (Wu and Dean 2023). An attractive alternative explanation is that the timing and efficiency of Exosc10's inactivation in oocytes is critical for the fertility phenotype's severity, since Demini et al. 2023 employed a Gdf9-Cre construct in their cKO experiment, and reported disrupted oogenesis accompanied by an altered proteome relevant for meiotic differentiation and oocyte maturation (Demini et al. 2023). The authors suggest a role for EXOSC10 in maintaining the ovarian reserve, which is corroborated by a very recent study that associates a homozygous human EXOSC10 missense variant with premature ovarian insufficiency (Kline et al. 2026).
Exosc10 is important for forebrain development and cortical architecture
An analysis by Ulmke et al. (2021) aimed at generating and characterizing telencephalon-specific (FoxG1-Cre) and cortex-specific (Emx1-Cre) Exosc10 targeted deletion models using the COIN method (Ulmke et al. 2021). The authors observed strong phenotypes, including loss of neurons and reduced cortical layers, that appeared to be the consequence of pervasive apoptosis in embryonic day E13.5 Emx1-Cre embryos. Further analysis of cortical samples from wild-type versus mutant E12.5 embryos by RNA-sequencing revealed altered levels for 1875 genes enriched in loci important for various aspects of brain development. This result likely reflects direct and indirect effects of disrupted cortical tissue development in the absence of Exosc10. To further obtain insights into the molecular underpinnings of the phenotype, the authors carried out RNA-immunoprecipitation sequencing (RIP-seq) of E12.5 wild-type mouse cortex samples and found 3159 transcripts bound by EXOSC10, including 144 RNAs that were upregulated in mutant Emx1-Cre embryos and that are enriched for the Gene Ontology (GO) term “neuron death” (Ulmke et al. 2021). This led them to investigate if Exosc10 is directly involved in controlling programmed cell death. Indeed, disrupting EXOSC10 activity during early cortical development increases the expression of Tp53-related genes, such as Aen and Bbc3, thereby triggering apoptosis at a level comparable to that caused by Tp53 overexpression. The authors conclude that the precise interplay between the nucleolar RNA exosome complex and TP53 signaling is critical for the viability of neurons and cortical development (Ulmke et al. 2021).
In a recent follow-up study, Ulmke et al. (2025) reported five novel human EXOSC10 alleles (the catalytically inactive de novo missense variant EXOSC10.pD295A and de novo microdeletions Del1p36.32p36.13, Del1p36.21p36.23, Del1p36.32p36.21, and Del 1p36.32-p36.15) in patients diagnosed with microcephaly, whose genomes lacked known pathogenic variants in all genes that have been associated with the disorder (Ulmke et al. 2025). The authors tested their hypothesis that altered EXOSC10 activity can cause microcephaly and abnormal cortical development by phenotypically analyzing homozygous (Exosc10cKO) and heterozygous (Exosc10Het) mutants, generated by crossing mice bearing Exosc10 COIN alleles or the Emx1-Cre transgene. Their results revealed roles for the enzyme in establishing cortical architecture and promoting neuronal differentiation at least in part by inhibiting the Shh (Sonic hedgehog) signaling pathway, which is critical for brain development (see Ulmke et al. 2025 and references therein).
Lack of Exosc10 activity in the principal cells of the epididymis’ initial segment does not affect sperm maturation and fertility
Zhou et al. (2024) investigated the role of Exosc10 in the initial segment (IS) of the epididymis, which is critical for sperm maturation and male fertility, by crossing homozygous floxed mice (Exosc10F/F) with a strain bearing Lcn9-Cre that expresses the recombinase in principal cells of the IS, which are an epithelial cell type that secretes proteins and ions into the luminal fluid, from day postpartum (dpp) 17 onward. They report that cKO mice produce morphologically normal and motile sperm at typical concentrations, capable of undergoing the acrosome reaction (a process critical for the spermatozoid's ability to penetrate the oocyte's protective zona pellucida). Zhou et al. therefore suggest that EXOSC10 is not needed in the IS of the epididymis (Zhou et al. 2024). This may be the case; however, the authors do not elaborate on the fact that their findings are inconsistent with all previous reports of global and targeted deletion experiments that revealed critical functions for Exosc10 in male and female germ cells and somatic tissues in vivo and cultured cells in vitro; for review, see Stuparevic et al. (2021). An alternative explanation may be that the cKO mice expressed sufficient residual levels of the enzyme in principal cells and other cells to sustain IS function, akin to earlier work based on partial siRNA-mediated depletion of EXOSC10 mRNA, which led the authors to suggest that the protein is not essential for HEp-2 cancer cell growth and division (although we now know that it is critical for the process) (van Dijk et al. 2007; Blomen et al. 2015; Davidson et al. 2019). Yet another mechanism might be, like possibly in the case of oocytes, compensation via DIS3. In this context, it is noteworthy that targeted deletion of the gene in the IS in a Dis3F/F Lcn9-Cre strain also yields no phenotype (Qiu et al. 2023), which suggests that EXOSC10 and DIS3 are (more or less) functionally redundant in principal cells of the epididymal IS. If this was indeed the case, a double Exosc10/Dis3 cKO experiment in epididymal principal cells should reveal an effect on male fertility, which, if it were true, raises the question of why EXOSC10 and DIS3 are not redundant in all cells.
Human EXOSC10 sumoylation and the prevalence of K168, K201, and K583 mutations in healthy and malignant tissues
Earlier studies have identified three lysines (K168, K201, and K583) that are sumoylated via SUMO1 in response to cold temperature, and this process was associated with a decreased cellular EXOSC10 level (Fig. 1C; Knight et al. 2016). Furthermore, the nucleolar ubiquitin-specific protease USP36 mediates sumoylation of EXOSC10 at K583, a posttranslational modification critical for the protein's function in rRNA processing and ribosome biogenesis but apparently unrelated to its stability (Chen et al. 2023). Recently, USP36 sumoylation activity was reported to be counteracted by SENP3, and hypoxia (oxygen deprivation) was found to trigger a translocation of EXOSC10 from the nucleolus to the nucleus, where the protein influences the expression of genes involved in the low-oxygen stress response (Filippopoulou et al. 2024). It is interesting that cultured cells bearing the SUMO-deficient allele EXOSC10K583R show altered expression of 31 genes, including loci involved in the response to oxygen deprivation, nutrients, and xenobiotics, which is a critical issue in cancer progression (van Dijk et al. 2007).
This finding may have clinical repercussions, given that the gnomAD database references EXOSC10K583Q (1-11079713-T-G; frequency of 6.20 × 10−7) and EXOSC10K583E (1-11079713-T-C; frequency of 6.20 × 10−7) alleles that may also fail to be sumoylated. Moreover, COSMIC identifies the EXOSC10K583E allele as present in a form of bone cancer (Ewing sarcoma/peripheral primitive neuroectodermal tumor; COSV58667799 [legacy ID COSM4576104]; COSMIC annotated reference [Crompton et al. 2014]). It is conceivable that EXOSC10 alleles, which affect the protein's subcellular localization pattern, notably its ability to shuttle from the nucleolus to the nucleus, contribute to tumor cell resilience by affecting stress response gene expression. We note that the recently developed AlphaMissense algorithm (https://alphamissense.hegelab.org) predicts EXOSC10K583E to be likely pathogenic (that is to say, altering the protein's function; score 0.611) (Cheng et al. 2023). Further work appears warranted to determine the possible effects of K583 alleles on protein subcellular localization, protein network interactions, and enzymatic activity and their relevance for cancer progression.
Human EXOSC10 expression and function during somatic cancer progression
EXOSC10 is differentially expressed in a variety of somatic cancers in comparison to normal tissues; for review, see Stuparevic et al. (2021). In this context, it is noteworthy that in a subset of these malignancies, RNA expression levels and outcome are significantly correlated. Data from GEPIA 2 suggest high (top quartile) EXOSC10 expression to be a negative prognostic factor for adrenocortical carcinoma (ACC), sarcoma (SARC), lower-grade glioma (LGG), and liver hepatocellular carcinoma (LIHC); the latter confirming a recent study (Fig. 3A; Meng et al. 2023).
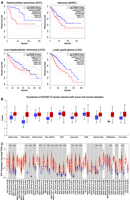
EXOSC10 expression and somatic cancer progression. (A) Color-coded KM plots from the GEPIA 2 database are shown. They illustrate associations that suggest EXOSC10 as a potential negative prognostic cancer biomarker. Legends for high and low expression color codes are shown in the top right corners. The log-rank and hazard score (HZ) P-values are shown, and the sample numbers for high/low expression (n) are given. (B) A whisker graph from UALCAN plots normal (blue) and tumor (red) samples (x-axis) against the Z-score transformed EXOSC10 protein concentration signal values (y-axis). Horizontal bars are the median. Sample annotation is spelled out or shown as clear cell RCC (renal clear cell), UCEC (uterine corpus endometrial carcinoma) and PAAD (pancreatic adenocarcinoma). (C) A whisker graph from TIMER 2 plots normal (blue) and tumor (red) samples (x-axis) against the log2-transformed transcript per million (TPM) signal values for E2F1 (y-axis). Horizontal bars are the median. Asterisks indicate statistically significant differences in expression levels. Sample numbers (n) are indicated. The full names of normal and cancer sample acronyms are shown in Supplemental Figure S1.
One must bear in mind that mRNA expression data are a relatively unreliable measure for cellular protein levels (apart from processes where timing of induction and time of function precisely coincide, such as progression through the meiotic cell divisions). We therefore retrieved proteome data for normal versus tumor samples from UALCAN and observed that malignant tissues, including brain, colon, kidney, and liver cancers, typically contain higher levels of EXOSC10 protein than normal controls (Fig. 3B; Zhang et al. 2022). Consequently, it appears justified to assume that increased EXOSC10 mRNA levels tend to coincide with elevated protein concentrations in cancer cells, which likely promotes processes such as DNA repair and ribosome biogenesis and helps maintain genome integrity by controlling R-loop formation (Marin-Vicente et al. 2015; Pefanis et al. 2015; Davidson et al. 2019; Domingo-Prim et al. 2019). This leads one to the somewhat puzzling question why elevated levels of EXOSC10 mRNA are not negatively correlated with patient survival in all malignant tumors. Perhaps increased gene expression does not systematically lead to elevated protein levels or enzymatic activity, or the majority of cancer cells have vulnerabilities or lack cofactors that prevent them from exploiting beneficial effects stemming from high EXOSC10 expression.
Wang et al. (2024) carried out a CRISPRi (interference or knockout) screen targeting the kinome (the complete set of kinases encoded in the human genome) that associates EXOSC10 with TGF-β signaling in cultured HeLa and HaCaT cells (Wang et al. 2024). The TGF-β pathway is involved in a wide variety of pathological processes, such as fibrosis, immune-disorders, and notably tumor invasiveness and formation of metastases; for review, see Gallo-Oller et al. (2025). Curiously, the kinome CRISPRi library employed by the authors that was originally published by Doench et al. (2016) (Brunello, Addgene 1000000083 [Doench et al. 2016]) identified EXOSC10 as a kinase, which, to the best of our knowledge, is not supported by published data. Wang et al. (who do not address the question of wrongly attributed enzymatic activity) report rather weak effects of decreasing EXOSC10 on TGF-β pathway target gene expression (SERPINE1|PAI-1, CCN2|CTGF, and CDKN1A|p21), which might be due to partial siRNA/CRISPRi-mediated depletion of EXOSC10. Moreover, the standard deviations are not consistently shown for the qPCR data points (see Figs. 3K,L, 4F–H; Wang et al. 2024). While these initial results are perhaps intriguing, time will tell if future studies by the authors and other laboratories confirm and extend these findings (Wang et al. 2024).
Another report by Tu et al. (2019) relevant for EXOSC10’s roles in molecular oncology reveals that the protein competes for binding to mRNAs encoded by DNA damage related genes, such as NBS1 and BRCA1, with programmed death ligand 1 (PD-L1|CD274). The latter is an immune checkpoint transmembrane protein that interacts with the programmed cell death protein 1 (PD-1) receptor expressed on activated immune cells and that also localizes to the cytoplasm (Tu et al. 2019). Related work by Sun et al. (2021) implicates EXOSC10 in enhancing the JNK (c-Jun N-terminal kinase) pathway in colon cancer cells via its ability to degrade the mRNA encoding the CYLD deubiquitinase, which can act as a suppressor of JNK signaling (Sun et al. 2021). This effect involves competition for target transcript binding between EXOSC10 and PD-L1|CD274 that, when nuclear under oxygen deprivation, acts as a transcriptional coactivator of the regulator STAT3 mediating a form of programmed cell death called pyroptosis (Hou et al. 2020). Given that EXOSC10 competes with the cytoplasmic form of PD-L1|CD274 for binding (and degrading) transcripts that encode DNA damage response genes, altered levels of the exoribonuclease may influence the ability of cancer cells to repair drug-induced DNA lesions.
Another intriguing link between the nuclear RNA exosome/EXOSC10 and cancer progression is highlighted by work on chemical alterations of RNA, notably N6-methyladenosine (m6A), which is a pervasive mRNA modification controlled (in a manner analogous to epigenomic marks) by writer, eraser, and reader proteins; for review, see Frye et al. (2018). Liu et al. (2020) report that inactivation of the mouse m6A writer Mettl3 and the reader Ythdc1 in embryonic stem cells promotes open chromatin and activates transcription via an m6A-dependent mechanism. Critically, METTL3 deposits m6A modifications on, among others, promoter-associated RNAs and eRNAs, some of which—notably elements of the long interspersed element (LINE)–1 family—are primed by YTHDC1 for degradation by the nuclear RNA exosome (Liu et al. 2020). Moreover, noncoding RNAs important for immunoglobulin heavy-chain (IgH) locus class switch recombination are subject to m6A-mediated processing by the RNA exosome in B cells (Nair et al. 2021). A better understanding of the intricate connection between RNA modifications and controlled decay might allow for the development of therapeutic strategies targeting enhancer-associated pathways in cancer; for review, see Vasukutty et al. (2025). Finally, seminal work by Leeman-Neill et al. (2023) brought together the initial observation of yeast Rrp6's role in the maturation of 5.8S rRNA, and how this process is affected by pathological overexpression of ZCCHC7, a subunit of the human TRAMP (Trf4/5–Air1/2–Mtr4 polyadenylation)–like complex essential for the RNA exosome, in lymphoma (Briggs et al. 1998; Leeman-Neill et al. 2023).
Exploring transcriptional regulatory networks that interact with the EXOSC10 core promoter
The mechanism for transcriptional control of EXOSC10 is relatively poorly understood, which is conceivably due to its cell type–independent fundamental roles that suggest it to be a ubiquitously expressed housekeeping gene. Its mRNA appears to be cycling during mitotic cell division, but the protein concentrations are not varying cell cycle stage specifically to a level detectable by a single-cell proteogenomics approach (Mahdessian et al. 2021; Stuparevic et al. 2021). Deng et al. (2025) report that E2F1, a DNA binding transcription factor that regulates genes involved in cell cycle progression and that promotes DNA damage repair, directly controls the expression of EXOSC10 by binding to the gene's promoter region in hepatocellular carcinoma cells (Manickavinayaham et al. 2020; Deng et al. 2025). The authors propose that E2F1-mediated EXOSC10 expression promotes tumor cell proliferation, which provides a straightforward explanation for the negative prognosis in LIHC and perhaps other cancers (Deng et al. 2025).
It is consistent with this model that E2F1 expression is significantly induced (or the mRNA's stability is increased) in 20 out of 23 tumor-versus-control sample pairs provided by TIMER 2, including different types of kidney cancer and liver cancer (Fig. 3C, see Supplemental Fig. S1 for TCGA cancer sample annotation). This raises the question if increased E2F1 expression (or transcript stability) in cancer cells is a cause for, or a consequence of, accelerated mitotic divisions. Given that this transcription factor contributes to the regulation of mitotic cell division, its dysregulation might affect cancer cell proliferation rates, but high E2F1 transcript levels might also at least be in part a consequence of altered epigenetic and genetic control of transcription or mRNA processing; for reviews of E2F1 with emphasis on its role in DNA replication and checkpoint control of the mitotic cell cycle or metabolic functions, see Denechaud et al. (2017) and Fouad et al. (2020). Such considerations also have to take into account that E2F1 is regulated at the posttranslational level via ubiquitination by the anaphase promoting complex/cyclosome (APC/C); for review, see Dubrez (2017).
The transcriptional regulation of EXOSC10 in normal somatic cells, cancerous cells, and germ cells is a potentially interesting, yet rather understudied problem; for review, see Stuparevic et al. (2021). Given the gene's ubiquitous expression profile, one might be tempted to assume that open chromatin in combination with a constitutive promoter should suffice to provide cells with appropriate levels of EXOSC10 during growth, development, and stress responses. However, given the various roles the exoribonuclease plays in many different somatic and reproductive tissues at pre- and postnatal stages of development, it might be worthwhile to explore the regulatory networks involved in the gene's expression.
To facilitate investigating human regulatory regions in hepatocarcinoma cells, and to allow for easily creating a graphical output of the data, we built the HepG2 Promoter Analysis Tool (HEPATo) using OpenAI's GPT-5 (ChatGPT) for R code syntax and development. This downloadable application (https://github.com/JulianPrim/HEPATo) combines ENCODE's candidate cis-regulatory elements (cCREs) with data from ChIP-Atlas (https://chip-atlas.org, as of September 2025) for transcription factors (TFs and others [33368 data sets]), epigenetic histone marks (histone [36073]), and chromatin accessibility (assay for transposase-accessible chromatin with sequencing, ATAC-seq [48822]; see Supplemental Fig. S2 for a brief HEPATo user guide) generated with a widely studied liver cancer cell line (HepG2 [1766]).
We used HEPATo to retrieve 182 transcriptional regulators and chromatin factors that bind the EXOSC10 locus (as annotated by Ensembl, www.ensembl.org [Harrison et al. 2024]; Supplemental File S1). Next, we employed a tool available via the String database to generate a physical subnetwork (based on experiments only and at medium confidence [0.400]) comprised of 37 DNA binding transcription factors (including E2F1 [Deng et al. 2025]), which belong to a wide variety of different families (Fig. 4A; Supplemental File S1). We then asked if any of these regulators were potential prognostic biomarkers for liver hepatocellular carcinoma (LIHC; the data were generated by UALCAN, Supplemental File S1) and identified a subnetwork of 11 transcription factors binding the EXOSC10 core promoter regions in vivo (Fig. 4B,C).
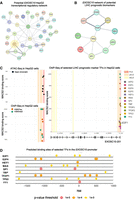
The liver regulatory network binding the EXOSC10 promoter. (A) A physical subnetwork generated using String is shown. Nodes are proteins, and edges represent direct binding. The edge's thickness indicates the level of confidence. E2F1 is highlighted in yellow. (B) A String subnetwork of 11 selected transcription factors (TFs) is shown as indicated at the top. (C) Diagrams generated with HEPATo plot genome annotation coordinates (x-axis) of EXOSC10 (encoding isoform 201) against the MACS2 binding score (−10 × log10 q-value; in the narrowPeak format from 0 to 1000), which is a significance metric that combines the level of enrichment magnitude with the variability of the background (y-axis). The values are not quantitatively comparable across independent SRX studies. We note that HEPATo plots midpoints and not the peak summit. Data from a chromatin accessibility assay (ATAC-seq, black dots) and a genome-wide DNA binding assay for epigenetic marks and transcription factors (ChIP-seq, colored dots) are shown as indicated at the top. E2F1 is included as a reference for promoter binding. Legends define the color code of a promoter-like signature (PLS) and proximal/distal enhancer-like signatures (pELS/dELS) as defined by ENCODE (www.encodeproject.org), two histone markers (red and green), and the color code of dots that represent the MACS2 values attributed to the TFs. (D) A schematic plots the genome coordinates (x-axis, genome assembly Hg38) against the color-coded positions of motifs for selected TFs as indicated (y-axis). The display window was set from −1000 to 100 bp relative to the EXOSC10 TSS, indicated by a light gray arrow. The JASPAR Core 2018 Vertebrates transcription factor motif library was employed for motif prediction in EPD. A legend indicates the color code of P-value thresholds (10−5 in red, 10−4 in orange, and 10−3 in yellow) for predicted motifs. Data exported from EPD and the code are available via the GitHub repository (https://github.com/JulianPrim/HEPATo).
The 5′-proximal promoter of human EXOSC10 is clearly defined by DNA accessibility and epigenetic marks associated with open chromatin (Fig. 4C). We note that highly transcribed promoters can yield artefactual DNA-binding signals called “phantom peaks” (Jain et al. 2015). Furthermore, so-called high occupancy target (HOT) regions containing atypically dense clusters of DNA binding motifs are often located within housekeeping promoters across species and can generate false-positive binding signals (Wreczycka et al. 2019). Consequently, ChIP-seq data must be interpreted with caution, especially at constitutively active promoters. To at least partially mitigate this issue, we employed the promoter motif search tool of the EPD (https://epd.expasy.org/epd/) that provides predicted binding sites upstream of and downstream from EXOSC10’s transcription start site (TSS) for eight prognostic TFs and E2F1 (Fig. 4D).
Next, we validated the predicted LIHC prognostic biomarker properties of these regulators using the RNA-seq pan-cancer panel of KM Plotter (https://kmplot.com) at default settings (Overall survival, auto select best cutoff) and confirmed the UALCAN predictions for six cases (Supplemental Figs. S3–S5): TBP is a TATA-box binding general transcription factor that mediates global gene expression and thereby influences cell proliferation and homeostasis; for review, see Johnson et al. (2003). E2F4 has a variety of roles as a corepressor and coactivator not only in cell cycle progression but also in stem cells and cancer, reviewed by Hsu and Sage (2016). E2F5 is a prognostic factor in esophageal squamous cell carcinoma and acts as a tumor suppressor in breast cancer cells (Ishimoto et al. 2013; To et al. 2024). Interestingly, TFDP1 forms a heterodimer with E2F family members and was found to promote the growth of liver tumor cells (Rogers et al. 1996; Yasui et al. 2003); for review, see La Thangue (1994). HEY1 is a Notch-signaling dependent repressor involved in embryogenesis, cell differentiation, and tissue homeostasis, and was suggested to be involved in liver tumor initiation (Lau et al. 2016); for review, see Weber et al. (2014). YY1 mediates gene expression via enabling promoter–enhancer interactions and might thereby help explain why EXOSC10 is expressed in such a wide variety of tissues at all developmental stages (Weintraub et al. 2017).
Further analyses of expression/survival data for TFs potentially regulating the expression of EXOSC10 in normal and cancer cells, followed by functional assays in cultured cells and in vivo models, are clearly warranted.
CONCLUSIONS AND FUTURE PERSPECTIVES
The conserved 3′-5′ exoribonuclease EXOSC10 is needed for normal mitotic growth and division of mammalian embryonic blastomeres, a variety of somatic cells and male/female germ cells. The human enzyme exerts its function predominantly in the nucleolus where it processes rRNAs to promote ribosome function, and under certain conditions that involve a posttranslational regulatory mechanism, EXOSC10 is able to shuttle into the nucleus to promote stress response gene expression. The protein also contributes to ribosome function and the regulation of long noncoding RNAs involved in recombination, DNA damage repair, and genome stability. These combined effects provide a compelling explanation of why high expression of EXOSC10 is typically associated with worse outcomes in multiple somatic cancers, and may pave the way for developing novel targeted therapies. Such approaches may involve detecting alleles that influence protein activity, localization, and network interactions in tumors, and targeting biological processes relevant for cancer progression that are positively regulated by EXOSC10 in a concentration-dependent manner. Finally, a better understanding of the regulatory networks that drive EXOSC10 expression in normal and various malignant cell types may lead to treatments that target DNA binding regulators important for the gene's transcriptional control.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Florence Demay in the laboratory and Igor Stuparević at the University of Zagreb for critical reading of the manuscript. We apologize to all authors whose data we were unable to refer to in this review owing to space restrictions. Our laboratory is in part funded by ANR (ANR-22-CE12-0038) to M.P. and also receives support from Inserm, the University of Rennes and EHESP.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080981.126.
-
Freely available online through the RNA Open Access option.
- Received January 30, 2026.
- Accepted February 6, 2026.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








