
SMDesigner: a program to design sequence mutations to assess RNA structure
- Corresponding author: jonathan.perreault{at}inrs.ca
-
Handling editor: Peter Stadler
Abstract
The structure of RNA is critical to its function. The advancement of structure prediction algorithms and deep sequencing technology has led to the discovery of numerous conserved RNA structures. However, functional analysis of these sequences is lagging behind the rate of novel RNAs’ predictions. Traditionally, mutations are designed to alter the structure of RNA and tested individually to assess function. We developed a program for the large-scale characterization of the structure/function relationship in multiple RNAs. Structure Mutation Designer (SMDesigner) automatically selects both disruptive and compensatory mutations according to inputted structural information. As proof of concept, we designed mutations for riboswitches with SMDesigner and experimentally assessed six of these riboswitches and their mutant sequences using an in-line probing assay to verify the effects on their structure and function. The in-line probing results show expected changes in five of six sequence structure patterns, confirming that SMDesigner can be useful to explore RNA structure and subsequent function. SMDesigner can be download at: https://github.com/lilihou/SMDesigner_0.1/tree/main/dist.
Keywords
INTRODUCTION
Noncoding RNAs (ncRNAs) have been found to have diverse molecular mechanisms and important biological functions. Indeed, small interfering RNAs (siRNAs), microRNAs (miRNAs) (Huntzinger and Izaurralde 2011), and piwi-interacting RNAs (piRNAs) (Aravin and Tuschl 2005) are all associated with RNA silencing, snoRNAs are involved in eukaryotic ribosome biosynthesis and other posttranscriptional RNA modifications (Kiss 2002), and riboswitches, which are noncoding regions of mRNAs, regulate gene expression by forming complex structures that bind to metabolites and other small molecules (Barrick and Breaker 2007; Garst et al. 2011; Serganov and Nudler 2013). The discovery and functional study of ncRNAs have increased our understanding of cellular processes and disease mechanisms. With the development of sequencing technologies and the inference of new ncRNAs with comparative genomics and bioinformatic tools, genome annotations have improved significantly and are becoming publicly available for an increasing number of species.
The systematic discovery of ncRNA structures has largely occurred through comparative genomics, which predicts the function of evolutionarily conserved sequences by looking at the functional correlation of these sequences across species (Rivas and Eddy 2001; Margulies et al. 2003; Alfoldi and Lindblad-Toh 2013). To accurately predict conserved ncRNA structures, it is important that the sequences from alignments display evolutionary variations (Pace et al. 1989; Rivas et al. 2017, 2020).
There are many search methods for ncRNA structures based on conservation of secondary structure, such as RNAz (Gruber et al. 2010), Evofold (Pedersen et al. 2006), CMfinder (Yao et al. 2006), which create covariance models (CMs) from multiple sequence alignments, and QRNA (Rivas and Eddy 2001).
By using these prediction tools, many structural ncRNAs have been discovered in different genomes. For example, the Breaker group discovered many structured ncRNAs, which include ribozymes and riboswitches in bacteria (Weinberg et al. 2007, 2010). In 2015, Li et al. (2015) found variant HDV self-cleaving ribozymes and atypical snoRNAs in fungal genomes. In the human genome, a variety of algorithms have been used for the discovery of structural ncRNAs. For example, in 2005, Washietl et al. (2005) used RNAz to discover 78 conserved noncoding elements across five mammals, while in 2006, Pedersen et al. (2006) found >40,000 candidate RNA structures across eight vertebrate species using Evofold. In 2008, Torarinsson et al. (2008) used CMfinder to find structural ncRNAs from 16 vertebrate genomes in the ENCODE project and found 6587 candidates. In 2011, Parker et al. (2011) used EvoFam to search for ncRNAs in 29 mammals and found 725 structures. In 2013, Smith et al. (2013) used RNAz and SiSSiz to make structural ncRNA predictions for 35 mammalian genomes and found >4 million RNA structures. In 2017, Seemann et al. (2017) used CMfinder to predict RNA structures from 17 mammalian genomes and found >500,000 conserved RNA structures, 23 of which were assessed through RT-qPCR and one through structure probing. In 2021, Hou et al. (2021) found >17,000 conserved structures in human by using CMfinder, two of which were assessed by RT-PCR.
The structure of ncRNAs is critical to their function (Cech and Steitz 2014; Fabbri et al. 2019; Ganser et al. 2019). Riboswitches are a good example of this as nucleotides need to be precisely positioned to form binding pockets that are highly specific for their cognate ligand (Barrick and Breaker 2007; Garst et al. 2011; Serganov and Nudler 2013). Similarly, ribozymes fold into complex structures that properly position a nucleophile and phosphate, leading to an RNA-catalyzed reaction and ensuring precise cleavage or splicing (Doherty and Doudna 2000). Another example of the importance of secondary structures is the specific recognition of pre-microRNA by Dcr-1 in flies through three structural features: 3′-overhang structure, a single-stranded loop, and correct stem size (Tsutsumi et al. 2011).
Targeted mutations can be used to understand the structure of ncRNAs and its impact on function. The presence of a base pair in the active structure can be tested by introducing mismatches; reduced activity as a result of these mutations lends supports to the importance of the RNA's structure for its function. The rescue of activity through the introduction of additional mutations to form alternate base pairs further supports this structure/function relationship (Anderson et al. 1994; Fedor 2000). This strategy has been used for the study of many classes of ncRNAs. Anderson et al. (1994) introduced different mutations to study the secondary structure of the hairpin ribozyme. Li and Breaker (2013) made structural disruption and restoration mutations to prove that the α–α′ stem structure formation is necessary for NCU01977 TPP riboswitch's alternative splicing regulation function. Thalalla Gamage et al. (2022) made disruptive and rescue mutations in stems I, II, III, and IV of SNORD13 to demonstrate how its secondary structure affects its processing and stability.
Although a growing number of new ncRNA structures have been discovered, their function remains unclear (Kalvari et al. 2021). Affordable synthesis of oligonucleotide library pools allows for the study of their function on a large scale. This technology was used in various instances to allow large-scale screens, for example, with reporter constructs (Strayer et al. 2024). At the same time, this technology gives a chance to characterize these newly discovered structures and investigate their functional roles in a high-throughput manner. The ability to synthesize pools containing more than 100,000 unique DNA oligos enables not only the genome-wide identification of noncoding DNA elements, such as identification of regulatory elements in conserved 3′ untranslated regions (3′ UTRs) (Oikonomou et al. 2014), but also the investigation of RNA function, such as identification of RNA nuclear enrichment sequences (Shukla et al. 2018) and of RNA-mediated features of RNA–protein interactions using parallel RNA assays (Lee et al. 2024). However, a key advantage of synthetic libraries is the ability to introduce mutants to assess the structure/function relationship. Mutations that disrupt base pairs combined with those that rescue these base pairs are the gold standard to confirm ncRNA structure and connect it with function (Fig. 1). Traditionally, such mutations have been created manually for select structures. However, such a design strategy for large libraries of structured RNAs is not a viable option.
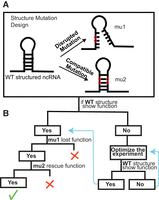
Gold standard to verify structured ncRNA function. (A) Structure mutation design of SMDesigner. Circles mark base pairs selected to make mutations: black for wild-type (WT) nucleotides and red for mutated nucleotides, which make disrupted and compatible mutations. (B) Analysis overview for structure assessment. Green tick mark indicates an assay that confirms the importance of the structure for the assessed function; red X indicates that the importance of structure for the function is not confirmed and likely infirmed. In most cases where no function is observed for WT (right), even after optimization, the mutants designed by SMDesigner will not help assess the importance of the structure for the RNA's function.
Many bioinformatic methods exist to analyze the effects of mutations on RNA secondary structure, such as RNAmute (Churkin and Barash 2006), SNPfold (Halvorsen et al. 2010), and RNAsnp (Sabarinathan et al. 2013a,b). These methods, along with others like remuRNA (Salari et al. 2013) and RNA2Dmute (Moss 2018), employ energy minimization techniques to predict and design mutant sequences for structured RNA. RNAmute predicts how single mutation affects RNA secondary structure, while SNPfold and remuRNA focus on the ensemble of RNA secondary structure changes. RNAsnp analyzes the effects on local structural change, and RNA2Dmute evaluates all possible single mutation effects on the predicted 2D structural ensemble.
Most of these tools evaluate the effects of mutations on predicted RNA structures from a single sequence and can generate mutant candidates aimed at disrupting specific structural features. However, they often lack support for identifying complementary rescue mutations to restore disrupted structures. While Barash and Gabdank (2010) employed energy minimization methods to design both disruptive and rescue mutations for TPP riboswitches, SMDesigner, proposed here, focuses on structured RNA mutation design based only on sequence alignment information. These different methods offer a comprehensive toolkit for RNA mutation analysis and design, each addressing specific needs and scenarios.
We developed SMDesigner, a program to automate mutation design for structured RNAs based on conserved structural features identified using covariance models. All conserved RNA structures with covariance model alignment information can utilize SMDesigner to design both disruptive and compensatory mutations, such as structures from Rfam (Kalvari et al. 2021). While Rfam is the most suitable database for SMDesigner, for the mutation design of riboswitches, other databases, such as RiboD (Mukherjee et al. 2019), RSwitch (Penchovsky et al. 2021), and Ribocentre-switch (Bu et al. 2024), are also useful as they incorporate structure predictions derived from both energy minimization and covariance model alignment.
We used SMDesigner to design mutations for a pool of riboswitch sequences and experimentally confirmed the expected structure disruption and restoration for the relevant mutants of several of these riboswitches. We also confirmed cognate ligand binding for some of these riboswitches in WT and compensatory mutants, but not for disruptive mutants. This study demonstrates that SMDesigner can be used to make mutations which can disrupt or rescue the secondary structure and evaluate if the structure is important to its function.
RESULTS
Gold standard to verify structured ncRNA function
The secondary structure of ncRNAs is critical to their function. One way of studying this relationship is to design mutations that will disrupt their structure and assess their impact on RNA function (Fig. 1). This also allows us to assess the validity-predicted structures. Given structured RNAs have been discovered through comparative genomics, the predicted structure is often the only information we have about a given RNA. Thus, it can become challenging to design experiments that will assess function. As shown in Figure 1, mutations that replace base pairs with mismatches will disrupt the structure and are expected to result in loss of function, while additional mutations that will form alternate base pairs are expected to rescue function.
SMDesigner workflow
We developed SMDesigner to automate design of mutations for structured RNAs (Fig. 1A). SMDesigner selects positions that maximize the potential impact on secondary structure while minimizing alterations to the primary sequence. Figure 2 shows a complete example of SMDesigner's workflow using the EEF1A2 motif and shows the input and output sequences. The input file is a Stockholm RNA sequence alignment with structural information. SMDesigner uses R2R to analyze RNA secondary structures and draw the consensus diagram (Fig. 2A, left). The program selects the most conserved stem, based on the number of covarying base pairs, and then chooses covarying positions closest to the middle of the stem to maximize its disruption (Fig. 2A, right). In this case, we introduced “mu1” mutations (two base pairs by default) to disrupt the ncRNA function and rescued the structure with “mu2” mutation, to restore the function to be similar to the WT RNA sequence (Fig. 2B, left). This is performed for each sequence in the alignment. We output designed sequences in a FASTA file (Fig. 2B, right).

SMDesigner input/output. (A, left) Secondary structure drawn by R2R; (A, right) portion of alignment motif EEF1A2 in Stockholm format. The “#=GC SS_cons” is the line describing secondary structure. (B) The output is generated for all sequences from the alignment. (Left) Secondary structure with two types of mutations; (right) portion of output sequences for Seq2 from the alignment in A. The output sequences corresponding to all the input alignment sequences are contained in the same FASTA file. The stars mark the base pairs located at the center of the stem chosen for mutation. The “mu1” is the disrupted mutant with the broken base pairs, and the “mu2” is the mutant with the restored base pairs. The boxed red nucleotides are altered nucleotides according to the mutation types.
Experimental validation of disrupted and rescued riboswitch structures
We designed mutant sequences for riboswitches annotated in Rfam (Kalvari et al. 2021) and selected six of them to test using in-line probing to demonstrate how structure and ligand binding are altered. While ligand binding alone does not represent the full functionality of riboswitches, which are also essential for transcriptional and translational regulation, the binding event often triggers specific structural shifts in the RNA. These conformational changes provide direct evidence of the riboswitch's structural and functional response to the ligand, forming a critical component of its regulatory mechanism.
SAM-III, located in the 5′ UTR of the metK gene of Enterococcus faecalis, is known to regulate translation by binding with S-adenosyl methionine (Majumder et al. 2022). SAM-III has three conserved stems (Fig. 3A) and binds directly with SAM at G18, G50, U51, A52, and G70 (Fig. 3A, bottom, “SAM” link; Lu et al. 2008). We took one sequence from Enterococcus sp. 3H8_DIV0648 (Fig. 3A, bottom) to verify whether the designed mutations have the desired outcome (i.e., that the mu1 mutation disrupts structure and ligand binding and that the compatible mutation [mu2] rescues the structure and ligand binding). As described, SMDesigner picked mutation base pairs located at the center of the stem and base pairs which have the most covariation (Fig. 3A, top, stars). The structure pattern from in-line probing indicates that the disruptive mutation (mu1) disrupted the structure while the compatible mutation (mu2) rescued the structure (Fig. 3B, circles). Moreover, when mapping some of the more prominent bands from the gel (Fig. 3B, circles) onto the structure, we can clearly see that both WT and mu2 fit the known SAM-III riboswitch structure with accessible degradation sites in predicted single-stranded regions. In contrast, positions corresponding to stem P2.2 are clearly accessible for degradation in the mu1 sequence (Fig. 3A,B, red circles). As for ligand binding, we can see similar band modulation patterns with increasing ligand concentrations (black or green square) between the wild-type and mu2 sequence, but not in the mu1-disrupted riboswitch. More experiments were performed to confirm these results and to evaluate the dissociation constant (Supplemental Fig. S1), with values of ∼0.5 µM for the WT and mu2 sequences, similar to KD known for other SAM riboswitches.

SAM-III riboswitch. (A, top) The consensus structure of the SAM-III riboswitch, see legend from Figure 2A. (A, bottom) The secondary structure of the sequence from Enterococcus sp. 3H8_DIV0648 used in in-line probing assay. The green nucleotides represent wild-type nucleotides, while the red nucleotides indicate the mutated nucleotide. The different colored circles show the structural changes among three different constructs, mapped from the gel result in B. (B) In-line probing gel patterns from the three different constructs of SAM-III and the modulation changes in the presence of SAM. The different colored rectangles indicate ligand-induced modulation. The arrows mark the positions used to calculate the KD. The stars mark the mutation positions. The black solid lines mark the stems of the secondary structure. The circles indicate the strongest bands corresponding to structural change among the wild-type, mu1, and mu2 constructs.
PreQ1-I riboswitch controls the translation of genes involved in queuosine biosynthesis and transport by binding to preQ1 (Kang et al. 2009; Klein et al. 2009; Eichhorn et al. 2014). We chose one sequence from Bacillus manliponensis to test whether the mutation designed by SMDesigner would have the expected effects as seen with SAM-III. The structural patterns observed from in-line probing assays agree with mu1 riboswitch disruption and mu2 rescue. We can see similar patterns from WT and mu2, but not from the disruptive mutation (mu1), which has a different pattern (Fig. 4B). Similarly, the WT and compensatory mutation (mu2) show ligand binding, as we see band modulation with increasing preQ1 concentrations, as indicated by rectangles in Figure 4B, while the disrupted mutant lost its binding ability. Indeed, the KD could be calculated for WT and mu2 (0.45 ± 0.18 µM and 0.32 ± 0.12 µM, respectively), and both KDs are close to the one reported for Fusobacterium nucleatum preQ1-I riboswitch (0.36 ± 0.02 µM) (Wintermans et al. 2024).

PreQ1-I riboswitch. (A, top) The consensus structure of preQ1-I riboswitch. (A, bottom) The secondary structure of the sequence from Bacillus manliponensis, which was used for in-line probing. Q1 refers to preQ1 molecule. (B) In-line probing gel patterns from the three different constructs of preQ1-I and the modulation changes in the presence of preQ1. The different colored rectangles indicate preQ1-induced modulation. The different colored circles indicate structural changes in the disrupted mutant. The arrows mark the positions used to calculate the KD in C. (C) Plot of the fraction of RNAs whose cleavage sites (marked in B with arrows) are modulated with increasing concentrations of preQ1. No curve could be inferred for mu1 KD calculation, as would be expected if preQ1 binding is abolished. Other legend details are as in Figure 3. We repeated the in-line probing assay with similar results (Supplemental Fig. S2). Note that the first concentration of preQ1 tested is not 10−7 as indicated, but rather 0, and this allows us to plot these data points on a log scale (note that we test two “0” for each RNA).
As for preQ1-I, the preQ1-III riboswitch regulates genes by binding with preQ1 in bacteria (Supplemental Fig. S3A; Liberman et al. 2015), but it has three conserved stems and two alternative stems. In-line assays with a sequence from Faecalibacterium prausnitzii (F. prausnitzii) (Supplemental Fig. S3A, right) showed both the expected structural changes and ligand-binding ability (Supplemental Fig. S3B) with KDs of 12.7 ± 4.8 nM for WT and 9.6 ± 0.96 nM for mu2 (Supplemental Fig. S3C). Again, no KD could be calculated for the disruptive mutation (mu1).
We also performed assays with two guanidine-I riboswitches (sequences from Burkholderia sp. and Methylorubrum extorquens). The results of in-line probing show the expected structural patterns (Supplemental Fig. S4). However, none of the constructs showed guanidine-induced modulation. In contrast with all other tested sequences, the three guanidine-III riboswitch constructs assayed showed very different structural patterns (Supplemental Fig. S5). While mu1 disrupted the structure as expected, mu2 did not restore it and, in fact, led to an alternative structure.
Five of six tested riboswitch sequences showed the expected structural changes with disruptive mutations leading to apparent modification of secondary structure and the compensatory mutations leading to structure rescue (Table 1). Moreover, the three riboswitches for which we confirmed function by assessing their ability to bind their cognate ligand displayed the expected functional changes, notably disruptive mutations led to loss of the ability to bind its ligand while the compensatory mutations restored the ligand binding. However, as indicated in Figure 1, if no function is observable in the WT sequence, in principle we cannot infer whether the structure is involved in the function. This is the case of the guanidine riboswitches for which the tested WT sequences did not modulate and thus for which no change in modulation could be observed for disruptive and compensatory mutations.
Summary of the experimentally assessed structure and function changes of the riboswitch sequences designed by SMDesigner
DISCUSSION
All disruptive mutations altered the RNA structure, while compensatory mutations rescued all the mutants with the exception of the guanidine-III riboswitch. One reason might be that the mutated positions can affect the tertiary structure as well. Indeed, the positions mutated in the guanidine-III riboswitch participate in a triplex (Supplemental Figs. S5 and S6; Huang et al. 2017). Positions within stems do not usually participate in tertiary interactions. This highlights the fact that, in spite of careful evaluation of the alignment, ultimately these designs rely on assumptions that are not always correct. Thus, as with any other prediction or design software, we should always remain critical of results. Even when carefully evaluating structures with folding software, results may vary. We have evaluated all the WT and mutant sequences for the six riboswitches that we tested and found that most predictions did fit the expected aptamer structure (Supplemental Figs. S7–S9) with the exception of one case where our results supported the expected aptamer structure for the compatible mutant (mu2), in spite of the fact the RNAfold predictions did not (Supplemental Fig. S10).
Another reason that could explain the guanidine-III results could have been that the wild-type construct did not form the correct structure. This may be the case for the guanidine-I riboswitch sequences that do not show structure changes. Riboswitches are regions of mRNAs that control gene expression; thus, when performing experiments, only a small region of the mRNA is used to probe structure. This region encompasses the conserved aptamer domain, but for optimal structure modulation some portions of the expression platform may not be useful, thus requiring preparation of different constructs. Our preQ1 and SAM constructs have shown modulation, but not those for guanidine-I and III riboswitches. While a thorough study of a given riboswitch may warrant assays with a dozen different constructs, we assayed only up to five constructs for each chosen riboswitches, and we thought that having five riboswitch examples with structure rescue and three riboswitch examples with observable modulation would suffice. Nevertheless, our results support that SMDesigner was successful in designing disruptive and compensatory mutations for these examples, and, by extension, for any type of structured RNA; it is not limited to riboswitches.
Many studies use randomized libraries to analyze relatively simple motifs, such as RNA aptamer, which can bind with specific molecules, like ATP (Sassanfar and Szostak 1993), GTP (Davis and Szostak 2002), and proline-rich RNA-binding protein (Hori et al. 2005), but these approaches are less suitable for evaluating more complex RNA structures. To study putative natural RNA structures, SMDesigner can be a valuable tool to design sequences to be synthesized in oligonucleotide pools that will allow for the study of function. For instance, Strayer et al. (2024) employed a synthetic oligonucleotide library of 11,088 sequences to investigate the regulation of potential cis-regulatory elements in translation. By introducing mutations, they simultaneously explored the effects of both sequence and secondary structure on translation efficiency. While their structural mutation design differs from SMDesigner, a similar approach was adopted in the RNA–protein interaction research of Lee et al. (2024), where structure-disrupting and compensatory mutations were designed to elucidate protein-binding secondary structure within a single experiment.
Conclusion
The aim of SMDesigner is to characterize the structure/function relationship of structured RNAs. It can be used to design structural mutants to study a single structure from a Stockholm alignment but is particularly useful for the design of such mutants for large libraries to screen structured RNAs. When exploring the function of one structured motif, most mutations are selected manually. However, with the major advances and improved cost-effectiveness of DNA synthesis services, a growing number of researchers are turning to synthesized oligonucleotide pools for their experiments. When exploring the function of structured motifs at large scale, an automated mutation design method becomes essential. The advent of RNA-seq has changed the study of gene expression, where RNA becomes a window to allow us to peer into living organisms at transcriptome-wide and single-cell levels. RNA-seq has also given us opportunities to study RNA structure on a large scale (Siegfried et al. 2014; Madden et al. 2020). Bioinformatics is essential to analyze data derived from such high-throughput methods. Yet, one of the most prominent contributions of bioinformatics to our understanding of RNA, and life in general, comparative genomics, has mostly been used to help define phylogeny and, in the case of RNA, predict conserved structural elements. Until now, the study of these elements was limited either to the study of RNAs individually or to simple metrics (is the RNA expressed or not). The combination of large-scale DNA synthesis and RNA-seq now makes it possible to compare WT and mutant sequences at unprecedented scales. We think SMDesigner could become instrumental in this endeavor and could help define functions for several putative ncRNAs, contributing to the next generation of functional genomics.
SMDesigner can help assess whether certain RNA structures are “real” and conserved for a function, using ligand binding as a proxy here. It can also be used to challenge the role of a given structure in an observed “function.” However, it cannot be used to definitively assess the absence of function (Fig. 1). While SMDesigner provides a scalable approach to disrupt and rescue mutation design for structured RNAs, it is important to acknowledge its limitations. The accuracy of predicted mutations relies heavily on the reliability of the alignment and predicted secondary structure, which may not always align with experimental results. Additionally, mutation sites are selected based on simple rules, which may not fully capture the complexities of RNA structure dynamics. SMDesigner's current applicability is also limited to structures based on covariance models and a predefined number of mutations, thus preventing the assessment for structures derived from a single sequence. Depending on the needs of users, future versions might include features such as allowing users to choose the number of mutations and include structural change evaluation for single-sequence structures. Despite these limitations, SMDesigner is a valuable tool for RNA structure and functional studies when used alongside experimental validation or advanced computational tools.
MATERIALS AND METHODS
Installation
This program has been packaged with a setup tool; you can find and download the package at https://github.com/lilihou/SMDesigner_0.1/tree/main/dist and use “pip install <package name>” to install the program on your computer. This program needs external software, R2R; if you do not have it, when you first run this program, it will install it automatically. If you already have R2R installed on your computer, you can install this program with pip and run it by typing <SMDesigner>. You can also produce single-sequence structures using: SMDesigner --run-r2r. SMDesigner has been tested and is compatible with Linux, macOS Sequoia, and Windows Subsystem for Linux or Cygwin on Windows. For details on how to install and use this program, please refer to README.md and README_INSTALL.md (https://github.com/lilihou/SMDesigner_0.1/blob/main/).
Implementation
This program is used to design mutations that are likely to disrupt the structure of covariance models for structured RNAs and can be applied to large sequence libraries. The source code is freely available at https://github.com/lilihou/SMDesigner_0.1. The SMDesigner program requires an input file consisting of multiple RNA sequence alignments with a consensus structure in Stockholm format. By default, SMDesigner uses another program R2R (Weinberg and Breaker 2011) to list the conserved base pairs within the stems from the structure alignment, more specifically, positions with covariation and compatible sequence variation.
The goal of SMDesigner is to create mutants to assess whether a predicted structure is important for function using mutations that disrupt a stem. It then makes a second mutation that restores that stem. We output multiple sequence files for wild-type (WT) and two mutant sequences in FASTA format. Two examples are given to show how SMDesigner selects mutation base pairs (Supplemental Fig. S11).
Algorithm: stem selection
Many structured RNAs have more than one stem with covarying base pairs. To select the most relevant stem, we evaluate the number of covariations, compatible variations, and conserved base pairs within each stem (Supplemental Fig. S11). The primary criterion is to choose the stem with the highest number of covariation base pairs. In the absence of covariation base pairs in the structure, the stem with the highest number of compatible variations is chosen. If the structure lacks both covarying and compatible base pairs, then we select the stem with the highest number of conserved base pairs. Structures submitted to SMDesigner will typically only be predictions, supported by conservation data, so this ensures that we choose the stem that is most likely to be accurate and essential for the function of the RNA structure. In cases where multiple stems contain an equal number of covariation base pairs, the longest stem will be chosen. SMDesigner requires that the structure has at least two base pairs in at least one stem.
Algorithm: selection of base pairs for mutations
Once the stem is chosen, if there are more than two covarying/compatible/conserved base pairs in the chosen stem, we need to select which base pairs will be used to make mutations. SMDesigner will select two base pairs closest to the center since they contribute more to stem stability and thus have a better chance of affecting RNA function (Supplemental Fig. S11C).
Algorithm: mutation designs
The consensus alignment structure is used to select which base pairs will be mutated, but mutations are designed for each sequence (Supplemental Fig. S11B,D). SMDesigner makes two types of mutations: a disruptive mutation that will prevent correct stem formation (mu1), and a mutation that keeps the mu1 mutation but restores the base pairs with a compensatory mutation on the other side of the stem. For example: a G-C base pair in the wild-type sequence would become C-C for the disruptive mutation and C-G for the compensatory mutation.
Assembly PCR
DNA templates of test sequences were amplified by assembly PCR, described in the study by Stemmer et al. (1995). For each sequence, we designed and synthesized three to four oligonucleotides including a T7 promoter sequence. The overlap between oligonucleotides is ∼20 nucleotides. The web server Primerize (Tian et al. 2015) was used for these designs. A list of full sequences and oligonucleotides is available in Supplemental Table S1.
In vitro transcription
The Assembly PCR products were used as templates to make RNA through in vitro transcription as described in Nielsen (2011). The product was purified with polyacrylamide gel electrophoresis (PAGE) (8% acrylamide and 8 m urea) gel. The concentration of purified RNA was quantified with a NanoDrop 2000C (Thermo).
Dephosphorylation
Around 20 pmoles of RNA were used for dephosphorylation with Antarctic Phosphatase (NEB) according to manufacturer's instructions.
5′ 32P end labeling RNAs
Half of the dephosphorylation reaction mix was used for 5′ end labeling with γ-32P ATP and T4 PNK (NEB) according to manufacturer's instructions (see details in Regulski and Breaker 2008). The labeled RNA was purified with PAGE (8% acrylamide and 8 m urea) gel.
In-line probing
5′ 32P-labeled RNAs were unfolded for 80 sec at 80°C and incubated with or without ligands for 35–45 h at room temperature in 50 mM Tris pH 8.3, 20 mM MgCl2, and 100 mM KCl. Three controls, RNase T1 (which cut at G positions), partial alkaline (OH)–digested RNA (cut at each nucleotide), and the nonreacted RNA (NR) were prepared in parallel (Regulski and Breaker 2008). The RNA products resulting from spontaneous cleavage during in-line, and controls, were loaded on denaturing (8 m urea) 8% polyacrylamide gel with 2× loading buffer (10 mM EDTA in formamide, a pinch of xylene cyanol, and a pinch of bromophenol blue). RNA was migrated on gel at 60 W for 2–3 h, and the gel was dried for 2 h at 75°C. It was then exposed in a Phosphor Imager cassette overnight, imaged with a Typhoon FLA9500 (Cytiva), and analyzed with ImageQuant (Cytiva).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank members of the Perreault laboratory for helpful comments and the following for financial support: China Scholarship Council, Natural Sciences and Engineering Council of Canada (NSERC) (RGPIN-2019-06403), Canadian Institutes of Health Research (CIHR) project, and Génome Québec.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080267.124.
- Received September 18, 2024.
- Accepted March 17, 2025.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Lijuan Hou is the first author of this paper, “SMDesigner: a program to design sequence mutations to assess RNA structure.” Lijuan is a PhD student in Professor Jonathan Perreault's laboratory at the Institute National de la Recherche Scientifique. Her research focuses on the conserved RNA structures.
What are the major results described in your paper and how do they impact this branch of the field?
In our paper, we developed a tool for large-scale mutation design of conserved RNA structures based on structure alignment information. This tool enables researchers to assess predicted secondary structures and the effects of these structures on RNA function. With the decreasing costs of synthesized oligonucleotides and advancements in deep sequencing technologies, our tool facilitates the simultaneous evaluation of structured RNA and exploration of its functional implications.
What led you to study RNA or this aspect of RNA science?
My journey into RNA science began during my undergraduate studies when I took a course on molecular biology. I was fascinated by the complexity of RNA molecules and their roles in gene expression and regulation. As I progressed through my master's and PhD programs, my interest deepened further as I explored noncoding conserved RNA structures and their significance in understanding various biological functions.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
The most landmark moment in my scientific journey was when I embraced the uncertainty inherent in the scientific process. I learned to appreciate the challenges and complexities of problem solving. This shift in mindset allowed me to enjoy my research more and to feel more composed when facing obstacles.
If you were able to give one piece of advice to your younger self, what would that be?
My advice to my younger self would be to have faith in yourself and your journey. It's okay to slow yourself down during this journey.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
The individuals who have significantly influenced my philosophy and approach to science are my two supervisors, Professor Li and Professor Perreault. Professor Li opened the door for me to the fascinating world of RNA science, especially regarding the beauty and power of RNA structures. Professor Perreault played a crucial role in my development as a scientist. He provided me with opportunities, invaluable guidance, and support, which helped me build confidence in my abilities and grow as a skilled researcher.








