
Bioinformatics-driven refinement of the commonly used TPI nonsense-mediated decay reporter system
- Laura Peter1,
- Lara Walotka1,2,
- Johannes Ptok,
- Caroline Meyer,
- Dominik Schüller,
- Heiner Schaal and
- Lisa Müller
- Institute of Virology, University Hospital Düsseldorf, Medical Faculty, Heinrich-Heine-University Düsseldorf, 40225 Düsseldorf, Germany
- Corresponding author: lisa.mueller{at}uni-duesseldorf.de
-
↵1 These authors contributed equally to this work.
Abstract
The cellular nonsense-mediated decay (NMD) pathway recognizes and degrades mRNAs with unusual structural features, such as long 3′ UTRs or overlapping reading frames, and therefore serves as a transcript quality control mechanism. A broad spectrum of today's knowledge about the nonsense-mediated mRNA decay pathway has been discovered using NMD reporter systems, mostly consisting of multiple exons, with a wild-type and a premature termination codon-containing variant. In a preliminary NMD study, we used the seven-exon triose phosphate isomerase (TPI) reporter and observed that in this well-known NMD reporter, surprisingly, not all splice sites are used constitutively, but additional cryptic splice sites are used. As this is more frequently observed in the construction of minigenes, especially when unknown splicing regulatory elements (SREs) are removed, for example, by shortening introns, this may affect the reliability of such reporters. To demonstrate how such minigenes can be improved in general with respect to constitutive splice site recognition, we restored an intron length in the TPI reporter or made bioinformatic adjustments to SREs or intrinsic strength of the splice sites themselves. As a result, this NMD reporter could be made more robust and specific for the evaluation of NMD sensitivity within a single transcript. The modifications of the TPI reporter shown here as examples can generally be used for the transfer of cellular multiexon transcripts to minigenes.
Keywords
INTRODUCTION
Nonsense-mediated mRNA decay (NMD) is a crucial mRNA surveillance system that eliminates transcripts with premature termination codons (PTCs) and prevents the production of dysfunctional or potentially harmful proteins, thus playing a critical role in the regulation of eukaryotic gene expression. It is vital for various cellular processes such as cellular homeostasis, cell cycle progression, stress response, development, differentiation, neural activity, and immunity (Lykke-Andersen and Jensen 2015; Ottens and Gehring 2016). This process, dependent on translation, involves core factors such as the RNA-dependent helicase and ATPase UPF1 (Carter et al. 1996; Thermann 1998; Karousis et al. 2016). UPF1 binds to transcripts sequence independently, regardless of whether the transcript is an NMD target (Hogg and Goff 2010; Kurosaki and Maquat 2013; Zund et al. 2013; Kurosaki et al. 2014; Lee et al. 2015). UPF1 interacts with UPF2 and UPF3B to enhance ATPase and helicase activities (Chamieh et al. 2008; Chakrabarti et al. 2011). Initially discovered in yeast and subsequently elucidated in humans (Chang and Kan 1979; Losson and Lacroute 1979; Leeds et al. 1991; Kugler et al. 1995), NMD operates through mechanisms including the exon junction complex (EJC)-dependent model and the long 3′-UTR-mediated model (Kolakada et al. 2024). The EJC-dependent NMD model identifies target mRNAs using EJCs positioned 20–24 nt upstream of exon–exon junctions during mRNA splicing (Le Hir et al. 2000, 2001; Bono et al. 2006). While EJCs are usually removed during translation, the presence of a PTC upstream of the last exon–exon junction causes downstream EJCs to persist, triggering EJC-dependent NMD that often occurs in proximity to the nucleus in the pioneer round of translation (Lykke-Andersen et al. 2000; Gehring et al. 2005). Effective NMD activation via this route is complex; PTCs within 50 nt upstream of the final exon–exon junction typically induce NMD (Wang et al. 2002; Buhler et al. 2006), whereas those more than 55 nt upstream often fail to do so (Knezevic et al. 1995; Nagy and Maquat 1998). Recognition of NMD targets without a downstream EJC is not well understood. The long 3′-UTR-mediated NMD model proposes degradation beyond the pioneer round of translation (Hosoda et al. 2005). EJC-independent NMD target recognition mainly relies on the distance between the stop codon and poly(A) tail. Shorter distances reduce UPF1 binding to the 3′-UTR and NMD sensitivity (Eberle et al. 2008; Hogg and Goff 2010; Kurosaki and Maquat 2013). Phosphorylated UPF1 accumulates in long 3′-UTRs, potentially triggering NMD by chance (Kurosaki et al. 2014; Imamachi et al. 2017).
Approximately 30%–35% of pre-mRNA splicing events in humans generate PTCs, activating NMD pathways (Mendell and Dietz 2001; Lewis et al. 2003; Pan et al. 2006, 2008; Mort et al. 2008; Weischenfeldt et al. 2012). Notably, one-third of inherited human diseases arise from mutations that lead to PTCs in transcripts. Even nonaberrant transcripts can be targeted by NMD, with 10%–20% of targeted transcripts falling into this category (Mendell et al. 2004; Yepiskoposyan et al. 2011; Hurt et al. 2013). Tissue-specific or developmentally regulated alternative splicing further complicates the NMD landscape as some PTC-containing transcripts can evade NMD in certain contexts (Yeo et al. 2004; Barberan-Soler et al. 2009). Alternative splicing coupled with NMD (AS-NMD) also serves as a homeostatic regulatory mechanism for mRNA isoform expression (Lewis et al. 2003; Lareau et al. 2007; Ni et al. 2007). NMD's broad target range and preference for unusual RNAs allow rapid responses to various stimuli, such as stress or viral infections (Balistreri et al. 2017; Contu et al. 2019).
Reporter plasmids are essential tools in molecular biology for studying gene expression and regulation, particularly in the assessment of NMD (Ghiasi et al. 2024). NMD reporter systems typically include multiple exon mini-genes in both wild-type (WT) and PTC variants. These systems provide a simplified evaluation method independent of the varying NMD sensitivities of endogenous transcripts. Commonly used NMD reporters include the three-exon β-globin, T-cell receptor beta, immunoglobulin μ, and seven-exon triose-phosphate isomerase (TPI) constructs (Boehm et al. 2016). These constructs have significantly contributed to our current understanding of the NMD pathway (Gehring et al. 2003; Buhler et al. 2006; Toma et al. 2015). The use of reporter plasmids allows detailed mechanistic studies under controlled conditions and the manipulation of specific features like PTCs and long 3′ UTRs to observe their effects. These studies also help elucidate the role of NMD in disease mechanisms, particularly in genetic disorders caused by PTCs. Additionally, NMD's regulatory roles in normal gene expression and cellular stress responses can be investigated, shedding light on processes such as AS-NMD. This research is pivotal for developing therapeutic interventions to modulate NMD in treating genetic disorders and enhancing antiviral responses.
Despite their advantages, using reporter plasmids in NMD studies can present the usual challenges of overexpressed reporters compared to endogenous transcripts, such as variations in plasmid copy number and transfection efficiency. These factors can complicate result interpretations. Additionally, NMD reporter plasmids should be preexamined for their splicing patterns, as alternative splicing is often observed in the construction of minigenes. Here, we describe the bioinformatically guided adjustment of the splicing behavior of a well-known reporter system that increases reporter sensitivity and makes the readout more reliable.
RESULTS AND DISCUSSION
To evaluate the reliability of the seven-exon TPI reporter for assessing NMD, we first reanalyzed the splicing pattern of the pCI-TPI-WT-4H reporter (Boehm et al. 2016) in the absence and presence of CHX. CHX, a translation inhibitor, is often used with NMD reporter assays. It halts translation, allowing the accumulation and easier detection of NMD-sensitive transcripts, thereby providing insights into the dynamics of this surveillance pathway (Medghalchi et al. 2001).
HeLa cells were either left untransfected or transiently transfected with TPI WT plasmid for 24 h. Prior to RNA extraction, the cells underwent treatment with CHX (50 µg/mL) for 6 h (+) or were kept untreated (−). Subsequently, total RNA was isolated and analyzed via RT-PCR. Unlike the endogenous TPI transcript (Fig. 1, lanes 1 and 2), transfection of the reporter plasmid surprisingly generated splice variants not observed in cTPI expression (Fig. 1, lanes 3 and 4). CHX treatment revealed that three of the alternatively spliced WT transcripts were NMD sensitive. All TPI splice variants were identified by isolation of the different PCR bands, reamplification, and sequencing. As transfection control, an expression plasmid for human growth hormone 1 (HGH) was co-transfected and amplified using a sequence-specific primer pair. Endogenous SRSF3 transcripts were used as CHX-specific controls regarding NMD inhibition, since one splice isoform contains a PTC that renders the transcript NMD sensitive (Fig. 1, bottom; Jumaa and Nielsen 1997; Anko et al. 2012).
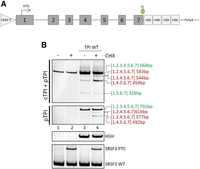
Multiple unexpected TPI splice isoforms derived from the TPI WT plasmid. (A) Schematic drawing of the used TPI WT reporter plasmid expressed under the control of a CMV promotor. The numbers indicate the exon numbers and the ATG and TC indicate the start and stop of the ORF. The four HBB repeats can be used as probe-binding sites for northern blot analysis. (B) RT-PCR results of untransfected HeLa cells (lanes 1 and 2) and HeLa cells transiently transfected with 1 µg of the pCI-TPI-WT plasmid for 24 h (lanes 3 and 4) that were either untreated (lanes 1 and 3) or treated with 50 µg/mL CHX for 6 h (lanes 2 and 4). The genes of interest were amplified with specific primer pairs (cTPI + pTPI: #5664/#5665, 26 cycles, pTPI: #4324/#5665, 18 cycles, HGH transfection control #1224/#1225 26 cycles, NMD control SRSF3 #4003/#4004 35 cycles), separated on a 10% PAA gel and visualized by EtBr staining and UV light exposure. The TPI bands were excised, reamplified, and sequenced. Numbers in brackets represent the exon numbers that were included in the splice isoform. Predicted NMD targets with frameshift mutations are highlighted in red letters, while green letters represent presumable not-NMD targets with intact ORFs.
Expanding upon the discovery that the plasmid-encoded TPI (pTPI) produced several transcripts, unlike the cellular-encoded TPI, we aligned and scanned the sequences for differences. Indeed, a comparison of exonic and intronic sequences revealed sequence differences between the pTPI and the cTPI, which might explain the differences in exon inclusion. The first major difference between the sequences of the pTPI versus the cTPI was found within exon 1. Within pTPI reporters, a different endogenous ATG codon, located 109 nt downstream from the cTPI start codon was used. These 109 upstream bases were not included in the pTPI sequence and the originally internal ATG was modified in position −3 by a G to A mutation. The second major difference between the pTPI and cTPI was found within the first intron. This intron lacked 250 nt which potentially might have resulted in the removal of splicing regulatory sequences. Minor splicing outcome unrelated differences included a single nucleotide exchange from C to G within intron 1 of the pTPI, 271 nt upstream of exon 2 as well as an artificially inserted probe-binding site within exon 7, directly downstream from the endogenous translational stop codon that consists of four repeats of a 100 nt sequence of the human β-globin gene (HBB). While the single nucleotide exchange within intron 1 and the alterations of the 3′ UTR most likely had no impact on the splicing process, the impact of the first two alterations from the cellular to the plasmid TPI were investigated regarding the splicing outcome. Therefore, the missing sequences of either exon 1 or intron 1 were amplified from isolated HeLa cell DNA and inserted into the TPI plasmid sequence. The splicing outcome was again monitored by transfection of HeLa cells with the newly generated modified TPI WT plasmids, and RT-PCR analysis using specific primers.
As a reference for the splicing pattern, HeLa cells were transfected with the original pCI-TPI-WT-4H plasmid, and the resulting alternative splice isoforms are shown in Figure 2, lanes 1 and 2. The insertion of the cTPI 250 nt sequence into intron 1 had no impact on the splicing outcome (Fig. 2, lanes 3 and 4 to lanes 1 and 2). Likewise, the insertion of the 109 nt sequence upstream of the plasmid-encoded ATG increased the PCR product sizes (labeled on the right-hand site) but had no further impact on the ratio of splice site usage and thus, exon inclusion (Fig. 2, lanes 5 and 6 to lanes 1 and 2). Hence, those sequence modifications, if at all, had only a minor impact on exon recognition within pTPI. Since the observed alternative pTPI splice isoforms were not found in the cTPI and were not reversed by adaptation of the plasmid TPI gene structure to the endogenous sequence, another approach was used.
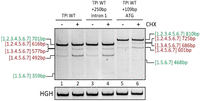
The pTPI splice isoforms are independent of Intron 1 and ATG alterations. RT-PCR results of HeLa cells transiently transfected with 1 µg of the pCI-TPI-WT plasmid or altered constructs for 24 h that were either untreated (lanes 1, 3, and 5) or treated with 50 µg/mL CHX for 6 h (lanes 2, 4, and 6). The genes of interest were amplified with specific primer pairs (pTPI: #4324/#5665, 18 cycles, HGH transfection control #1224/#1225, 26 cycles, NMD control SRSF3 #4003/#4004, 35 cycles), separated on a 10% PAA gel and visualized by EtBr staining and UV light exposure. The pTPI transcripts were amplified with specific primer pairs (#4324/#5665, 26 cycles), HGH served as transfection control (#1224/#1225, 26 cycles). The PCR products were separated on a 10% PAA gel and visualized by EtBr staining and UV light exposure.
To increase recognition and usage of exons 2 and 3 that display alternative splicing in the original plasmid TPI sequence, we employed established bioinformatic prediction tools to improve the splicing behavior of these two exons to restore a natural TPI splicing outcome. Here, we used the HBS and HEXplorer algorithms to amend the sequence to increase the likelihood of exon recognition within the pTPI sequence either by the adjustment of closely located splice regulatory elements (SREs) or by manipulating the intrinsic splice donor strength. Intrinsic splice site strength was calculated and adjusted using the HBS algorithm that is based on the duplex formation between mRNA and spliceosomal U1 snRNA that recognizes splice donor sites. Thus, the higher the complementarity between the two sequences, the higher the HBS (range 1.8–23.8). Here, we chose to increase the intrinsic strength of the splice donor of pTPI exon 3 (SD3, WT HBond score 16.9). The intrinsic strength was increased by the introduction of a single point mutation (gAGGTtAGTAg > gAGGTAAGTAg) that resulted in an increase of HBS 20.8 (Fig. 3A).
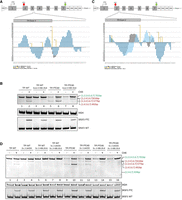
Combinatorial adjustments mitigate alternatively spliced transcripts. (A) Schematic drawing of the used TPI reporter construct. The 3′ end of exon 3 and the 5′ end of intron 3 were enlarged to show the introduced mutation and their effect on the HEXplorer plot. The SD was modified by a single point mutation (gAGGTtAGTAg > gAGGTAAGTAg, HBS 16.9 > 20.8). (B) RT-PCR results were obtained from the transfection of HeLa cells with the modified plasmids. HeLa cells were transiently transfected with the depicted reporter constructs for 24 h. Six hours before the total RNA was harvested, the cells were treated with 50 µg/mL CHX. The RNA was reverse transcribed and amplified with specific primer pairs (#4324/#5665 pTPI, 18 cycles). HGH (#1224/1225, 26 cycles) served as transfection control and SRSF3 (#4003/#4004, 35 cycles) as control for CHX treatment. The PCR products were separated on a 10% PAA gel, visualized by EtBr staining and UV light exposure. (C) Schematic drawing of the used TPI reporter construct. The 3′ end of exon 2 and the 3′ end of intron 2 were enlarged to illustrate the inserted mutations. The HEXplorer profile was modified by two silent point mutations within exon 2 (ACT-GGG-GAG-ATC-AG > ACC-GGA-GAG-ATC-AG), and two point mutations within intron 2 (CAG/GTGAGATCGAGGTGG > CAG/GTGAGATCTAGGCGG). (D) HeLa cells were transiently transfected with one of the depicted reporter constructs for 24 h. Six hours before the total RNA was harvested, the cells were treated with 50 µg/mL CHX. The RNA was reverse transcribed and amplified with specific primer pairs (#4324/#5665 pTPI, 20 cycles). HGH (#1224/1225, 26 cycles) served as transfection control and SRSF3 (#4003/#4004, 35 cycles) as control for CHX treatment. The PCR products were separated on a 10% PAA gel, stained with ethidium bromide, and visualized via UV light.
For the alteration of SREs regulating splice site usage, the HEXplorer tool was used. The HEXplorer is based on differences in hexamer frequencies between, up to 100 nt long, sequence segments upstream and downstream from annotated splice donor sites, within exonic or intronic sequences of the human reference genome. Ultimately, differential hexamer frequencies are taken into account to generate an average score for an index nucleotide by taking overlapping hexamers into account (HZEI). These calculations can be used to predict potential SREs that offer binding sites for splicing regulatory proteins (SRPs) that regulate splice sites in proximity. Generally, positive HEXplorer sequence plot areas can be interpreted as putative binding sites for SR(-like) proteins and negative areas as putative hnRNP(-like)-binding sites (Erkelenz et al. 2014; Müller et al. 2022). Following the concept of position dependency of splice sites (Erkelenz et al. 2013), we adjusted the overall SSHW, which is determined as the difference between the total upstream and downstream HZEI values, with higher SSHW indicating greater SRP binding potential and enhanced splice site usage, to be advantageous for splice donor 2 (SD2) recognition and thus, exon 2 inclusion (Fig. 3C).
In addition to the above used pCI-TPI-WT construct, also the pCI-TPI-PTC40 plasmid was included in the experimental setup. This isoform contains a premature termination codon in exon 2, making the full-length transcripts sensitive to NMD. The comparison of HeLa cells, which were transfected with the SD3-modified TPI plasmids showed fewer pTPI splice isoforms than HeLa cells, which were transfected with the original pCI-TPI, constructs. The results in Figure 3B showed that the single nucleotide substitution within spice donor 3 removed the TPI [1.2.4.5.6.7] and the TPI [1.4.5.6.7] splice isoforms from the pattern, both of which showed exon 3 skipping. The one alternative splice isoform that was still observable and more abundant upon SD3 modification was TPI [1.3.4.5.6.7], an isoform where exon 2 was skipped. The exclusion of the 124 nt long exon 2 resulted in a frameshift mutation that made the resulting mRNAs sensitive to degradation by the NMD machinery, which was nicely illustrated by the accumulation upon CHX treatment (Fig. 3B, lanes 3–4 and 7–8). Thus, the single-base modification of the pTPI SD3 resulted in increased recognition and inclusion of exon 3. However, this modification was not sufficient, as the remaining exclusion of TPI exon 2 from some splice isoforms still resulted in NMD-sensitive transcripts.
Therefore, the increased inclusion of TPI exon 2 was mediated by modification of the sequence surrounding of SD2, thereby increasing its SSHW. The aim of this modification was the modulation of the HEXplorer profile to generate a supportive SRE landscape as predicted by the HEXplorer algorithm for TPI SD2. This was accomplished by the mutation of 4 nt, two up and two downstream from TPI SD2 (Fig. 3C). The substitutions of the exon 2 nt upstream of SD2 were chosen as synonymous mutations with respect to the coding sequence. Within the intronic sequence, the mutations were chosen based on their impact on the SRE landscape. The overall impact of the modifications was monitored by the predicted change in the SRE capacity as indicated by the HEXplorer ΔHZEI of 35.3. The SRE modification of TPI exon 2 alone showed no difference in the splicing pattern in comparison to the pCI-TPI original plasmid (Fig. 3D, WT: lanes 1–4 and PTC40: lanes 9–12). However, the combination of both, the increased HBond score of the TPI SD3 together with the TPI exon 2 HEXplorer score modification finally resulted in a single splice isoform of pTPI. This splice isoform contained all constitutive TPI exons [1.2.3.4.5.6.7] and only the PTC-containing transcript showed an accumulation upon CHX treatment (Fig. 3D, lanes 7 and 8 [WT] and lanes 15 and 16 [PTC40]). To quantify the reduction of NMD-sensitive splice variants, qRT-PCR analysis was performed. We utilized an exon-junction primer at E1-4, specific for the most abundant NMD-sensitive splice variant, in comparison to Exon 2/3 inclusion as a measure of constitutively spliced transcripts, serving as a proxy for full-length transcripts (Fig. 4A). The results demonstrate that the HEXplorer-guided adjustments (Fig. 4B, “adj”) reduced exon 1–4 splicing events that generate NMD-sensitive transcripts compared to full-length transcripts where exons 2 and 3 are constitutively spliced.
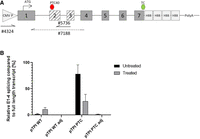
qRT-PCR analysis of relative exon usage. (A) Schematic drawing of the used TPI reporter constructs and primer spacing for qRT-PCR analysis. The numbers indicate the exon numbers, and the ATG and TC indicate the start and stop of the ORF. To distinguish between cellular and plasmid TPI, a forward primer specific for the plasmid promotor region was used (#4324). For amplification of the constitutive transcript containing exons 2 and 3, an exon junction primer was used (#5736). For amplification of the NMD-sensitive splice variant where exons 2 and 3 are skipped, an exon junction primer (#7188) was employed. (B) Results from qRT-PCR analysis are depicted in the bar graph showing the relative E1-4 splicing compared to E2/3 constitutively spliced full-length transcript variants (%) for the pTPI WT, pTPI WT adjusted, pTPI PTC, and pTPI PTC adjusted constructs. E1-4 splicing and E2/3 splicing were normalized to GAPDH (#3602/3603). Data are shown for both untreated and 50 µg/mL CHX-treated conditions. Error bars represent standard deviations from three independent replicates.
To further validate the enhanced specificity and consistency of the adjusted reporter set, caffeine was employed as an alternative NMD inhibitor alongside the commonly used CHX. Caffeine interferes with NMD activity by inhibiting UPF1 phosphorylation through the suppression of SMG1 (Keeling et al. 2013). In this experiment, HeLa cells were transfected with either the adjusted pTPI WT or the adjusted pTPI PTC plasmid, followed by treatment with 50 µg/mL CHX for 6 h (Fig. 5, lanes 2 and 4) or 10 mM caffeine for 4 h (lanes 6 and 8). For both the adjusted pTPI WT and the adjusted pTPI PTC plasmids, a single spliced band was observed, and alternative splicing was completely abolished. For the adjusted pTPI WT, neither an increase in transcript abundance nor variation in transcript profiles was detected upon treatment with either CHX or caffeine, indicating that the bioinformatically guided adjustments rendered the reporter completely independent of NMD. For the adjusted pTPI PTC plasmid, an increase in transcript abundance was observed following treatment with both CHX and caffeine, while no alternative spliced transcripts were detected. This supports the enhanced accuracy and robustness of the reporter system.
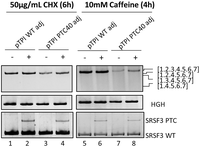
The adjusted variants of the reporter plasmids produce single bands upon treatment with different inhibitors. RT-PCR results of HeLa cells transiently transfected with 1 µg of the adjusted pCI-TPI-WT or PTC plasmid for 24 h that were either untreated (lanes 1, 3, 5, and 7) or treated with 50 µg/mL CHX for 6 h (lanes 2 and 4) or 10 mM caffeine for 4 h (lanes 6 and 8), respectively. The genes of interest were amplified with specific primer pairs (pTPI: #4324/#5665 18 cycles, HGH transfection control #1224/#1225 26 cycles, NMD control SRSF3 #4003/#4004 35 cycles), separated on a 10% PAA gel and visualized by EtBr staining and UV light exposure.
This reevaluation of the seven-exon TPI reporter underscored the necessity of aligning splicing patterns accurately between endogenous transcripts and reporter constructs in NMD studies. Initially, discrepancies in splice site utilization between the TPI reporter and native TPI transcripts hinted at cryptic splice site involvement—a common occurrence in minigene construction; however, further analysis revealed a differential splicing behavior of the plasmid TPI sequences compared to the cTPI. Ultimately, bioinformatically guided adjustments of the SREs around exon 2 in combination with the enhancement of intrinsic splice site strength of splice donor 3 effectively mitigated unwanted splicing variants, improving the TPI reporter's specificity in assessing NMD sensitivity within a single transcript. These modifications help eliminate NMD-sensitive transcript variants of the reporter plasmid that can affect transcript abundance, particularly in the WT construct when treated with an NMD inhibitor. Reducing the presence of such variants from the reporter plasmid minimizes transcript imbalances, ensuring more consistent and interpretable results. Using reporters to assess NMD compared to endogenous transcripts presents several pitfalls, which can affect the accuracy and reliability of the assessment. Reporter systems often use artificial sequence combinations that may not fully replicate the complexities of endogenous NMD substrates, leading to differences in NMD efficiency and regulation, as shown by the present study. To this end, also the method of transfection, transient versus stable, has been shown to influence the degradation efficiency of NMD targets. Transient transfections may not consistently mimic the stable expression of endogenous transcripts, leading to variability in NMD activity assessment (Gerbracht et al. 2017). Reporter assays often operate under nonphysiological conditions that might not accurately reflect the native cellular environment. It is well established that in the context of the minigene expression system several factors, including, for example, the type of cell line used for transfection, can vastly influence the outcome (Warzecha et al. 2012; Pereverzev et al. 2015), while endogenous transcripts are subject to a variety of regulatory mechanisms, including alternative splicing and the presence of multiple NMD-inducing features such as long 3′ UTRs and upstream open reading frames (uORFs). Thus, reporter systems might not capture this complexity, leading to oversimplified interpretations (Yepiskoposyan et al. 2011). A study investigating nonsense-associated altered splicing (NAS) in the TCR beta reporter demonstrates that alternative splicing is triggered by nonsense mutations, either through disruption of SREs or by introducing frameshift mutations that lead to PTCs (Chang et al. 2007). The results highlight the complexity of NAS, showing that both SRE disruption and reading frame disruption independently regulate alternative splicing patterns, ultimately affecting the transcript's susceptibility to NMD. The presence of NAS and alternative splicing in the TCR beta reporter affects its robustness by introducing variability in the splicing patterns, which can generate NMD-sensitive transcript variants. This variability complicates the interpretation of results, as it may lead to imbalances in transcript abundance and reduce the reliability of the reporter in accurately reflecting NMD activity, potentially distorting conclusions about NMD efficiency and regulation. Here, it might be beneficial to employ a prediction algorithm such as the HEXplorer to assess putative regulatory effects on SREs. In conclusion, while reporter assays provide a useful tool for studying NMD, it is crucial to be aware of their limitations and to complement and compare them with analyses of endogenous transcripts to obtain a more comprehensive understanding of NMD mechanisms and their regulation.
Concluding remarks
This meticulous approach emphasizes the critical need for precise representation of endogenous splicing patterns in NMD reporter systems. By refining the TPI reporter to better mirror natural splicing events, we have created a more robust tool for assessing NMD sensitivity. Looking ahead, this optimized reporter system can be instrumental in advancing our understanding of gene expression, as well as mRNA surveillance pathways.
MATERIALS AND METHODS
Expression plasmids
The pCI-TPI-WT-4H reporter and its PTC-containing variant, pCI-TPI-PTC40-4H, were obtained from Niels Gehring's laboratory (Boehm et al. 2016). To assess transfection efficiency, a plasmid encoding the human growth hormone HGH (pXGH5) was cotransfected in each experiment (Selden et al. 1986).
Oligonucleotides
All oligonucleotides used were obtained from Metabion GmbH.
Cloning
The pCI-TPI-WT-4H reporter and its PTC-containing variant, pCI-TPI-PTC40-4H, were obtained from Niels Gehring's laboratory (Boehm et al. 2016). Modifications to these reporter constructs involved the cloning of PCR products using specific forward and reverse primer pairs. These alterations included the insertion of an additional 109 nt of exon 1, plus the original start codon (#5744/#5673), and 250 nt of intron 1 (#5743/#5673), both sourced from cellular TPI (cTPI) (HeLa DNA). Additionally, the splice donor of TPI exon 3 (HBond score 16.9) underwent alteration through the introduction of a single point mutation (gAGGTtAGTAg > gAGGTAAGTAg, HBS 16.9 > 20.8; #5672/#5673, Q5 SDM, NEB). The inclusion of exon 2 was facilitated by modifying its surrounding nucleotides, leading to an increase in its splice site HEXplorer weight (SSHW) (Erkelenz et al. 2014): ACT-GGG-GAG-ATC-AG > ACC-GGA-GAG-ATC-AG (#5607/#5683, #5682/#5665).
Cell culture and RNA isolation
HeLa cells (ATCC CCL-2, mycoplasma-free) were cultivated in T75 flasks with 12 mL Dulbecco's high-glucose modified Eagle's medium (Gibco 41966), supplemented with 10% fetal calf serum (PAN Biotech P30-3031) and 1% penicillin/streptomycin (Gibco 15140-122). Transient transfection experiments were performed with six-well plates at 2.5 × 105 cells per well by using TransIT-LT1 Transfection Reagent (Mirus Bio, LLC US MIR2305), according to the manufacturer's instructions. Total RNA was isolated 24 h posttransfection. The cells were washed with PBS to remove medium residues and lysed in 500 µL solution D (4 M guanidinium thiocyanate, 25 mM sodium citrate, 0.5% sarcosyl, and 0.1 M β-mercaptoethanol). Adherent cells were detached by scraping and transferred into a tube. RNA isolation continued with phenol/chloroform extraction. Therefore, the tubes were supplied with β-mercaptoethanol (Sigma-Aldrich M3148), 2 M sodium acetate (pH 4.0), phenol (Roth A980.3), and chloroform/IAA (24:1). After vortexing and centrifugation, the watery phase was transferred to a new tube with isopropanol (VWR 20842.330) for RNA precipitation. RNA pellets were washed with ethanol (70%, Sigma-Aldrich 1.07017), dried, and resuspended in DEPC-ddH2O. RNA concentration was measured using NanoDrop 1000. Isolated RNA was stored at −80°C.
Inhibitors
Cycloheximide (CHX) was used as the primary translation inhibitor during this work. CHX was first reported as a naturally occurring fungicide. In eukaryotic cells, it rapidly blocks translation elongation by binding to the E-site of the 60S large ribosomal subunit and interference with the residing deacetylated tRNA. This interference blocks the dissociation of the tRNA within the E-site and therefore the movement of the ribosomal subunit (Schneider-Poetsch et al. 2010). The used CHX stock solution had a concentration of 100 mg/mL and was stored at 4°C (Sigma-Aldrich, C4859-1ML). For translation inhibition, the stock solution was added to the cell culture medium 18 h posttransfection, to an end concentration of 50 µg/mL and incubated at 37°C and 5% CO2 for 6 h.
Caffeine was described to reduce the NMD activity by interference with the UPF1 phosphorylation cycle, by inhibition of SMG1 (Keeling et al. 2013). A 100 mM caffeine stock solution (Roth N815.1) was prepared in serum and antibiotic-free DMEM (Gibco). For translation inhibition, the stock solution was added to the cell culture medium 18 h posttransfection, to an end concentration of 10 mM and incubated at 37°C and 5% CO2 for 4 h.
RT-PCR and qRT-PCR analysis
For RT-PCR analysis, RNA was reverse transcribed using Superscript III Reverse Transcriptase (Invitrogen 18080–085) and oligo(dT) primer (Roche 10814270001). cTPI was analyzed using primer pair #5664/#5665, while plasmid TPI was amplified with primer pair #4324/#5665. The transfection control plasmid encoding HGH was detected using primer pair #1224/#1225, and the cellular NMD control SRSF3 was detected using primer pair #4003/#4004. PCR products were separated on nondenaturing 10% polyacrylamide gels.
Quantitative PCR (qPCR) reactions were carried out in a final reaction volume of 20 μL. Each reaction mixture consisted of 2 μL of synthesized cDNA, 10 μL of Forget-Me-Not EvaGreen qPCR Master Mix (Low ROX, Biotium), 1 μL of forward primer (10 pmol, Metabion), 1 μL of reverse primer (10 pmol, Metabion), and 6 μL of nuclease-free water (ddH2O). The qPCR was performed on a QuantStudio 5 (Applied Biosystems) under the following thermal cycling conditions: initial denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 10 sec, annealing and combined extension for short PCR products at 56°C for 30 sec. Data collection and analysis were conducted using the accompanying software provided by QuantStudio 5. To ensure data reliability, all qPCR reactions were performed in triplicate. Negative controls (no-template controls, NTC) were included in each run to rule out contamination.
Isolation, reamplification, and sequencing of DNA bands from PAA gels
The DNA bands were excised from the PAA gel using a clean scalpel (Feather) and transferred to 1.5 mL SafeSeal reaction tubes (Sarstedt). The gel slices were incubated with 100 µL diffusion buffer (0.5M NH4Ac, 10 mM MgAc, 1 mM EDTA, 0.1% SDS) for 30 min in a heating block at 50°C (Thermomixer comfort, Eppendorf), followed by centrifugation for 1 min at 13,000 rpm (Eppendorf Centrifuge 5430). The supernatant was mixed with isopropanol and processed using the QIAquick Gel Extraction Kit (Qiagen), according to the manufacturer's instructions. To reamplify the DNA eluted from the PAA gel, 4 µL of the DNA served as the template for a subsequent PCR reaction using the same primer pair, with the cycle number adjusted to 34. The PCR products were analyzed on a 1% agarose gel, and the bands of interest were excised and purified using the QIAquick Gel Extraction Kit (Qiagen), according to the manufacturer's protocol. The DNA concentration was measured using a NanoDrop spectrophotometer (ND-1000 Version 3.7.0), and the samples were prepared for sequencing. For PCR products smaller than 300 bp, 1 ng/µL and for products larger than 300 bp, 5 ng/µL were sent to Eurofins (Eurofins Genomics Germany GmbH) in an end volume of 15µL for sequencing.
Bioinformatics tools
For the adjustment of splicing regulatory elements (SREs) in the proximity of splice sites, the HEXplorer tool was employed (https://rna.hhu.de/HEXplorer/) (Erkelenz et al. 2014). For the calculation of intrinsic splice site strength, the H-bond score (HBS) tool was used (https://rna.hhu.de/HBond/).
ACKNOWLEDGMENTS
We thank Niels Gehring and colleagues for providing the original TPI reporter constructs. We would like to extend our gratitude to Yvonne Dickschen, Alexandra Graupner, and Björn Wefers for their excellent technical assistance throughout the project. This work was supported by the Jürgen Manchot Foundation (to L.P., L.W., and C.M.).
Author contributions: L.W., L.M., and H.S. conceptualized the study and designed the experiments. L.P., L.W., C.M., and D.S. performed the experiments. J.P. performed bioinformatic calculations. L.P. and L.M. wrote the manuscript, and all authors reviewed and edited the manuscript. H.S. and L.M. provided supervision.
Footnotes
-
Handling editor: John Woolford
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080134.124.
-
Freely available online through the RNA Open Access option.
- Received June 20, 2024.
- Accepted September 21, 2024.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHORS


Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Laura Peter and Lara Walotka are co-first authors of this paper, “Bioinformatics-driven refinement of the commonly used TPI nonsense-mediated decay reporter system.” Laura is a PhD candidate at the Institute of Virology at the University Hospital Düsseldorf, with a focus on RNA splicing mechanisms and their regulatory implications in the context of viral gene expression and regulation. Lara obtained her PhD in 2022 from the Institute of Virology at the University Hospital Düsseldorf. Her research focused on the interactions between viral transcripts and cellular RNA decay mechanisms, particularly investigating how viruses manipulate RNA stability to evade cellular defenses.
What are the major results described in your paper and how do they impact this branch of the field?
In our paper, we present a bioinformatics-driven refinement of the seven-exon triose-phosphate isomerase (TPI) reporter system used to study nonsense-mediated mRNA decay (NMD). We found that the initial TPI reporter construct generated unintended NMD-sensitive splice variants, which could affect the reliability of results. By utilizing bioinformatic tools like the HEXplorer, we optimized the reporter to fully eliminate these additional splice variants. Consequently, these improvements enhance the reliability of NMD assessments, providing a more consistent tool for studying nonsense-mediated mRNA decay. Our findings emphasize the necessity of careful optimization of reporter constructs to ensure accurate results in NMD research.
What led you to study RNA or this aspect of RNA science?
LW: I have always been fascinated by the detailed, complex, and intertwined regulatory mechanisms of RNA processing. Observing how viruses like HIV-1 interfere with these pathways further fueled my curiosity and strongly influenced my decision to investigate mRNA decay during my PhD.
LP: My fascination with RNA biology began during my undergraduate studies when I first encountered the complexity of RNA splicing in HIV-1. The fact that a virus co-opts the cellular splicing machinery as a genome maximization strategy was particularly intriguing. The intricate regulatory mechanisms of the splicing process and their highly coordinated interactions ultimately inspired me to explore splicing mechanisms in greater depth during my PhD.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
The focus of the paper emerged from an unexpected result: The discovery of additional NMD-sensitive splice variants in the original TPI reporter system, compared to the endogenous TPI, raised significant questions about the reliability of initial results and underscored the importance of validating experimental designs. During the bioinformatic optimization process, challenges arose in refining the reporter system to eliminate these splice variants. This experience highlighted the complexity of RNA splicing and its regulatory mechanisms, emphasizing the need for adaptability in research and meticulous analysis to ensure constructs yield accurate and reliable data.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
LP: A key moment that shaped my scientific journey was my master's project focusing on RNA splicing. This project raised many questions and challenged my understanding. For the first time, I witnessed how a scientific project develops and learned the importance of adaptability. It's crucial not to get stuck on one idea. I enjoy being challenged to rethink concepts and uncover what happens at the molecular level. While this process was sometimes frustrating, it became the most rewarding part of my work—developing a theory and, ideally, seeing it validated.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
LW: I always highly appreciated the collaborative and friendly research environment in our lab group and enjoyed that we were always able to be productive and thriving without missing out on fun and friendship.
What were the strongest aspects of your collaboration as co-first authors?
The strongest aspects of our collaboration were the complementary nature of our research focuses and our shared working group. Lara's PhD project focused on nonsense-mediated mRNA decay as a defense mechanism against viral infections, while Laura's research centered on splicing regulatory elements and their effects on splice site choice and export. Although our broader research topics differ, they intersected in this project, allowing us to bring different perspectives and expertise to the table. The collaborative environment helped integrate our contributions into a cohesive study.
How did you decide to work together as co-first authors?
The decision to work together as co-first authors arose naturally from the supportive team atmosphere in our working group and our shared interest in the research topic. The project brought our expertise together, leading us to collaborate and publish the findings jointly.








