
Depurination of sarcin/ricin loop 25S rRNA is signaled through the small ribosomal subunit during translation
- Corresponding author: hudak{at}yorku.ca
-
Handling editor: Marina Rodnina
Abstract
In addition to their function in protein synthesis, translating ribosomes serve as sensors that communicate the presence of aberrant messenger RNAs (mRNAs); however, how they recognize damage to their ribosomal RNA (rRNA) remains poorly understood. The conserved sarcin/ricin loop (SRL) of the 25S rRNA is a component of the GTPase center essential for ribosome movement during translation. In this study, we expressed an RNA N-glycosylase called pokeweed antiviral protein (PAP) in yeast Saccharomyces cerevisiae to specifically damage rRNA by hydrolysis of a purine base from the SRL. 25S rRNA depurination inhibited translation elongation, as shown by reduced incorporation of a methionine analog and binding of eukaryotic elongation factor 2 (eEF2) to ribosomes. PAP expression altered sucrose gradient profiles, increasing free subunits and 80S peaks and reducing polysomes without causing ribosome collisions. We discovered depurinated rRNA associated with 80S monosomes and polysomes, suggesting that cells would detect damage to rRNA during active translation. These ribosomes were ubiquitinated by E3 ligases Mag2 and Hel2, elements of the 18S nonfunctional rRNA decay (NRD) pathway involved in recognizing slow-moving ribosomes. Furthermore, mass spectrometry analysis revealed ubiquitination of ribosomal protein uS3, characteristic of 18S NRD. Even though the SRL is a component of the large ribosomal subunit, its depurination is signaled by ubiquitin ligases that recognize damage to the small subunit. We suggest that slow translation elongation is the factor that communicates SRL depurination to E3 ubiquitin ligases, which extends our understanding of how rRNA integrity is surveilled in yeast.
Keywords
- sarcin/ricin loop
- pokeweed antiviral protein
- rRNA depurination
- ribosome quality control
- nonfunctional ribosome decay
INTRODUCTION
Translational control of gene expression plays a central role in cellular homeostasis. Recent advances describe how ribosomes monitor translation and signal defects in this process that influence cell fate and survival (Kim and Zaher 2022; Monaghan et al. 2023; Ford et al. 2024; Inada and Beckmann 2024). Most of our knowledge of these responses comes from investigating effects of mRNA characteristics on translation in yeast. For example, mRNAs with nonoptimal codons, secondary RNA structures, or chemical modifications, such as alkylation or oxidation, will slow the rate of elongation, either by transiently pausing or stalling ribosomes, with the latter potentially leading to ribosome collisions (Doma and Parker 2006; Letzring et al. 2010; Simms et al. 2014; Yan and Zaher 2021; Veltri et al. 2022). Depending on the frequency and nature of these ribosome states, different surveillance pathways may be triggered. Stalled or collided ribosomes are recognized by ribosome quality control (RQC) pathways that are generally signaled by regulatory ubiquitination of 40S ribosomal proteins by the E3 ubiquitin ligase Hel2, leading to mRNA decay, nascent peptide degradation, and ribosome subunit recovery (Matsuo et al. 2017; Simms et al. 2017; Ikeuchi et al. 2019; Best et al. 2023; Miścicka et al. 2024). Ribosome stalls or collisions that persist and overwhelm RQC pathways can elicit an integrated stress response (ISR) (Yan and Zaher 2021). This response is best described for starving cells deficient in amino acids, and hence charged tRNAs required for translation (Hinnebusch 2005). ISR activation downregulates global translation initiation via phosphorylation of the translation factor eIF2α (Hinnebusch 1984; Dever et al. 1992; Krishnamoorthy et al. 2001). In addition to reducing further rounds of ribosome collisions by inhibiting initiation, eIF2α phosphorylation increases translation of Gcn4, a factor that enhances transcription of stress-responsive genes (Natarajan et al. 2001; Harding et al. 2019; Inglis et al. 2019).
Defects of the ribosome itself, such as mutations introduced to the 18S or 25S rRNA that impact either the decoding or the peptidyl transferase center, respectively, are also subject to ubiquitin-dependent quality control. 18S nonfunctional rRNA decay (18S NRD) results from monoubiquitination of the 40S subunit by the E3 ligase Mag2, and subsequent polyubiquitination by the E3 ligase Hel2 or Fap1, depending on ribosome state (Sugiyama et al. 2019; Li et al. 2022; Matsuo and Inada 2023). Specifically, Fap1 targets defective ribosomes stalled at initiation codons and does not bind to collided ribosomes (Li et al. 2022; Matsuo and Inada 2023). Studies on 25S nonfunctional rRNA decay (25S NRD) in yeast with mutant 25S rRNA have shown that E3 ligase Rtt101 targets the 60S subunit for polyubiquitination (Fujii et al. 2009). The importance of Rtt101 in 25S NRD is well documented although the ribosomal protein that it ubiquitinates is currently unknown. Activation of NRD results in ribosome dissociation, providing subunits as substrates for proteasomal degradation required for associated rRNA decay (Fujii et al. 2012).
Though considerable insight into downstream signaling has been gained from ribosomes with designed mutations to rRNA, our knowledge of the impact of damage to wild-type ribosomes during ongoing translation is limited. Perhaps the most evolutionarily conserved sequence of RNA belongs to the sarcin/ricin loop (SRL) of 25S rRNA, named after toxins that target this short sequence (Endo et al. 1983; Endo and Tsurugi 1987). The SRL is an essential component of the ribosomal GTPase center and is required for binding of translation elongation factors and facilitating ribosome movements during translation (Voorhees et al. 2010; Maracci and Rodnina 2016; Das et al. 2023; Cheng et al. 2025). Modification of this loop, either by endonucleolytic cleavage or removal of bases, inhibits translation in vitro (Brigotti et al. 1989; Osborn and Hartley 1990); however, the signaling arising from this damage has not been well characterized. In this study, we expressed pokeweed antiviral protein (PAP), a plant-derived RNA N-glycosylase that depurinates the SRL in Saccharomyces cerevisiae (Mansouri et al. 2006; Di and Tumer 2015). We show that ribosomes with depurinated 25S rRNA are tagged by ubiquitination during active translation by factors that recognize slow-moving ribosomes. Curiously, ubiquitination occurs on a 40S ribosomal protein; therefore, damage to the SRL loop is communicated through the small ribosomal subunit even though depurination occurs within the large subunit.
RESULTS
PAP depurinates 25S rRNA
To identify how yeast cells detect damage to rRNA, we first conditionally expressed PAP and monitored evidence of its enzyme activity on the SRL of 25S rRNA. The BY4741 strain was transformed with plasmids encoding PAP, its inactive mutant PAPx, or an empty vector (EV). PAPx bears a single amino acid substitution (E176V) at its active site and was used as a negative control for wild-type PAP enzyme activity (Hur et al. 1995). Expression of both proteins was under the control of a Gal-1 promoter and immunoblot analysis of total cell lysate illustrated PAP and PAPx accumulation over the 6 h time course (Fig. 1A). Three bands were visible, corresponding to the expected mature (29 kDa), precursor (35 kDa), and partially processed (33 kDa) forms of PAP. PAP is naturally processed by cleavage of 22 and 29 amino acids from its N and C termini, respectively, to produce the mature form (Hur et al. 1995).
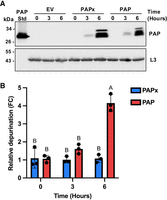
PAP accumulates in WT BY4741 cells and depurinates 25S rRNA. (A) Time course of PAP expression from WT BY4741 cells transformed with cDNAs of PAP, PAPx, or EV at 0, 3, and 6 h postinduction with 2% galactose. Total cell lysates (60 µg) were separated through 12% SDS PAGE, transferred to nitrocellulose and probed with a PAP-specific antibody (1:10,000). Purified PAP (29 kDa) was used as a standard. RpL3 (43 kDa) was detected using an antibody against RpL3 (1:5000) as a loading control. The blots shown are representative of three independent biological replicates (n = 3). (B) Depurination levels of SRL upon PAP and PAPx expression at 0, 3, and 6 h postinduction with 2% galactose. Total RNA (500 ng) was isolated from cells, and RT-qPCR was performed to measure “reference” and “target” cDNA levels as illustrated in Supplemental Figure S1. Fold changes for PAP and PAPx are shown relative to EV. Differences in depurination levels were tested for statistical significance by two-way ANOVA with Tukey's multiple comparisons test. Bars represent means ± SEM of three independent biological replicates (n = 3). Different letters on bars represent statistically significant differences at P < 0.05.
To assess the enzyme activity of PAP, we performed a rRNA depurination assay based on RT-qPCR (Supplemental Fig. S1). The assay relies on the binding of two primer pairs, one flanking the depurination site of the SRL (target) and the other at the 5′ end of the 25S rRNA (reference) where depurination would not occur (Pierce et al. 2011). If the SRL was depurinated, the reverse transcriptase would stall at the abasic site and produce a truncated cDNA, preventing subsequent PCR. The amount of PCR product measured from the 5′ end reference primers served as a control for total 25S rRNA. Therefore, we used the ratio of target relative to reference PCR product as a measure of PAP activity. The levels of rRNA depurination increased during the 6 h induction of PAP expression (Fig. 1B). Based on absolute copy number data, we quantified the percentage of ribosome depurination and found that 59% ± 2.82 (SEM) of ribosomes were depurinated in PAP-expressing yeast at 6 h postinduction compared with 5% ± 1.89 (SEM) for PAPx-expressing cells. PAPx was previously described as an inactive mutant (Hur et al. 1995), and we attribute its current low level of activity to the sensitivity of our assay. We continued to use PAPx in comparison to PAP, to evaluate the extent of depurination needed to observe a biological effect. Our results show that PAP significantly depurinated rRNA when expressed in yeast for 6 h.
To test whether PAP also depurinated other RNAs during our time course in yeast, we performed a series of northern blots using probes that specifically detected 18S, 5.8S, and 5S rRNA, in addition to 25S rRNA, along with a selected tRNA (tyrosyl-tRNA) and mRNA (ribosomal protein L3). Total RNA was isolated from cells expressing PAP, PAPx, or EV for 6 h, and equal amounts of RNA (10 µg) from each sample were either treated with aniline or left untreated. As PAP is a glycosylase rather than a nuclease, it will remove a base from RNA, leaving the phosphoribose backbone intact. Subsequent treatment with aniline will cleave the RNA at the abasic site, allowing fragments to be visualized through denaturing gel electrophoresis (Bass et al. 1992; Hudak et al. 1999; Küpfer and Leumann 2007). We first examined the general integrity of total RNA separated through denaturing agarose gel and stained with SYBR Gold. The migration pattern of RNA prior to aniline treatment was similar for samples from PAP-, PAPx-, and EV-expressing cells and illustrated the expected bands for 25S, 18S, 5.8S, and 5S rRNAs (Supplemental Fig. S2A). Examination of samples following aniline treatment showed two fragments consistent in size with cleavage of 25S rRNA at the sarcin/ricin loop of only PAP-expressing cells, as indicated by black arrows in Supplemental Figure S2A. Specifically, depurination of A3027 followed by aniline-mediated cleavage will generate fragments of 3027 and 369 bases from 25S rRNA. To verify that these fragments resulted from cleavage of 25S rRNA, we examined the same samples by northern blotting, using a biotinylated oligonucleotide specific for the 3′ end of 25S rRNA. We observed full-length 25S rRNA in all samples as expected, along with a single fragment in the sample expressing PAP following aniline treatment, which was of the expected size of the 3′ portion of 25S rRNA following cleavage at A3027 (Supplemental Fig. S2B). The absence of fragments by northern blotting of 18S, 5.8S, and 5S rRNA and tyrosyl-tRNA and L3 mRNA following aniline treatment indicates that PAP expression in yeast did not result in detectable depurination of these RNAs (Supplemental Fig. S2B–G).
25S rRNA depurination reduces translation elongation
To examine the effect of PAP expression on translation, we measured nascent protein synthesis through incorporation of AHA (L-azidohomoalanine), which is a methionine analog, in BY4741 cells expressing PAP or PAPx for 6 h. AHA contains an azido group that will form a covalent linkage with an alkyne-containing fluorophore, added to cell lysates following PAP or PAPx expression. This fluorophore allows quantification of newly translated proteins (Jha and Mapa 2025). Significantly less AHA was incorporated in PAP-expressing cells compared to cells expressing PAPx (Fig. 2A). This decrease in protein synthesis is consistent with earlier in vitro experiments showing that rRNA depurination inhibits binding of eEF2 to ribosomes, thereby preventing the translocation step of translation (Montanaro et al. 1975; Gessner and Irvin 1980; Brigotti et al. 1989; Mansouri et al. 2006). To test whether this was likely the case in vivo, we probed ribosomes isolated from PAP- and PAPx-expressing cells with antibody for eEF2. Figure 2B shows less eEF2 associated with equal amounts of ribosomes from PAP-expressing cells compared to PAPx and EV, despite abundant eEF2 in total cell lysate, supporting data that PAP inhibits translation elongation.
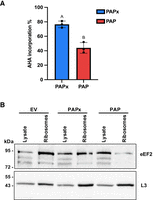
25S rRNA depurination reduces translation and binding of eEF2 to ribosomes. (A) WT BY4741 yeast cells transformed with PAP, PAPx, and EV cDNAs were induced with 2% galactose for 5 h. The methionine analog AHA (L-azidohomoalanine; 50 µM) was then added to cultures for 1 h. Protein synthesis was measured by incorporation of AHA into newly synthesized proteins. Following the covalent binding of a fluorescent alkyne with AHA, incorporated labeled proteins were quantified by fluorescence. Fluorescence values were normalized to OD600 of each sample, and AHA % incorporation values for PAP and PAPx are shown relative to EV values. Bars represent means ± SEM of three independent biological replicates (n = 3). The different letters on bars represent statistically significant differences at P < 0.05. (B) Association of eEF2 with ribosomes of cells expressing PAP, PAPx, or EV was assessed by immunoblot. Total protein and ribosomes were isolated from WT BY4741 cells transformed with cDNAs of PAP, PAPx, or EV following induction with 2% galactose for 6 h. Total cell lysate and ribosomes (20 µg) were separated through 12% SDS PAGE, transferred to nitrocellulose, and probed with eEF2 (1:10,000) and RpL3 (1:5000) antibodies. Immunoblot illustrates eEF2 (93 kDa) and L3 (43 kDa) in total cell lysates and ribosomes.
Monosomes and polysomes have depurinated 25S rRNA
Since PAP decreased the level of protein synthesis, we investigated whether rRNA depurination altered distribution of ribosomes on mRNAs during translation. We compared sucrose gradient profiles of BY4741 cells transformed with PAP, PAPx, or EV constructs at 6 h postinduction. The sucrose gradient profiles of cells expressing PAP showed increased 40S, 60S, and 80S peaks and decreased levels of polysomes relative to the profiles of PAPx and EV cells (Fig. 3A). Because the differences in profiles between PAP-, PAPx-, and EV-expressing cells were not remarkable, we quantified the area under the curves of polysomes and monosomes. The polysome to monosome ratio for PAP-expressing cells was 0.67 ± 0.03, compared with 0.96 ± 0.05 and 1.06 ± 0.04 for PAPx- and EV-expressing cells, respectively. These values are means ± SEM for three sucrose gradient profiles for each type of cell transformation. We observed a modest but statistically significant decrease in this ratio in cells expressing PAP compared with PAPx and EV. This pattern suggests that ribosomes of cells expressing PAP could not efficiently translate. Given that depurinated SRL rRNA inhibits translation, we tested whether fractions collected from these profiles contained depurinated rRNA or whether damaged ribosomes were somehow sequestered from active translation. We discovered a significant enhancement of depurinated rRNA associated with polysomes relative to monosomes in PAP-expressing cells (Fig. 3B). As expected, significantly lower levels of depurination were observed in PAPx-expressing cells. Given that ribosomes with depurinated rRNA were found on polysomes and to a lesser extent, on monosomes, we hypothesized that it would be during translation that cells would detect damage to their ribosomes and initiate a signaling response.
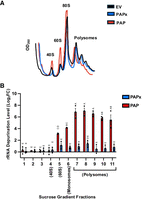
25S rRNA depurination alters sucrose gradient profiles and is present in polysome and monosome fractions. (A) Representative sucrose gradient profiles of WT BY4741 cells transformed with cDNAs encoding PAP (red), PAPx (blue), and EV (black) following induction with 2% galactose for 6 h. Cell lysates were separated by a 20%–50% sucrose gradient, and the absorbance of collected fractions was measured at A260. (B) Total RNA (500 ng) was isolated from each sucrose gradient fraction from WT BY4741 cells transformed with PAP, PAPx, and EV cDNAs following induction with 2% galactose for 6 h. RT-qPCR was performed to measure “reference” and “target” cDNA levels as illustrated in Supplemental Figure S1. Fold changes for PAP and PAPx are shown relative to EV. The statistical significance of differences in depurination levels was determined by two-way ANOVA with Tukey's multiple comparisons test. Bars represent means ± SEM of three independent biological replicates (n = 3). The different letters on bars represent statistically significant differences at P < 0.05.
25S rRNA depurination does not activate ISR
To investigate how rRNA depurination was signaled in yeast, we tested whether the ISR pathway was activated. Previous research showed that an intermediate level of antibiotic concentration inhibited translation, which elicited a Gcn2-mediated ISR in yeast resulting in phosphorylation of eIF2α and downstream activation of Gcn4 to promote survival (Wu et al. 2020; Yan and Zaher 2021). Given that PAP also inhibits translation, we firstly tested whether its expression increased phosphorylation levels of eIF2α; however, we did not detect eIF2α phosphorylation in PAP-, PAPx-, and EV-expressing cells (Fig. 4A) As a positive control, WT BY4741 cells were treated with 30 mM 3-aminotriazole (3-AT) for 90 min, and eIF2α phosphorylation was observed. To ensure that subtle changes in eIF2α phosphorylation level due to PAP expression were not missed by immunoblotting, we quantified Gcn4 translation levels. To do this, we cotransformed WT and Δgcn2 BY4741 with our PAP and PAPx-encoding plasmids along with a β-galactosidase reporter fused to Gcn4 construct (Hinnebusch 1985). If rRNA depurination activated ISR, we would anticipate an increased Gcn2-dependent expression of our Gcn4 construct. Though β-galactosidase levels from WT BY4741 cells expressing PAP were statistically higher than cells transformed with EV, they were similar to levels in PAPx-expressing cells, suggesting that the downstream signal for Gcn4 activation was independent of the level of rRNA depurination. β-Galactosidase activity was significantly lower than our positive control 3-Amino-1,2,4-triazole (3-AT), which inhibits histidine synthesis, resulting in depletion of His-tRNA, and is used as an activator of ISR (Fig. 4B; Natarajan et al. 2001; Yan and Zaher 2021). A low level of β-galactosidase activity was measured in cells lacking functional Gcn2 as anticipated, indicating that Gcn4 translation is primarily controlled by Gcn2. We conclude that high levels of rRNA depurination in PAP-expressing cells were not signaled primarily through the ISR pathway.
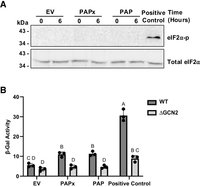
25S rRNA depurination does not activate integrated stress response. (A) Immunoblots of eIF2α (38 kDa) expression and phosphorylation level from WT BY4741 cells transformed with cDNAs of PAP, PAPx, or EV at 0 and at 6 h following induction with 2% galactose. As a positive control for eIF2α phosphorylation, WT BY4741 cells were treated with 30 mM 3-aminotriazole (3-AT) for 90 min. Total cell lysates (60 µg) were separated through 12% SDS PAGE, transferred to nitrocellulose, and probed with total eIF2α (1:1000) and phospho-eIF2α (1:1000) antibodies. The blots shown are representative of three independent biological replicates (n = 3). (B) WT and Δgcn2 BY4741 cells were cotransformed with plasmids encoding cDNAs of PAP, PAPx, or EV, along with p180 and pRS313 and were grown in 2% galactose for 6 h. 3-AT (30 mM) served as a positive control and was added to cells cotransformed with EV, p180, and pRS313 and incubated in 2% galactose for 90 min. β-Galactosidase enzyme activity was quantified using ONPG (O-nitrophenyl β-galactoside) as a substrate. A two-way ANOVA with Tukey's multiple comparisons test was used to determine the statistical significance of differences in β-galactosidase activity. Bars represent means ± SEM of three independent biological replicates (n = 3). The different letters on bars represent statistically significant differences at P < 0.05.
25S rRNA depurination correlates positively with ubiquitination of ribosomes
Given that ubiquitination of ribosomal proteins regulates different ribosome surveillance pathways (Fujii et al. 2012; Sugiyama et al. 2019; Yan and Zaher 2021; Li et al. 2022; Ford et al. 2024; Monem and Arribere 2024), we explored whether rRNA depurination resulted in increased ubiquitination of ribosomes. Following a 6 h induction of PAP and PAPx expression, we isolated ribosomes and probed changes in ubiquitination level by immunoblot. WT BY4741 cells expressing PAP showed increased levels of ribosome ubiquitination relative to cells transformed with the PAPx and EV constructs (Fig. 5A). We also probed fractions collected from sucrose gradient profiles of these cells by immunoblot for ubiquitin. Both 80S monosomes and polysomes were enriched for ubiquitination in PAP-expressing cells relative to cells expressing PAPx or EV (Fig. 5B). We anticipated that polysomes would be ubiquitinated given their level of rRNA depurination and resulting inhibition of elongation. Ubiquitination of 80S monosomes suggests that a proportion of the 80S ribosomes were elongating as single ribosomes on a transcript (Heyer and Moore 2016). To more clearly evaluate the connection between 25S rRNA depurination and ribosome ubiquitination, we quantified the level of rRNA depurination within monosome and pooled polysome fractions of cells following 6 h of PAP or PAPx expression (Supplemental Fig. S3A). In addition, we assessed the level of ubiquitination in the same monosome and pooled polysome fractions, using equal amounts of protein for immunoblotting (Supplemental Fig. S3B). Comparison between the two results illustrates a higher level of rRNA depurination and ubiquitination in polysome fractions of PAP-expressing cells, suggesting that it is rRNA depurination that leads to ubiquitination of ribosomes. The lack of rRNA depurination and ubiquitination observed in PAPx-expressing cells supports the idea that expression of an active glycosylase is required for subsequent ubiquitination.
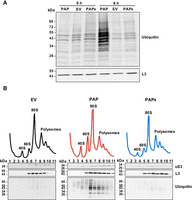
PAP expression causes ubiquitination of monosomes and polysomes. (A) Immunoblot of ubiquitination of ribosomes isolated from WT BY4741 cells transformed with cDNAs of PAP, PAPx, or EV at 0 and 6 h postinduction with 2% galactose. Ribosomes (20 µg) were separated through 12% SDS PAGE, transferred to nitrocellulose, and probed with a polyclonal ubiquitin antibody (1:1000). A monoclonal antibody against ribosomal protein L3 (43 kDa; 1:5000) was used as a loading control. The blots shown are representative of three independent biological replicates (n = 3). (B) Sucrose gradient profiles of WT BY4741 cells transformed with cDNAs of PAP, PAPx, or EV at 6 h postinduction with 2% galactose are illustrated. Fractions of these profiles were separated through 12% SDS PAGE, transferred to nitrocellulose, and probed with a polyclonal ubiquitin antibody (1:1000), or monoclonal antibodies against ribosomal proteins L3 (43 kDa; 1:5000) and uS3 (27 kDa; 1:1000). The blots shown are representative of three independent biological replicates (n = 3).
25S rRNA depurination activates Mag2- and Hel2-dependent ubiquitination
To investigate the ribosome surveillance pathway triggered from 25S rRNA depurination, we tested if ubiquitination of depurinated rRNA is dependent on E3 ubiquitin ligases active in 25S NRD, RQC, and 18S NRD pathways. Firstly, we chose to assess whether 25S NRD is involved because the SRL target of PAP is located in the large ribosomal subunit. During 25S NRD, Rtt101 mediates ubiquitination of large subunit ribosomal proteins (Fujii et al. 2009, 2012). Secondly, we considered RQC, as ribosomes treated with translation inhibitors cause ribosome stalling and/or collisions that are recognized by the ligase Hel2 that ubiquitinates uS10 (Matsuo et al. 2017; Miścicka et al. 2024). The third pathway we examined was 18S NRD, as damage to the large ribosomal subunit may be detected through the small subunit. Two types of 18S NRD are described: Fap1-mediated polyubiquitination of uS3, which is specific for ribosomes that are individually stalled, or Mag2-mediated monoubiquitination of uS3, which is followed with polyubiquitination by Hel2 that specifically targets slow-moving ribosomes (Sugiyama et al. 2019; Li et al. 2022; Matsuo and Inada 2023). To test these, we examined ubiquitination levels of ribosomes from WT BY4741, Δrtt101, Δhel2, Δmag2, and Δfap1 strains at 6 h postinduction of PAP expression. Ubiquitination levels in Δrtt101 and Δfap1 strains increased at 6 h similar to WT (Fig. 6A), indicating that deleting these E3 ubiquitin ligases did not impact the ubiquitination levels of ribosomes. Therefore, 25S NRD and Fap1-mediated 18S NRD are likely not involved in recognizing damage to the SRL of 25S rRNA. In contrast, the ubiquitination levels of ribosomes did not increase in Δhel2 and Δmag2 strains at 6 h postinduction, and the levels were lower than WT, Δfap1, and Δrtt101 strains at 6 h (Fig. 6A). These data suggest that Hel2 and Mag2 play a role in detecting 25S rRNA depurination, possibly activating components of 18S NRD.
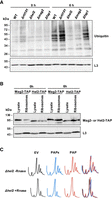
PAP activates 18S NRD without causing ribosome collisions. (A) Immunoblot illustrating ubiquitination of ribosomes from WT, Δrtt101, Δhel2, Δmag2, and Δfap1 BY4741 cells transformed with a cDNA encoding PAP at 0 and 6 h postinduction with 2% galactose. Ribosomes (20 µg) were separated through 12% SDS PAGE, transferred to nitrocellulose, and probed with a ubiquitin antibody (1:1000). A monoclonal antibody against RpL3 (43 kDa; 1:5000) was used as a loading control. The blots shown are representative of three independent biological replicates (n = 3). (B) Immunoblot of TAP-tagged Hel2 (93 kDa) and Mag2 (96 kDa), in total cell lysates and ribosomes isolated from Hel2-TAP and Mag2-TAP tagged BY4741 cells transformed with a cDNA encoding PAP at 0 and 6 h postinduction with 2% galactose. Total cell lysate and ribosomes (40 µg) were separated through 12% SDS PAGE, transferred to nitrocellulose, and probed with a polyclonal TAP (1:1000) antibody. A monoclonal antibody against RpL3 (43 kDa; 1:5000) was used as a loading control. The blots shown are representative of three independent biological replicates (n = 3). (C) Evaluation of disome presence in sucrose gradient profiles of PAP-expressing cells. Representative sucrose gradient profiles of Δhel2 BY4741 cells transformed with cDNAs encoding PAP, PAPx, and EV following induction with 2% galactose for 6 h. Lysates were treated with or without RNase-I before separation on a 20%–50% sucrose gradient. Absorbance of collected fractions was measured at A260.
If Hel2 and Mag2 are involved in detecting 25S rRNA depurination, we would anticipate their preferential association with ribosomes of PAP-expressing cells. Due to a lack of good antibodies for these proteins in yeast, we expressed PAP in strains of BY4741 with TAP-tagged Hel2 or Mag2. Isolation of ribosomes followed by immunoblotting with a TAP-specific antibody revealed a higher level of Hel2-TAP associated with equal amount of ribosomes from cells at 6 h postinduction of PAP compared with 0 h (Fig. 6B). In addition, Mag2-TAP association with ribosomes increased following 6 h of PAP expression (Fig. 6B). TAP signal in total cell lysates verified Hel2 and Mag2 expression in each tagged strain. The enhanced ribosome association supports the Hel2- and Mag2-dependent ubiquitination of ribosomes from PAP-expressing cells.
Since Hel2 is involved in resolving ribosome collision stress (Matsuo et al. 2017; Ikeuchi et al. 2019; Yan and Zaher 2021), we assessed whether 25S rRNA depurination initiated ribosome collisions. To do this, we treated cell lysates of PAP, PAPx, and EV expressing Δhel2 cells with ±RNase and generated sucrose gradient profiles. We anticipated that if ribosome collisions occurred upon PAP expression, they would accumulate in the Δhel2 strain. We did not detect a significant difference in the profiles from PAP compared with PAPx- and EV-expressing cells treated with RNase (Fig. 6C). A disome peak was visible in all profiles, indicating some degree of ribosome collision; however, there was no PAP-dependent increase in this peak suggesting that rRNA depurination did not enhance ribosome collisions. In addition, we noticed that polysomes were maintained in Δhel2 cells expressing PAP (unlike WT cells shown in Fig. 3A), suggesting that Hel2 is involved in recovering 40S subunits from ribosomes with damaged 25S rRNA.
To identify which ribosomal protein was specifically ubiquitinated, we conducted LC-MS/MS of ribosomes isolated from WT BY4741 cells expressing PAP, PAPx, or EV using diglycine tag as a marker. Our results revealed that Rps3 (uS3) was ubiquitinated upon 25S rRNA depurination (Supplemental Table S1). To support the ubiquitination of uS3 and its dependency on Hel2 and Mag2, we immunoprecipitated uS3 from total cell lysates of WT, Δhel2, and Δmag2 BY4741 cells at 6 h postinduction of PAP and probed proteins eluted from beads with our ubiquitin-specific antibody. Supplemental Figure S4 illustrates increased ubiquitination of eluates from WT compared with Δhel2 and Δmag2 cells. Thus, both the absence of detectable Rps20 (uS10) ubiquitination and ribosome collisions indicate that RQC was not activated in response to 25S rRNA depurination. Rather, our data showing reduced protein synthesis and less binding of eEF2 suggest that depurinated ribosomes elongate slowly and are recognized by Mag2 and Hel2, which ubiquitinate uS3 and are components of the 18S NRD (Fig. 7).
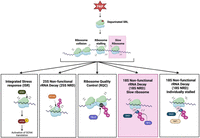
Model depicting 25S rRNA depurination leading to slow-moving ribosomes that are signaled by Mag2- and Hel2-dependent 18S NRD. The primary pathway activated in response to rRNA depurination is highlighted in pink. Other pathways, including the Gcn2-mediated ISR, Rtt101-mediated 25S NRD, Hel2-mediated RQC, and Fap1-mediated 18S NRD, did not play a major role in recognizing 25S rRNA depurination.
DISCUSSION
Our rationale for this work was to investigate how damage to rRNA is detected and signaled. We chose to investigate this process in yeast as ribosome surveillance pathways are best characterized in this model (Fujii et al. 2012; Sugiyama et al. 2019; Yan and Zaher 2021; Li et al. 2022). Yeast also provided inducible and tightly controlled PAP expression. Unlike previous studies that introduced sequence mutations to rRNA (Fujii et al. 2009, 2012), we expressed an enzyme that targeted ribosomes once they were already assembled and engaged in translation (Mansouri et al. 2006). The prevalence of rRNA in cells and the essential role of the SRL in controlling GTPase-dependent movement of ribosomes predicted that base removal from this loop would elicit a significant response. We show that ribosomes with depurinated rRNA were ubiquitinated on Rps3 by the E3 ubiquitin ligases Mag2 and Hel2. This modification occurs during translation and is indicative of slow-moving ribosomes that were detected by elements of the 18S NRD pathway even though the SRL is a component of the large ribosomal subunit. We suggest that decrease in translation elongation, rather than location of damage, is the factor required for sensing problems with protein synthesis and determining cellular responses.
As anticipated, PAP expression in yeast reduced the incorporation of a methionine analog into nascent protein chains and reduced the binding of eEF2 to ribosomes, consistent with rRNA depurination inhibiting translation elongation. Effects of SRL damage were visible by altered sucrose gradient profiles from PAP-expressing cells. We observed a decrease in polysome abundance and accumulation of ribosomal subunits and 80S monosomes. Typically, elongation inhibitors trap ribosomes in polysomes resulting in a shortage of free 40S and 60 subunits (Liu and Qian 2016). We speculate that ubiquitination of Rps3 in polysome and monosome fractions tags 40S subunits for recovery from ribosomes with depurinated 25S rRNA, which would explain the reduction in polysomes we observed. Accumulation of 80S ribosomes usually indicates inhibition of initiation; however, high 80S peaks have also been attributed to slow elongation rates (Heyer and Moore 2016). We suggest that the enhanced 80S peak we observed in PAP-expressing cells may be ribosomes at start codons that elongate slowly. Accordingly, we would anticipate an accumulation of free subunits as well. Alternatively, these monosomes may also represent some final ribosomes following polysome dissociation. Our data indicate that damage to rRNA largely occurs during protein synthesis and is detected during active translation.
Despite the substantial damage to ribosomes caused by rRNA depurination, ISR was not triggered. ISR is typically associated with a frequency of damage that cannot be resolved by ribosome quality control (Wu et al. 2020; Yan and Zaher 2021; Nanjaraj Urs et al. 2024); therefore, we anticipated that depurination of ∼59% of rRNA would be signaled to cells by ISR. However, we did not detect an increase in eIF2α phosphorylation or Gcn4 activation upon PAP expression. One possible explanation for the lack of ISR involvement is that damage to the SRL increases the number of ribosomes in a pretranslocation state with the peptidyl-tRNA occupying the A site (Mansouri et al. 2006), which is not the conformation favored by ISR. Studies using various translation inhibitors that trap ribosomes in different conformations have shown that ISR activation requires an empty A site (Yan and Zaher 2021). Moreover, we speculate that PAP expression may suppress ISR because SRL depurination inhibits the binding of eEF1A in addition to eEF2, and excess free eEF1A may bind Gcn2, which is known to reduce its activation (Visweswaraiah et al. 2011). Therefore, ISR does not appear to be the primary way that cells respond to depurinated SRL.
We initially reasoned that 25S rRNA damage would be recognized by the ubiquitin ligase Rtt101 dependent 25S NRD, as it specifically targets nonfunctional ribosomes containing mutations within the rRNA of 60S subunits (Fujii et al. 2009, 2012). This was not the case, as the levels of ribosome ubiquitination did not change when PAP was expressed in the Rtt101 deletion strain. Rather, we identified Mag2- and Hel2-dependent ubiquitination of ribosomes, indicative of elements involved in 18S NRD. It is curious that 25S rRNA depurination was signaled from the small ribosomal subunit rather than the large subunit. Examination of the 25S NRD mechanism shows that degradation of mutant 60S subunits is not dependent on changes in translation rate (Cole et al. 2009), a factor that we suggest is important for recognition of SRL damage. Moreover, PAP expression causes translation defects that mimic ribosome states known to recruit cytoplasmic surveillance factors. For example, 60S subunits containing incompletely processed rRNA engage in translation but become stalled with tRNAs in hybrid conformation. These ribosomes are bound by Hel2 and proteins of the ribosome quality control trigger complex (Sarkar et al. 2017). These factors typically act upon the 40S subunit, illustrating that damage to the 60S can be signaled through the 40S. In a similar manner, we suggest that translation inhibition caused by SRL depurination, which also increases numbers of ribosomes in a pretranslocation state with A and P sites occupied, is recognized by factors that bind to the 40S subunit.
Our study does not describe whether ribosomes stalled due to SRL depurination; however, we did not detect collisions, which we suspect would result from stalling. Though recent results show that ribosome collisions are not essential for Hel2-dependent ubiquitination (Miścicka et al. 2024), Hel2 is the primary factor that binds collided ribosomes and ubiquitinates Rps20 to initiate subunit dissociation (Matsuo et al. 2017; Juszkiewicz et al. 2018, 2020; Ikeuchi et al. 2019). This process is a key element of RQC, which removes stalled ribosomes to permit upstream ribosomes to continue elongating. Our finding that polysome peaks were retained in the Hel2 deletion strain expressing PAP supports the idea that Hel2 may be involved in ribosome dissociation upon SRL depurination. Though ubiquitination of ribosomes from PAP-expressing cells was dependent on Hel2, Rps3 was ubiquitinated rather than Rps20. Both proteins are substrates of Hel2; however, ubiquitination of Rps3 is also associated with Mag2, which monoubiquitinates Rps3 of slow-moving ribosomes (Li et al. 2022; Matsuo and Inada 2023; Ford et al. 2024). Once Rps3 is monoubiquitinated, it may also become polyubiquitinated by Fap1, if the ribosome is solitary, as shown by decoding-deficient mutants that stalled at initiation codons as noncollided, 80S monosomes (Li et al. 2022). Therefore, the sequential ubiquitination of Rps3 by Mag2 followed by Fap1 is required for 18S NRD of ribosomes unable to elongate beyond start codons. We did not observe Fap1-dependent ubiquitination of ribosomes with depurinated 25S rRNA, suggesting that the majority of ribosomes were not stalled at initiation codons despite the increased 80S monosome peak observed in PAP-expressing cells. Our model of how SRL depurination is signaled is best described by detection of slow-moving ribosomes by components of the 18S NRD pathway. In this scenario, we suggest that Mag2 monoubiquitinates Rps3, which is followed by Hel2-dependent polyubiquitination. Subsequent work will identify how 60S subunits are degraded; however, our results show that recognition of SRL damage is signaled through the small ribosomal subunit and may be dependent on reduced translation elongation.
MATERIALS AND METHODS
Plasmids, yeast strains, and time-course induction for PAP expression
Plasmids encoding PAP cDNA, its enzymatic inactive mutant, PAPx (Hur et al. 1995), or an empty vector (EV), all under the control of a galactose-inducible (Gal-1) promoter in the Yep351 backbone, were transformed into various yeast strains: the wild-type (WT) S. cerevisiae strain BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0), BY4741 derivatives with deletions of the protein kinase Gcn2 (Δgcn2), and E3 ubiquitin ligases Rtt101 (Δrtt101), Hel2 (Δhel2), Mag2 (Δmag2) and Fap1 (Δfap1), as well as TAP-tagged strains of Hel2 (Hel2-TAP) and Mag2 (Mag2-TAP). Transformed yeast cells were grown in SC-Leu containing 2% raffinose to an OD600 of 0.5 and then transferred to SC-Leu containing 2% galactose to induce PAP expression. Samples were collected by centrifugation of aliquots at 0, 3, and 6 h postinduction, for protein and RNA extractions.
Protein extraction, immunoblotting, and immunoprecipitation
Cell pellets were resuspended in buffer (25 mM Tris-HCl, pH 7.5, 1 mM DTT, 1 mM EGTA, 5% glycerol, 1 mM PMSF), lysed with 0.5 mm glass beads, and centrifuged at 16,000g for 20 min to separate cell debris from soluble lysates. The protein concentration of the lysates was quantified using Bradford assay. For protein extraction from sucrose gradient fractions, trichloroacetic acid (final concentration of 20%) was added to precipitate proteins from each fraction. Samples were centrifuged at 16,000g for 30 min, and protein pellets were dissolved in water. A Bradford assay was performed to quantify proteins, and samples were mixed 1:1 with 2× SDS sample buffer.
For immunoblots, protein samples were separated by 12% SDS-PAGE and transferred to nitrocellulose membranes for 30 min at 230 mA. The membranes were blocked in 5% nonfat milk in PBS-T or 5% BSA in TBS-T for 2 h at room temperature, then incubated with primary antibodies overnight (16 h) at 4°C: polyclonal anti-PAP (1:5000), anti-total eIF2α (1:1000; provided by Dr. T.E. Dever), anti-phospho eIF2α (1:1000; Abcam 131505), monoclonal anti-ubiquitin (1:1000; Santa Cruz 8017), monoclonal anti-Rps3 (1:1000; Cell Signaling 9538), monoclonal anti-RpL3 (1:5000; provided by Dr. J.R. Warner), polyclonal anti-eEF2 (1:10,000; Kerafast ED7002), and polyclonal anti-TAP tag (1:1000; Novus Biologicals 31052). Membranes were washed four times with 1× PBS-T or 1× TBS-T for 10 min each wash, followed by incubation with HRP-conjugated secondary antibodies in either 5% milk/PBS-T or 5% BSA/TBS-T for 2 h at room temperature. Membranes were washed as described, and protein bands were visualized by chemiluminescence detection system.
Cell lysates from WT, Δhel2 and Δmag2 BY4741 strain transformed with PAP cDNA were collected at 6 h postinduction with 2% galactose. Immunoprecipitation was performed according to the NEB protocol for Protein A/G Magnetic Beads (NEB S1425S). Briefly, 5 µg of monoclonal anti-Rps3 (Cell Signaling 9538) antibody was added to 200 µg of lysate and incubated for 3 h at 4°C with rotation. Prior to the addition of the antibody, an aliquot was collected and saved as input control. Subsequently, 25 µL of Protein A/G magnetic beads were added to the cell lysate antibody mix, and samples were rotated for 2 h at 4°C. Beads were separated magnetically, washed three times with immunoprecipitation buffer (150 mM NaCl, 10 mM Tris-HCl [pH 7.4], 1 mM EDTA, 1 mM EGTA [pH 8.0], 0.2 mM sodium orthovanadate, 0.2 mM PMSF, 1% Triton X-100, 0.5% NP-40) and eluted in 25 µL 3× SDS sample buffer. Eluted proteins and input samples were incubated at 70°C for 5 min and separated by SDS-PAGE, followed by immunoblotting with monoclonal anti-ubiquitin and anti-Rps3 (loading control) antibodies, as described above.
Isolation of total RNA and quantification of rRNA depurination
Total RNA was isolated from cell pellets collected at 0, 3, and 6 h postinduction of PAP, PAPx, or EV in 2% galactose by first resuspending in AE buffer (50 mM NaOAc pH 5.2, 10 mM EDTA), 1% SDS, and equal volume of acidic PCI (pH 5.2). Samples were incubated at 65°C for 10 min with intermittent vortexing and chilled on ice for 5 min. After centrifugation at 16,000g for 15 min, samples were washed with chloroform. Contaminating DNA was digested with 2U of DNase I (NEB M0303S). Samples were incubated at 37°C for 1 h, extracted with acidic PCI (pH 5.2), and RNA was precipitated in 0.3 m NaOAc and 2.5 volumes of 100% ethanol. Following centrifugation at 16,000g for 30 min, the pellets were washed with 70% ethanol and resuspended in dH2O. For RNA isolation from sucrose gradients, samples were extracted in an equal volume of acidic PCI followed by chloroform extraction. RNA was precipitated in 0.3 m NaOAc and 2.5 volumes of 100% ethanol. Following centrifugation, pellets were washed in 70% ethanol, air-dried, and resuspended in dH2O. RNA concentrations were measured using a NanoDrop spectrophotometer.
Reverse transcription was performed using Ref RT and SRL RT primers to synthesize cDNA for reference and target PCRs, respectively (Supplemental Table S2). For rRNA depurination analysis of individual sucrose gradient fractions, monosomes (fraction #6) and pooled polysomes (fractions #7–10), cDNA synthesis was conducted using 500 ng of RNA. The reverse transcriptase reaction used MashUp-RT (Alekseenko et al. 2021), which stalls at the depurination site in the SRL. Subsequently, qRT-PCR was performed to measure reference and target rRNA levels. Each 20 μL RT-qPCR reaction was performed in a QIAGEN RotorGene Q thermocycler using 1 μL cDNA, 0.3 μM of forward and reverse primers each, and 10 μL GB-Amp 2X SYBR Green Master Mix (GeneBio Systems P2092). The qPCR was conducted under the following conditions: 10 min polymerase activation at 95°C, followed by 40 cycles of 15 sec at 95°C and 45 sec at 64°C.
To quantify rRNA depurination levels, a modified qRT-PCR-based method was used as described by Pierce et al. (2011). The locations of the reference and target rRNA primers are illustrated in Supplemental Figure S1, and their sequences are listed in Supplemental Table S2. Relative depurination levels were calculated using the Pfaffl method with modifications, generating a ratio of reference to target rRNA levels. In addition, absolute quantification of rRNA depurination levels was measured based on standard curves for both reference and target 25S rRNA generated using known concentrations of 25S rDNA template. Percent depurination was calculated as the ratio of absolute copy number of target relative to reference. To determine statistical significance of differences in depurination levels, a two-way analysis of variance (ANOVA) followed by Tukey's multiple comparisons test was conducted.
Northern blotting of cellular RNAs
Total RNA was isolated as described above, from WT BY4741 strain transformed with EV, PAPx, or PAP, following 6 h of induction with 2% galactose. RNA samples were quantified, and 10 µg of each sample was treated with 1 m aniline acetate (pH 4.5) for 30 min on ice. An equal amount (10 µg) of each sample was left untreated in water. All samples were precipitated in 0.3 m NaOAc and 2.5 volumes of 100% ethanol. The RNA was centrifuged at 16,000g for 30 min, washed in 70% ethanol, and resuspended in dH2O. Treated and untreated RNA (2.5 µg; ± aniline) was separated on a 1.5% formamide agarose gel along with a combined ssRNA ladder (NEB N0364S and N0362S). To assess RNA integrity following separation, the gel was stained with 10 µL of 10,000× SYBR gold in 50 mL 1× TAE for 20 min and destained for 10 min with RNase-free water. A gel image was acquired by a UV transilluminator and served as loading and quality control for subsequent northern blots. The RNA was then transferred onto a positively charged nylon membrane (Hybond N+) for 3 h at 0.23 mA. The membrane was UV cross-linked for 2 min at 1200 mJ/cm3 and dried in a gel dryer at 80°C for 30 min. The subsequent steps were followed according to Hwang et al. (2025). Prehybridization of the membrane was performed using 15 mL of hybridization buffer (5× SSC, 5× Denhardt's Solution, and 0.5% SDS w/v) for 2.5 h at 60°C, followed by overnight (16 h) hybridization at 60°C. Hybridization was performed using 60 µL of a probe mixture in 15 mL of hybridization buffer, prepared from the following 5′ biotinylated RNA probes stock solutions: 25S (10 μM), 18S (10 μM), 5.8S (10 μM), 5S (10 μM), tyrosyl-tRNA (40 μM), and ribosomal protein L3 mRNA (RPL3; 40 μM). The sequences of the RNA probes are listed in Supplemental Table S3. Following hybridization with the probe, the membrane was washed with Wash Solution I (2× SSC and 0.1% SDS w/v) and Strep wash buffer (1× NB salt buffer and 0.1% SDS w/v), then blocked in blocking buffer pH 7.4 (1× NB salt buffer and 2% SDS w/v) for 30 min. The membrane was then incubated with horseradish peroxidase (HRP) conjugated streptavidin antibody (0.5 µg/mL) in blocking buffer for 1 h. Additional washes with Strep wash buffer were performed, and the membrane was developed using a chemiluminescence detection system. The same membrane was subsequently stripped twice with Wash Solution II (0.1× SSC and 0.1% SDS w/v). Reblotting of the membrane was performed by cross-linking, blocking, and reprobing with different 5′ biotinylated probes as described above.
Click chemistry–based protein labeling and fluorescence measurement
WT BY4741 yeast cells transformed with PAP, PAPx, and EV cDNAs were induced in SC-Leu medium containing 2% galactose for 5 h. Cells were incubated for 1 h with 50 µM of Click-IT AHA (L-azidohomoalanine, Invitrogen C10102) followed by lysis. The click chemistry reaction was initiated by the addition of 10 mM THPTA, 2 mM CuSO4, 30 mM sodium ascorbate, and 1 µM Alexa Fluor 488 Alkyne (Invitrogen A10267). The samples were incubated in the dark for 30 min. The labeled proteins were precipitated by the addition of chloroform:methanol (1:4) and collected at the interface following centrifugation at 16,000g for 5 min. Precipitated proteins were washed in methanol and resuspended in water. Fluorescence measurements were quantified for each sample using a BioTek Synergy H4 Hybrid reader with excitation at 495 nm and emission at 519 nm. Fluorescence values were normalized to OD600 of each sample, and values are shown relative to EV values. The statistical significance of differences in fluorescent protein levels was determined by unpaired two-tailed t-test.
Quantification of Gcn4 translation levels using β-gal activity assay
WT and Δgcn2 BY4741 strains were cotransformed with plasmids encoding PAP, PAPx cDNAs or EV, p180 (encoding Gcn4-LacZ fusion; Hinnebusch 1985), and pRS313 (containing a histidine selection marker). The strains were induced in SC-Leu-Ura-His medium containing 2% galactose, and aliquots were collected at 0 and 6 h postinduction. For the positive control, WT and Gcn2 BY4741 strains transformed with EV + p180 + pRS313 were grown and induced under the same conditions, with an additional treatment of 30 mM 3-aminotriazole (3-AT), and aliquots were collected at 0 and 90 min postinduction. β-galactosidase (β-gal) activity was quantified using ONPG (O-nitrophenyl β-D-galactopyranoside) as a substrate. The activity of each sample was normalized relative to its respective 0 h time point, calculated as the difference in β-gal activity between the 6 and 0 h time points. The statistical significance of differences in β-gal activity was determined by two-way ANOVA with Tukey's multiple comparisons test.
Isolation of ribosomes
WT BY4741 cells transformed with plasmids encoding EV, PAPx, and PAP, and deletion strains (Δrtt101, Δhel2, Δmag2, and Δfap1) transformed with a plasmid encoding PAP, were induced in SC-Leu medium containing 2% galactose for 6 h. One hundred milliliters of aliquots was collected at 0 and 6 h postinduction, and cells were pelleted and ground in liquid N2. Buffer A (200 mM Tris-HCl, pH 9.0, 200 mM KCl, 200 mM sucrose, 25 mM MgCl2, 25 mM EGTA, 25 mM 2-mercaptoethanol) was added, and the mixture was centrifuged at 16,000g for 20 min. The supernatant was layered over a cushion of 1 m sucrose in 25 mM Tris-HCl pH 7.6, 25 mM KCl, and 5 mM MgCl2. Ribosomes were pelleted by centrifugation at 240,000g for 3.5 h at 4°C. The pellets were resuspended in 25 mM Tris-HCl (pH 7.6), 25 mM KCl, and 5 mM MgCl2 and quantified. The concentration of ribosomes was determined by measuring the absorbance at 260 nm, using a conversion factor of 1 OD260 = 20 pmol/mL of ribosomes (Chiou et al. 2008).
Sucrose gradient profile and collection of fractions
WT BY4741 cells transformed with plasmids encoding PAP, PAPx cDNAs or EV were induced in SC-Leu medium containing 2% galactose for 6 h. Cultures were then incubated in cycloheximide for 10 min (100 µg/mL), harvested by centrifugation, washed, and pelleted. Cell pellets were ground in liquid N2 and resuspended in Polysomal Lysis Buffer (20 mM Tris-HCl pH 7.5, 50 mM KCl, 10 mM MgCl2, 100 µg/mL cycloheximide, 1 mM DTT, 0.0067% Halt Protease Inhibitor Cocktail, 6.7 U RNase Inhibitor; 5 mL buffer per gram of grindate). The suspension was vortexed and clarified by centrifugation at 16,000g for 10 min. Supernatants (26 OD260 units) were diluted in 500 µL of Polysomal Lysis Buffer containing 0.2 mg/mL heparin and loaded onto 20%–50% sucrose gradients. For RNase I treatment, yeast grindate of Δhel2 strain was resuspended in buffer lacking RNase I inhibitor followed by clarification as described above. Five hundred micrograms of total RNA was incubated with 100 U of RNase I at 37°C for 30 min. Gradients were centrifuged at 220,000g for 4 h at 4°C. The sucrose gradient profile was obtained, and 11 fractions (1 mL each) were collected using a Brandel fractionator (sensitivity 1.0, baseline coarse 2, speed 60 cm/h, and rise time 2.5 sec). Polysome to monosome (P/M) ratios were determined by quantifying the area under the 254 nm absorbance curve using ImageJ software. One-way ANOVA followed by Holm–Sidak's multiple comparison test was performed to determine statistical significances in areas among samples.
Identification of ubiquitinated peptides by mass spectrometry
Ribosomes (90 µg) of PAP, PAPx, and EV expressing WT BY4741 strain for 6 h were adjusted to a final volume of 100 µL in 50 mM ammonium bicarbonate (pH 8.5). To reduce disulfide bonds, 5 µL of 200 mM DTT in 100 mM ammonium bicarbonate was added, and samples were heated at 95°C for 10 min. Subsequently, 8 µL of 0.5 m iodoacetamide in 100 mM ammonium bicarbonate was added, and the samples were incubated in the dark for 45 min. Trypsin (1 µg per 50 µg protein) was added, and the samples were incubated at 37°C overnight for proteolysis. The cleanup and MS analysis was performed by BioZone Mass Spectrometry Facility at the University of Toronto, Canada. After digestion, peptides were cleaned using ZipTip OMIX tips, washed with 1:1 acetonitrile:water and 0.1% TFA. Peptides were eluted with 95% acetonitrile/0.1% formic acid and vacuum dried. For MS analysis, peptides were resuspended in 0.1% formic acid, sonicated, and filtered. The analysis was performed using a Thermo Scientific Easy nLC 1000 coupled to a Thermo Q-Exactive, with a custom C18 column and a 5 µL injection. Data were processed using the X!Tandem search algorithm with the S. cerevisiae proteome database. For identification of ubiquitination, a potential modification of GlyGly (monoisotopic mass of 114.042927) at Lys residues was incorporated into the search parameters.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
Mass spectrometric analysis was performed by BioZone Mass Spectrometry Facility at the University of Toronto, Canada. This study was supported by a Discovery Grant to K.A.H. from the Natural Sciences and Engineering Research Council of Canada.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080559.125.
-
Freely available online through the RNA Open Access option.
- Received April 25, 2025.
- Accepted September 8, 2025.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue could introduce themselves and their work to readers of RNA and the RNA research community. Tanya Prashar is the first author of this paper, “Depurination of sarcin/ricin loop 25S rRNA is signaled through the small ribosomal subunit during translation.” Tanya is a PhD candidate in the Hudak Lab at York University. The main focus of her research is the signaling induced by damage to the sarcin/ricin loop of the ribosome and its downstream effects on the cell.
What are the major results described in your paper and how do they impact this branch of the field?
In this study, we demonstrate that depurination of the sarcin/ricin loop (SRL) in 25S rRNA impairs translation elongation and triggers ubiquitination of the 40S ribosomal protein uS3 during active translation. We show that these depurinated ribosomes are targeted for ubiquitination by the E3 ubiquitin ligases Mag2 and Hel2. Our findings support a model in which SRL damage is sensed through the slow movement of the ribosomes and subsequently recognized by components of the 18S nonfunctional rRNA decay (NRD) pathway.
What led you to study RNA or this aspect of RNA science?
I became interested in this area of RNA science after writing a review early on during my PhD on how abasic RNAs can act as signaling molecules during stress. This work sparked my curiosity about how damage to a highly conserved ribosomal RNA sequence like SRL depurination affects translation and led me to explore the stress signaling pathways activated in response to this damage in my current research.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
During our experiments, we were surprised to find that although the damage caused by depurination occurs in the 25S rRNA of the large 60S subunit, the cell detects this through the small 40S subunit. We initially focused on 25S nonfunctional rRNA decay (NRD), since the damage is localized to the 60S subunit. We also considered the integrated stress response (ISR), given that the majority of ribosomes were depurinated, and ribosome quality control (RQC), as depurination could lead to ribosome collisions. However, none of these pathways appeared to be activated. These unexpected findings shifted our focus toward alternative mechanisms involving ubiquitination on the 40S subunit and how the cell signals ribosome damage through this route.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My approach to science has been shaped by both my family and my PI, Dr. Kathi Hudak. My family taught me to stay curious, ask questions, and persevere during challenging times. My PI has built on that foundation by teaching me how to think critically, design experiments thoughtfully, stay open to unexpected results, and appreciate the importance of digging into the literature. This combination has been helpful in tackling new research questions and navigating unfamiliar scientific topics.








