
High-resolution RNA tertiary structures in Zika virus stem–loop A for the development of inhibitory small molecules
- 1Department of Pharmacology and Toxicology, The University of Texas Medical Branch, Galveston, Texas 77555, USA
- 2Department of Biochemistry and Molecular Biology, and Sealy Center for Structural Biology, The University of Texas Medical Branch, Galveston, Texas 77555, USA
- 3Department of Molecular and Cellular Biochemistry, Indiana University, Bloomington, Indiana 47405, USA
- Corresponding author: kaychoi{at}iu.edu
-
Handling editor: Eric Westhof
Abstract
Flaviviruses such as Zika (ZIKV) and dengue virus (DENV) are positive-sense RNA viruses belonging to Flaviviridae. The flavivirus genome contains a 5′ end stem–loop promoter sequence known as stem–loop A (SLA) that is recognized by the flavivirus polymerase NS5 during viral RNA synthesis and 5′ guanosine cap methylation. The crystal structures of ZIKV and DENV SLAs show a well-defined fold, consisting of a bottom stem, side loop, and top stem–loop, providing unique interaction sites for small molecule inhibitors to disrupt the promoter function. To facilitate the identification of small molecule binding sites in flavivirus SLA, we determined high-resolution structures of the bottom and top stems of ZIKV SLA, which contain a single U- or G-bulge, respectively. Both bulge nucleotides exhibit multiple orientations, from folded back on the adjacent nucleotide to flipped out of the helix, and are stabilized by stacking or base triple interactions. These structures suggest that even a single unpaired nucleotide can provide flexibility to RNA structures, and its conformation is mainly determined by the stabilizing chemical environment. To facilitate discovery of small molecule inhibitors that interfere with the functions of ZIKV SLA, we screened and identified compounds that bind to the bottom and top stems of ZIKV SLA.
Keywords
- Zika virus
- Flavivirus
- RNA structure
- stem–loop A
- RNA promoter
- small molecule inhibitor
- X-ray crystallography
INTRODUCTION
RNA molecules are key regulators of various biological processes. In viruses, RNA molecules act as promoters to regulate genomic replication (Barton et al. 2001; Lee et al. 2021), signaling molecules to facilitate viral assembly (Gottlieb et al. 1991; Patel et al. 2017), and mediators to control life cycle progression (Phuphuakrat and Auewarakul 2005; Jayaraman et al. 2014). With this large array of functions, efforts have begun shifting toward determining the potential of both coding and noncoding RNA as druggable targets for the design of small molecule inhibitors (Warner et al. 2018; Falese et al. 2021; Lundquist et al. 2021). However, the dynamic nature of RNA molecules makes chemical spaces within the molecule difficult to target. To aid in the identification of low molecular weight chemical fragments that can bind to a specific target molecule, structure-guided fragment-based techniques are often used (Li 2020; Wang et al. 2021). Chemical fragments have the advantage of interacting within small binding pockets that are normally inaccessible to larger molecules. After the identification of hit fragments, optimization strategies such as linking and merging can be used to design a small molecule inhibitor with high specificity and affinity (Bancet et al. 2020).
Zika virus (ZIKV) is a positive-sense RNA virus in the family Flaviviridae, and is closely related to dengue (DENV), West Nile (WNV), and Japanese encephalitis virus (JEV) (Song et al. 2017). Since its emergence in 2007, ZIKV has become a major threat to global health in both Americas, the South Pacific, and Micronesia (Weaver et al. 2016). ZIKV can be transmitted sexually, through blood transfusions, and trans-placentally, the latter leading to severe birth defects such as microcephaly (Basurko et al. 2009). In adults, it has been linked to the development of Guillain–Barré syndrome, an autoimmune disorder of the peripheral nerves that can lead to paralysis and death (Shahrizaila et al. 2021). Despite the severity of ZIKV infection, there are currently no specific targeted treatments against ZIKV, highlighting the need for the development of safe and effective therapeutics (Knyazhanskaya et al. 2021). The 11 kb ZIKV genome contains three functional domains: an open reading frame (ORF) and 5′ and 3′ untranslated regions (UTRs) responsible for the regulation of viral replication (Fig. 1A; Chambers et al. 1990). The ORF encodes three structural proteins (capsid, premembrane, and envelope proteins) that form viral capsids and seven nonstructural proteins (NS1–NS2A–NS2B–NS3–NS4A–NS4B–NS5) that are involved in viral replication (Lindenbach et al. 2007). Among NS proteins, NS3 and NS5 have enzymatic activities. NS3 consists of an N-terminal protease and a C-terminal helicase domain. NS5 consists of an N-terminal methyltransferase (MTase) and a C-terminal RNA-dependent RNA polymerase (RdRp) domain (Potisopon et al. 2014; Wang et al. 2017). The 5′ and 3′ UTRs of the viral genome are also required for replication. Synthesis of viral RNA requires an RNA promoter element called stem–loop A (SLA) at the 5′ end of the viral genome that is used by viral polymerase NS5 (Fig. 1A; You and Padmanabhan 1999; Bujalowski et al. 2017; Liu et al. 2017b). Flavivirus genome replication begins when NS5 recognizes SLA within the 5′ UTR and synthesizes negative-strand RNA from the genomic positive-strand RNA (You and Padmanabhan 1999). SLA is essential for viral replication, as deletions or mutations within the SLA region abolish viral replication entirely (Filomatori et al. 2011). The SLA structure is also required for NS5 MTase function, mediating 5′ cap methylation at the N7 position (Zhou et al. 2007; Issur et al. 2009). The predicted secondary structures of flavivirus SLAs are highly conserved, making SLA an ideal antiviral target (Choi 2021).
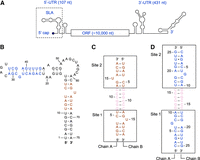
Schematic of the self-complementary bottom and top stem constructs of ZIKV SLA. (A) Schematic representation of Zika virus genome. The genome contains one open reading frame (ORF), flanked by a highly structured 5′ untranslated region (UTR) and a 3′ UTR. The stem–loop A (SLA) structure is located at the 5′ terminus of the 5′ UTR (boxed). (B) The secondary structure of ZIKV SLA based on the 3D structure (Lee et al. 2021). The SLA sequences used for the bottom stem (brown) and top stem (blue) constructs are indicated. (C) Design of bottom stem construct. The GGGCCC linker sequence (pink) connects the U13 and A62 nucleotides of the bottom stem in ZIKV SLA. The RNA construct dimerizes with two identical bottom stem sites, outlined in boxes. (D) Design of top stem construct. The GGGCCC linker sequence (pink) connects the A31 and U36 nucleotides of the top stem in ZIKV SLA. The construct dimerizes with two top stem sites, outlined in boxes.
Previous structural studies have shown that RNA secondary structure motifs (i.e., internal loops and bulges) and noncanonical base-pairing interactions (i.e., wobble base pairs) provide chemical spaces for small molecule interaction, whereas double-stranded Watson–Crick base paired segments do not (Disney et al. 2008, 2020; Ursu et al. 2020). The tertiary structures of two flavivirus SLAs (ZIKV and DENV) have recently been determined, both of which show a well-defined three-way junction consisting of a bottom stem, side loop, and top stem–loop (Lee et al. 2021). The bottom and top stems contain a U-bulge and G-bulge, respectively, which provide unique interaction sites that can be targeted by small molecule inhibitors. In an effort to develop specific small molecule inhibitors against the ZIKV SLA, we determined high-resolution structures of the bottom and top stems using self-complementary single-stranded RNAs (ssRNAs) that dimerize giving rise to two identical stem sites within each structure. We found that the same bulge sequences can adopt various conformations and are stabilized by noncanonical nucleotide interactions both within the same molecule and with neighboring symmetry-related RNA strands. To test their potential to act as drug targets, small molecules known to bind RNA motifs within the bottom and top stem of ZIKV SLA were first identified using the Inforna database (Disney et al. 2016). Several chemical fragments and compounds were then screened for their ability to bind the ZIKV SLA stems using a gel shift assay. Small molecules interact differently with the bottom and top stems, highlighting the potential for the development of a specific inhibitor against ZIKV SLA that can interfere with genomic replication.
RESULTS
Design and crystallization of ZIKV stem structures
The crystal structure of the full-length ZIKV SLA, determined to 3.8 Å resolution, identified the single-nucleotide bulges, internal loops, a three-way junction, and noncanonical base pairs in the molecule (Fig. 1B; Lee et al. 2021). However, structure-based drug design requires high-resolution structures that can provide detailed information of the local chemical environment within the molecule. Thus, high-resolution structures of the bottom and top stems of ZIKV SLA were determined to 2.1 Å and 1.5 Å, respectively. Due to the flexibility of RNA molecules around RNA substructures such as bulges, we wanted to capture at least two sites of interest to observe any conformational differences that exist within these flexible regions. Thus, ZIKV bottom and top stem constructs were designed as single-stranded, self-complementary RNA molecules with a GGGCCC linker sequence to promote dimerization into double-stranded RNA (dsRNA) containing two identical stem sequences. The bottom stem RNA (19 nt) was designed by linking the 8UGAUCU13 and 62AGUAUCA68 sequences with a GGGCCC linker (Fig. 1C). The top stem (25 nt) was designed by linking the 22CAGACUGCGA31 and 36UCGAGUUUG44 sequences with a GGGCCC linker (Fig. 1D). Predicted free energies of bottom and top stem dimerization were calculated at −28.07 and −39.79 kcal/mol, respectively, while the free energies of stem–loop formation from the ssRNAs were at −4.22 and −9.76 kcal/mol, respectively. Thus, the RNA constructs are likely to form a dsRNA, rather than a ssRNA stem–loop (Fig. 1B). Both constructs readily crystallized, and structures were determined to 2.1 Å and 1.5 Å, respectively. Data collection and refinement statistics are shown in Table 1.
Data collection and structure determination
Incorporation into base triples stabilizes the bottom stem U-bulge
The 19 nt bottom stem construct crystallized in the space group P212121 and contained a single dsRNA in the asymmetric unit. The RNA duplex contains two identical bottom stem sequences, each with a U15 single-nucleotide bulge (Fig. 2A). The RNA duplex was arranged by head-to-tail stacking in the crystal lattice, forming a pseudo-continuous helix. The structure was well-ordered and had a clear electron density throughout the entire dsRNA molecule. The bottom stem duplex is continuously stacked except for the U15 single-nucleotide bulges which are flipped out of the dsRNA helix (Fig. 2A). Structural similarity of the two bottom stem sites was evaluated by superposition of the ssRNA chains and dsRNA stem sites (Fig. 2B,C). The two ssRNA chains (A and B) superposed with an RMSD of 1.8 Å (Supplemental Table S1). This structural similarity is carried over into the dsRNA stem sites (sites 1 and 2) in which superposition of the stem sites without the GGGCCC linker sequence (271 atoms) resulted in an RMSD of 1.6 Å (Supplemental Table S1). The major source of structural deviation was observed with the U15 nucleotides (Fig. 2B,C). Upon exclusion of the U-bulge and its flanking pyrimidine nucleotides, 14GUA16, this incongruency is improved as the two stem sites can be superposed with an RMSD of 0.7 Å (Supplemental Table S1).
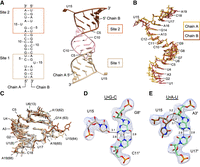
Structure of ZIKV bottom stem. (A) Overall structure of bottom stem of ZIKV SLA. The asymmetric unit contains two ssRNA chains (A and B) that contain two bottom stem sites. Sites 1 and 2 are shown in tan and brown, respectively, and the GGGCCC linker is shown in pink. The 5′ and 3′ termini of chains A and B are labeled. (B) Superposition of bottom stem ssRNA chains. Chains A and B are shown in yellow and dark brown, respectively. (C) Comparison of bottom stem sites with the full-length SLA. The bottom stem sites 1 and 2 (tan and brown, respectively), are superposed onto that of the full-length SLA (gray). (D) The U15•G8′-C11′ base triple interaction. The U15 bulge at site 1 (tan) interacts with the G8-C11 base pair along the minor groove of the 7GGGCCC12 linker of the adjacent symmetry-related RNA molecule (light green). The 2Fo − Fc map surrounding the base triple is contoured at 1.0 σ. Hydrogen bond distances are listed in angstroms (Å). (E) The U15•A3′-U17′ base triple interaction. The U15 bulge at site 2 (brown) interacts along the minor groove of the symmetry-related A3′-U17′ base pair (light green). Hydrogen bond distances are listed in angstroms (Å). The 2Fo − Fc map surrounding the base triples is contoured at 1.0 σ.
The two flipped-out U15 bulges interact with neighboring duplexes via base triple interactions, where the unpaired U base forms hydrogen bonds with a Watson–Crick base pair. In site 1, the U15 bulge interacts along the minor groove of the G8′-C11′ base pair of the neighboring dsRNA molecule, forming a U•G-C base triple (Fig. 2D). Specifically, the O4 of the U15 base forms a hydrogen bond with the N2 of G8′. The frequency of similar U•G-C base triple interactions was determined using the WebFr3D server, which identifies specified RNA tertiary interactions from published RNA structures (Sarver et al. 2008; Abu Almakarem et al. 2012). Twenty-five U•G-C base triples in the database share similar interactions to the U15•G8′-C11′ base triple. In site 2, the U15 interacts with the A3′-U17′ base pair of another neighboring symmetry-related duplex also through the minor groove, forming a U•A-U base triple (Fig. 2E). In this interaction, the O2′ and O3′ of U15 ribose form hydrogen bonds with the N3 and O2′ of A3′, respectively. The U•A-U base triple has been observed more frequently than the U•G-C base triple within site 1. Thirty-five U•A-U base triple interactions were identified with similar geometries to the U15•A3′-U17′ via WEbFr3D.
We next investigated whether the formation of the base triples in the bottom stem affected the A-form dsRNA helical structure using the 3DNA webserver. 3DNA assesses helical structure according to nucleotide shift, slide, and rise, which describe the distances between two bases across the x, y, and z axes, as well as tilt, roll, and twist, which describe the angular relationship between two bases along the x, y, and z axes (Lu and Olson 2008; Li et al. 2019). To accommodate U15 into base triple interactions while preserving the overall dsRNA helical structure, the flanking G14 and A16 nucleotides undergo drastic compensatory changes (Fig. 3). For instance, the helical rise from G14 to U15 deviates from the typical A-form helix of ∼3 Å to −7.0 Å and −8.8 Å at sites 1 and 2, respectively (Fig. 3). This deviation in helical structure is immediately offset from U15 to A16 with a helical rise of 11.3 Å and 12.5 Å for sites 1 and 2, respectively (Fig. 3). This conformational adjustment allows the dsRNA duplex to retain its normal A-form helical structure with stable stacking interactions. This deformation of the helical structure from G14 to U15 and compensatory adjustment in the following U15 to A16 is also seen with the nucleotide shift, tilt, roll, and twist (Fig. 3). Furthermore, sugar puckering plays a role in alleviating the rigidity of the helical structure (Steffen et al. 2020). Within site 2, the ribose of G14 exists in the C2′-endo conformation, as opposed to the C3′-endo conformation more commonly found in A-form RNA. This C2′-endo conformation at G14 allows the O3′ to extend farther away from the dsRNA helix, facilitating U•A-U base triple interactions (Fig. 2C, dark brown).
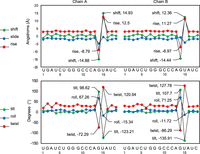
Analysis of bottom stem helical structure. Base step parameters for each RNA chain (shift, slide, rise, tilt, roll, and twist) were analyzed using a 3DNA webserver. Note the largest deviations in the helical structure are found around the U-bulge, from G14 to U15 and from U15 to A16.
The top stem G-bulge is extensively stabilized by base triple interactions, hydrogen bonding, and base stacking
The top stem construct of ZIKV crystallized in the space group P1 and contains two dsRNA molecules per asymmetric unit. Thus, the structure contains four top stem sites (sites 1–4), each containing a single G nucleotide bulge at position 7 (Fig. 4A). A clear electron density was observed throughout the structure with the exception of the G7 nucleobases of sites 2 and 4 (chains B and D), which were thus omitted from the final model. Hence, the top stem structure showed continuously stacked base pairs and two well-defined G7 single-nucleotide bulges within sites 1 and 3. The G7 bulge at site 1 is positioned within the helix and the G7 bulge at site 3 is flipped outside of the helix (Fig. 4A–C). The structural similarity of the top stem was evaluated by superposition of the individual ssRNA chains and dsRNA stem sites (Fig. 4B,C). The four ssRNA molecules superpose with RMSD values ranging from 1.6 Å to 3.3 Å, with the largest deviation seen between chains A and C (Supplemental Table S2). Similarly, the dsRNA stems superpose with RMSD values ranging from 0.9 Å to 2.3 Å, with the largest deviation seen between sites 1 and 3 (Supplemental Table S2). This deviation can be traced back to the G7 bulge and its flanking U6 and C8 pyrimidine nucleotides, as chains A and C contain the G7 bulges of sites 1 and 3. The exclusion of the 6UGC8 nucleotides improved the RMSD value between sites 1 and 3 to 1.1 Å (Supplemental Table S2).
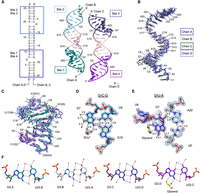
Structure of ZIKV top stem. (A) Overall structure of the top stem of ZIKV SLA. The asymmetric unit contains four ssRNA molecules (A–D). The four top stem sites, 1, 2, 3, and 4 are shown in teal, light blue, indigo, and purple, respectively. The GGGCCC linker is shown in pink. The G7 base of sites 2 and 4 did not contain a well-defined electron density and were thus omitted from the model. (B) Superposition of top stem ssRNA chains. Chains A (blue), B (gray), C (black), and D (lavender) are colored. (C) Comparison of top stem sites with that of the full-length ZIKV SLA. The four top stem sites, colored as in A, are superposed onto that of the full-length SLA (gray). (D) The G7•C8-G19 base triple interaction. The G7 bulge at site 1 is positioned inside the dsRNA helix and interacts with the C8-G19 base pair within the same plane (teal). Additionally, the G7 nucleotide is stabilized by interactions with water and a sulfate molecule. The 2Fo − Fc map surrounding the base triple is contoured at 1.5 σ. Hydrogen bond distances are listed in angstroms (Å). (E) The G7•U6′-A20′ base triple interaction. The G7 bulge at site 3 (indigo) interacts with both sugar edges of the U6′-A20′ base pair (light green). The Watson–Crick face of G7 is additionally stabilized by a glycerol, a water, and ribose sugar of the preceding U6 nucleotide. The 2Fo − Fc map surrounding the base triple is contoured at 1.5 σ. (F) Wobble G-U base-pair interactions. The four G-U bases at sites 1, 2, 3, and 4 are stabilized by hydrogen bonding with water molecules (red spheres) along the Watson–Crick, Hoogsteen, and sugar edges of the nucleotides. A glycerol molecule replaces a water molecule in the G-U wobble pair at site 4 (purple). Hydrogen bonds <3.1 Å are designated by solid lines.
These flexible G7 bulge nucleotides require extensive stabilization within the crystal by incorporation into base triples, hydrophilic solvent interactions, and/or hydrophobic stacking interactions. In site 1, the G7 bulge nucleotide interacts along the same plane of the subsequent C8-G19 base pair through the major groove, forming a G•C-G base triple (Fig. 4D). The helical rise between G7 and C8, drops to −1.8 Å, indicating the planar orientation of the two bases (Fig. 5). Here, the sugar edge of the G7 nucleotide interacts along the Hoogsteen edges of both nucleotides of the C-G base pair. Specifically, N3 and N2 of G7 base form hydrogen bonds with N4 of C8 and O6 of G19, respectively (Fig. 4D). Furthermore, the G7 base forms hydrogen bonds with a sulfate molecule via its N1 and N2 atoms, and water molecules via its O6 and N7 atoms (Fig. 4D). Lastly, G7 is stacked on U6 with a helical rise of 3.1 Å and a slide of 0.7 Å (Fig. 5). Interestingly, these various interactions are conserved in six geometrically similar G•C-G base triples identified using WebFr3D (Supplemental Fig. S1), all of which contain various hydrophobic stacking and hydrophilic solvent interactions that stabilize the G-bulge.
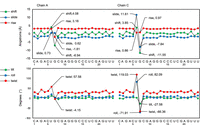
Analysis of top stem single-stranded helical structure. Base step parameters for each RNA chain (shift, slide, rise, tilt, roll, and twist) were analyzed using a 3DNA webserver. Note the largest deviations in the helical structure are found around the G-bulges within chains A and C, from U6 to G7 and G7 to C8.
In site 3, the G7 bulge nucleotide is flipped out of the helix, with its purine base in the higher energy syn conformation (Fig. 4E). Similar to the bottom stem U-bulges that flipped out of the helix, the compensatory behavior of ssRNA nucleotides (shift, slide, tilt, roll, and twist) around this G7 bulge nucleotide was observed, as measured by 3DNA (Fig. 5). The G7 nucleotide is stabilized along its sugar, Hoogsteen, and Watson–Crick edges via incorporation into a G•U-A base triple and interactions with solvent molecules (Fig. 4E). G7 interacts along the minor groove of a U6′-A20′ base pair from a neighboring symmetry-related molecule. Here, the O2′ of G7 ribose interacts with the O2′ and N3 of A20′, and the N7 of G7 interacts with the O2′ of U6′ (Fig. 4E). A glycerol and water molecule additionally form hydrogen bonds with N1, N2, and O6 of the G7 base (Fig. 4E). Interestingly, no geometrically similar G•U-A base triples were identified via WebFr3D. This may be due to the rotation of G7 into the higher energy syn conformation, thus necessitating the extensive stabilization seen (Murthy et al. 1999; Sokoloski et al. 2011).
The top stem additionally contains four G3-U23 wobble base pairs that create solvent-accessible cavities along both major and minor grooves (Fig. 4F). The N1 and O6 of G3 form hydrogen bonds with N3 and O2 of U23, respectively. Thus, the O4 of U23 protrudes into the major groove, and the N2 of G3 protrudes into the minor groove of the RNA helix (Fig. 4F). In all four G-U wobble pairs, three water molecules coordinate with the O4 of U23 base and the N7 and O6 of G3 base in the major groove (Fig. 4F). In the minor groove, water molecules coordinate the N2 and/or N3 of G3, and the O2 of U23 (Fig. 4F). In site 4, an additional glycerol molecule forms hydrogen bonds with N2 and N3 of G3. These hydrogen bonds along the major and minor grooves represent potential binding sites for the design of specific small molecule inhibitors.
The full-length ZIKV SLA also exhibits flexibility around the bulge nucleotides
The bottom and top stem structures were compared to the respective segments in the full-length ZIKV SLA structure (Figs. 2C and 4C). As expected, the overall structures of the bottom and top stem were similar to those of the full-length SLA except around the bulge nucleotides. In the bottom stem of the full-length ZIKV SLA, the U64 bulge points toward the dsRNA helix, as opposed to the corresponding U15 bulges that flipped out of the dsRNA helix in the isolated bottom stem (Fig. 2C). The bottom stem sites 1 and 2 superposed onto the full-length structure with an RMSD of 1.9 Å and 2.3 Å, respectively (Fig. 2C; Supplemental Table S3). The exclusion of U15 along with the flanking purine nucleotides (14GUA16) in sites 1 and 2 improves RMSD values to 0.9 Å and 1.0 Å for the respective superposition onto the full-length SLA structure, demonstrating structural flexibility of the single-nucleotide U-bulge (Supplemental Table S3). In the top stem of the full-length ZIKV SLA, the G28 bulge points toward the dsRNA helix in a different conformation from those observed in the isolated top stem structures (Fig. 4C). The four isolated top stem sites can be superposed onto that in full-length ZIKV SLA with RMSD values ranging from 1.5 Å to 2.3 Å, suggesting overall structural similarity (Fig. 4C; Supplemental Table S3). The exclusion of G-bulge and its flanking pyrimidine nucleotides, 6UGC8, improves the superposition with RMSD values ranging from 1.3 Å to 1.6 Å (Supplemental Table S3).
Small molecules differentiate between bottom and top stem ZIKV SLA for binding
The structural motifs of bulges, base triples, and wobble pairs observed within the bottom and top stem structures provide chemical spaces to which small molecules can specifically bind. To investigate the potential chemical spaces within the ZIKV SLA bottom and top stems, a total of 12 candidate molecules were tested for their RNA-binding ability (Figs. 6A and 7A). The workflow of generating these candidates is outlined in Figure 6A. First, the Inforna ligand database (Disney et al. 2008, 2016) was used to search for small molecules that are known to bind secondary structures present within ZIKV SLA. The database contains over 1900 RNA motifs and associated small molecule binders that were identified experimentally using small molecule microarrays (Disney et al. 2008, 2016). The SLA sequence of ZIKV returned 23 potential binding compounds, four of which matched the exact target sequence motif of the bottom stem U-bulge (5′GUA/3′UC) and one which matched the exact sequence motif of the top stem G-bulge (5′UGC/3′GA) (Fig. 6B). The similarity search feature in PubChem (Kim et al. 2023) was then used to expand the five compounds into 10 (Supplemental Fig. S2). Next, fragmentation of the 10 compounds by eMolFrag, an open-source molecular fragmentation software (Liu et al. 2017a), was used to generate a list of 17 chemical fragments, which were expanded to 23 fragments using the PubChem structure similarity tool. Eight of the 23 chemical fragments were included in the final list of molecules to screen for RNA-binding potential (Fig. 7A). Additionally, four compounds were manually selected. d-glucose was one of the expanded fragments, and thus, sucrose was included in the list to compare RNA-binding effects of monosaccharides and disaccharides (Fig. 6A). Since many of the compounds identified by Inforna included aminoglycoside antibiotics, kanamycin and two candidates from other antibiotic drug classes, ampicillin and chloramphenicol, were also included to assess for RNA-binding ability (Fig. 6A).
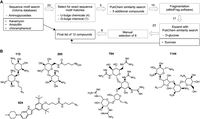
Selection of molecules for RNA-binding assay. (A) Workflow of generating the list of 12 molecules for RNA-binding assay. The number of chemicals selected after each step is listed on the arrows. Manually selected compounds are shown in black boxes. (B) Compounds identified by Inforna. Five compounds identified by Inforna matched the exact sequence motifs of the bottom stem U-bulge (Inforna ID: 112, 295, 784, 924) or top stem G-bulge motifs (Inforna ID: 1144).
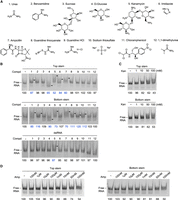
Screening for the RNA-binding potential of selected chemicals. (A) Twelve chemicals assayed for their ability to bind the isolated ZIKV SLA stem constructs. (B) Gel shift assay. The chemicals listed in A (50 mM) were incubated with top and bottom stem RNA and the negative control dsRNA (150 nM), and the mixtures were resolved via native PAGE. Band shifts are indicated by a triangle arrow to the right side of the lane. Band intensities of the remaining RNA were quantified and normalized against the control lane. Interaction between the chemical and RNA was identified via deviation of the band intensity by more than 10% from that of the control lane. (C) Gel shift assay with kanamycin. Top and bottom stem RNA (1 µM) was incubated with an increasing concentration of kanamycin from 1 to 100 mM. Quantified band intensities for dsRNA are shown below the gel. (D) Gel shift assay with ampicillin. Top and bottom stem RNA (1 µM) was incubated with an increasing concentration of ampicillin from 100 nM to 100 mM.
To test for RNA-binding ability, 300 nM of top and bottom stem ssRNAs (150 nM when duplexed into dsRNA) were incubated with 50 mM of each of the 12 selected molecules in water and resolved using 12% native polyacrylamide gel electrophoresis (PAGE) (Fig. 7A,B). As a negative control, a dsRNA, generated from fully complementary ssRNAs lacking secondary substructures, was tested for compound interaction. The dsRNA control did not display a significant shift for any of the compounds tested except kanamycin (Fig. 7B). The RNA bands were quantified using ImageJ and normalized against the control lane. RNA band intensities that deviated more than 10% from that of the control lane were taken as positive binding interactions between the chemical and RNA. Three compounds, sucrose (3), chloramphenicol (11), and 1,1-dimethylurea (12) did not exhibit binding to either top or bottom RNA. The remaining compounds showed different preferences between the bottom and top stems. Urea (1), d-glucose (4), kanamycin (5), and ampicillin (7) bound to both stems. d-glucose (4) increased the intensity of the lower migrating RNA band (ssRNA) in both constructs, suggesting disruption of the dsRNA duplex into ssRNA (Fig. 7B). This shift was more prominent in the top stem. Imidazole (6) bound only the top stem. Benzamidine (2), guanidine thiocyanate (8), guanidine HCl (9), and sodium thiosulfate (10) bound only the bottom stem, and increased the intensity of the dsRNA band, while reducing the intensity of the ssRNA band, suggesting that these chemicals stabilize the dsRNA molecule (Fig. 7B). All selected RNA-binding compounds contain polar atoms, positive charges, or planar rings, all of which have been implicated in the ability for small molecules to interact with RNA (Warner et al. 2018; Rizvi et al. 2020). For example, urea, d-glucose, guanidine, and thiosulfate likely bind RNA via hydrogen bonding or electrostatic interactions. Additionally, the planar structure of benzamidine and imidazole could interact with RNA via intercalation in addition to potential electrostatic or hydrogen bonding interactions.
Among the antibiotic compounds tested, kanamycin (5) and ampicillin (7) bound both RNA stems, but chloramphenicol (11) did not. Chloramphenicol lacks an amine group, which is present in both kanamycin and ampicillin, suggesting that the positively charged amine likely plays a role in binding the top and bottom stem RNAs (Fig. 7A). To further investigate the ability for these antibiotics to bind the ZIKV RNA stems, 1 µM of each RNA was incubated with kanamycin (5) in concentrations from 1 mM to 100 mM or ampicillin (7) in concentrations from 100 nM to 100 mM in water (Fig. 7C,D). Kanamycin displayed a band shift with both the bottom and top stem between 1 and 10 mM (Fig. 7C). Ampicillin displayed a band shift with the bottom stem starting at 100 µM and the top stem at 10 mM (Fig. 7D). Furthermore, there is a progressive decrease in the intensity of the lower ssRNA band as the ampicillin concentration increases for both RNAs, suggesting that ampicillin may stabilize the dimerized dsRNA duplex. This effect occurs more rapidly with the bottom stem (Fig. 7D). Although antibiotic groups are shown to interact with off-target protein and RNAs (Zapp et al. 1993), the control dsRNA only bound kanamycin with weaker interactions than that seen with the bottom and top stem RNAs (Fig. 7B). Thus, the compounds corresponding to the shifts seen in the bottom and top stem RNAs represent chemicals that bind RNA bulges, and further substantiate the potential to use a fragment-based approach in building a small molecule to target specific RNA molecules.
DISCUSSION
RNA can form complex 3D structures through canonical Watson–Crick base-pairing and other tertiary interactions mediated by noncanonical bonds. These RNA structures are as intricate and stable as protein structures and similarly recognize RNA, protein, and ligands with high affinity and specificity (Rother et al. 2011). However, a major hindrance in the development of small molecule inhibitors against RNA molecules is the lack of sufficient information on how to create “drug-like” RNA-binding small molecules (Rizvi et al. 2020). Small molecule-binding sites in RNAs are considered to be more shallow, hydrophilic, and conformationally flexible than those of proteins (Hermann 2016). Thus, the chemical structures of small molecules that bind RNA may be different from those that bind proteins, making the design of small molecule compounds that target RNA difficult. Furthermore, despite databases such as Inforna storing over 1900 RNA-small molecule interactions, little information is available on how these small molecules actually interact with their targets (Disney et al. 2016). Because of this, we wanted to use X-ray crystallography to obtain high-resolution structures of RNA targets and identify potential sites for small molecule interactions. Since the previous ZIKV SLA structure is limited to 3.8 Å resolution (Lee et al. 2021), we determined the structures of isolated bottom and top stems of ZIKV SLA to high resolution (2.1 Å and 1.5 Å, respectively) as a step toward identifying small molecule binders to the flavivirus RNA promoter.
We designed self-complementary, ssRNA molecules whose dimerization was promoted by the incorporation of a GGGCCC linker sequence. This design generates two identical stem sites per dsRNA molecule for a structural analysis of RNA dynamics. We found that the single-nucleotide U- and G-bulges in the bottom and top stems of ZIKV SLA are dynamic and their bulge motifs (14GUA16 or 7UGC9, respectively) adopt various conformations to stabilize the unpaired nucleotide. Non-Watson–Crick interactions such as base triples play a major role in stabilizing the single-nucleotide bulges. We observed U•G-C, U•A-U, G•C-G, and G•U-A base triple interactions via Watson–Crick, Hoogsteen, and sugar edges of the unpaired U or G nucleotide. These base triple geometries exist in other RNA structures with varying degrees of frequency (25 U•G-C, 35 U•A-U, and six G•C-G base triples), suggesting that specific interactions mediating base triple geometries are conserved. Since all four bases are shown to form a base triple with Watson–Crick base pairs, base triple interactions may be more prevalent than previously reported (Abu Almakarem et al. 2012). This has implications on the stabilization of viral RNA genomes. Recent RNA sequencing and SHAPE analysis have shown that viral RNA genomes form many secondary structures (Rausch et al. 2018; Boerneke et al. 2019; Huber et al. 2019; Schroeder 2020), and thus, these secondary structures may further be stabilized by incorporation of unpaired nucleotides into base triples within cells and virions.
These RNA structures also provide targets for small molecule interactions, which are likely within solvent-accessible cavities in RNA structural motifs, such as bulges, hairpins, and three-way junctions, and in non-Watson–Crick base pairs, such as wobble pairs. Small molecules that target RNA would require unique site-specific recognition mediated by specific stacking, hydrogen bonding, and electrostatic interactions (Vicens and Westhof 2001, 2002; Disney et al. 2014). We observe similar chemical interactions and arrangements of solvent molecules for G7 bulges and G-U wobble pairs in our structures and in other RNA structures (U•A-U or U•G-C base triple interactions) (Fig. 4D–F). Thus, small molecules that can mimic these observed interactions can be designed to target the chemical spaces within RNA. To determine the feasibility of designing a small molecule inhibitor against SLA, we tested the RNA-binding ability of 12 different chemicals against the isolated bottom and top stems of ZIKV SLA. To understand the chemical properties of small molecules that bind to RNA, we identified known RNA binders using Inforna and fragmented them to assess the binding of their individual chemical components. We found that even these compounds discriminate between the two RNA structures (Fig. 7B). For example, ampicillin binds to the bottom stem at 10 µM, whereas it does not show band dispersion with the top stem until at 10 mM (Fig. 7D). Furthermore, we show the utility of using fragment-sized chemicals in exploring the chemical spaces of RNA, as sucrose failed to exhibit any binding, whereas its monomer component d-glucose displayed a strong RNA-binding interaction (Fig. 7B). The design of small molecule inhibitors against RNA structures is a growing area of research. Thus, an understanding of RNA structure and the chemical interactions mediating the stabilization of secondary motifs is essential in unlocking the druggable potential of RNA. The high-resolution structures of the ZIKV SLA stems provide a starting point for the identification of hit fragments that bind within chemical spaces of RNA structures. Future structural studies of SLA and small compounds will help facilitate the design of a small molecule inhibitor that interferes with the SLA promoter-NS5 polymerase interaction.
MATERIALS AND METHODS
Design of bottom and top stem ssRNA construct
The short stem–loop structures of the top and bottom stem of ZIKV SLA could open up and form a duplex via their complementary sequence, leading to a heterogeneous population. We thus designed a self-complementary ssRNA that contains two identical stem sequences. To enforce a duplex structure, we added a GGGCCC linker sequence between the two sites. This construct design eliminates the need to synthesize two separate ssRNAs to form a duplex. Further, incorporating two identical sites within the duplex would allow us to compare the structures in the same chemical environment (crystallization condition) to address RNA dynamics. The sequence of the bottom stem construct is 5′-UGAUCUGGGCCCAGUAUCA-3′ (19 nt) and the sequence of the top stem is 5′-CAGACUGCGAGGGCCCUCGAGUUUG-3′ (25 nt), where the two complementary stem sequences are underlined and the single-nucleotide bulge is bolded. The designed ssRNAs were synthesized by IDT Technologies. The dimerization of each construct was predicted using the RNAcofold server and stem–loop formation was predicted using the RNAfold server within the ViennaRNA web services (http://rna.tbi.univie.ac.at) (Lorenz et al. 2011).
Crystallization and structure determination
The bottom and top stem RNAs were dissolved in molecular biology grade water at a concentration of 300 µM and subjected to crystallization trials using commercially available sparse-matrix crystallization screens followed by optimization of positive crystallization hits. Optimized crystals of both RNAs were grown using the hanging-drop vapor diffusion method at 18°C. Initial crystals for the bottom stem RNA were obtained from reservoir G10 of the Hampton Index-HT commercial screen. The final crystallization condition for bottom stem RNA contained 100 mM Bis-Tris propane pH 7.0, 200 mM MgCl2, and 18% polyethylene glycol (PEG) 3350. Crystals appeared overnight and grew to full size within 5 d. The crystals were cryoprotected using 20% glycerol, and diffraction data were collected at the Advanced Light Source (ALS) beamline 8.2.2. The crystals diffracted to 2.1 Å, and belong to the space group P212121 with a = 28.32 Å, b = 41.95 Å, and c = 97.66 Å. The asymmetric unit contains two ssRNA molecules. Initial crystals for the top stem RNA were obtained from reservoir A7 of the Molecular Dimensions Wizard 4 commercial screen. The top stem RNA crystallization solution was optimized to 100 mM sodium acetate pH 4.5, 100 mM MgSO4, 1.6 M Li2SO4, and 5% 2-propanol. Crystals appeared within 3 d and grew to full size within 10 d. The top stem crystals were cryoprotected using 20% glycerol, and the diffraction data were collected at the Advanced Photon Source (APS) beamline 21-ID-D. The crystals diffracted to 1.5 Å and belong to the space group P1 with a = 29.23 Å, b = 52.53 Å, and c = 53.67 Å and α = 63.6°, β = 88.1°, and γ = 85.9°. The asymmetric unit contains four ssRNA molecules. The bottom and top stem structures were determined using small dsRNA molecules (PDB 3ND3) (Mooers and Singh 2011) as search models for molecular replacement using Phaser within Phenix suite (Liebschner et al. 2019). The initial models were rebuilt using Coot and refined using the Phenix refinement application within the Phenix suite (Emsley and Cowtan 2004; Liebschner et al. 2019). Atomic coordinates for top and bottom stem RNAs are deposited to PDB with accession numbers 8TQX and 8TSV, respectively.
RNA structure analysis
The ssRNA chains and dsRNA stem sites were compared by calculating the RMSD of atomic positions using the align function within PyMOL (http://www.pymol.org/pymol). All the atoms were used in RMSD calculations. To compare the dsRNA stem sites, two sets of RMSD calculations were performed. In the first, the GGGCCC linker sequence was excluded prior to the superposition of the dsRNA stem sites. In the second, the respective U15 and G7 bulges and their flanking nucleotides (14GUA16 and 6UGC8) were excluded in addition to the GGGCCC linker sequence. The individual bottom and top stem sites were also superposed onto their respective sites within the full-length ZIKV SLA structure in the same manner (PDB code 7LYG) (Lee et al. 2021). The RNA helical structures were analyzed using the Web 3DNA webserver (http://web.x3dna.org/) (Lu and Olson 2008; Li et al. 2019). The “local base step analysis” within the 3DNA webserver was used to determine the changes in nucleotide shift, tilt, slide, roll, rise, and twist occurring from base to base along the ssRNA structure (Li et al. 2019). Tertiary interactions involving noncanonical Watson–Crick base-pairing interactions and solvent interactions were identified using the Chimera FindHBond function (Pettersen et al. 2004).
The WebFR3D server (https://www.bgsu.edu/research/rna/web-applications/webfr3d.html) was used to search published RNA structures for similar base triple interactions to those observed within the bottom and top stem structures (Sarver et al. 2008). A symbolic search was performed for the three bases in the RNA base triple, where the identity of bases and their interaction with one another were specified. Cis (c), near-cis (nc), trans (t), and near trans (nt) base interactions involving the different ribonucleotide edges, Watson–Crick (W), Sugar (S), and Hoogsteen (H) edges, were screened for. To identify U•A-U base triples, a search for cSS/ncSS between U1-A2 and cWW between A2-U3 was performed. To identify U•G-C base triples, a search for cWS/ncWS between U1-G2 and cWW between G2-C3 was performed. To identify G•C-G base triples, a search for cSH/ncSH between G1-C2(next), tSH/ntSH between G1-G3, and cWW between C2-G3 was performed. Finally, to identify G•U-A base triples, a search for tHS/ntHS between G1-U2, cSS/ncSS between G1-A3, and cWW between U2-A3 was performed. Constraints were set to permit the query to pull near exact interactions in addition to exact interactions as those seen in our structures. A resolution cutoff of 3.5 Å using X-ray crystallography and cryo-EM was applied to limit the structures to those determined to a high resolution.
Generation of a compound list for small molecule screening
The Inforna ligand database (Disney et al. 2016) was used to identify known small molecules that bind to secondary structures present within ZIKV SLA. The database mines RNA motifs inferred based on the target RNA sequence, and those secondary structures are then searched against a database of 1936 RNA-binding small molecules. The database search using the full-length ZIKV SLA resulted in 23 compounds, which were further filtered based on whether the targeted sequence motifs were an exact match with the U- and G-bulge sequences. Four compounds were identified for the 5′GUA/3′UC bulge and one compound for the 5′UGC/3′GA bulge. The output includes the chemical name, structure, SMILES notation, a fitness score, and PMID. The five small molecules were then expanded into 10 chemicals using the similarity search feature within PubChem (Kim et al. 2023). The open-source, molecular fragmentation software, eMolFrag was next used to generate a list of 17 chemical fragments from those 10 small molecules (Liu et al. 2017a). A Tripos mol2 file is used as the input and the software generates a list of nonredundant fragment “bricks” and “linkers.” The PubChem similarity search feature was used again to expand the 17 chemical fragments into 23. Kanamycin, ampicillin, chloramphenicol, and sucrose were manually selected to be included in the list of chemicals to screen for binding. The final list of chemicals tested includes eight of the 23 chemical fragments and the four chosen chemicals.
Screening of small molecules using a gel shift assay
Selected chemicals and RNA were prepared in molecular biology grade water. The bottom and top stem ssRNAs (final duplex concentration of 150 nM) were each mixed with 50 mM of each chemical for 15 min at room temperature. Additionally, a negative control dsRNA was designed as two fully complementary ssRNA molecules lacking any secondary substructures 5′-GCCUCGCUGCCGUCGCCA-3′ (18 nt) and 5′-UGGCGACGGCAGCGAGGC-3′ (18 nt). The negative control ssRNA molecules were refolded to form the dsRNA molecule (150 nM) and incubated with 50 mM of each chemical for 15 min at room temperature. The mixtures were resolved on a 12% polyacrylamide gel in 1× TBE running buffer (100 mM Tris-HCl, 100 mM boric acid, 2 mM EDTA) and run at room temperature for 10 min at 120 V. Gels were stained with GelRed nucleic acid stain and binding was assessed by RNA band shifts. Gel shift assays were performed in triplicate for each RNA. ImageJ was used to quantify RNA band shifts (Schneider et al. 2012). RNA-binding interactions of each chemical were determined by measuring the proportion of the remaining dsRNA band to the total RNA signal of the lane and normalized to the control. To determine concentration-dependent binding, 1 µM of the bottom and top stem RNAs were incubated with kanamycin at concentrations ranging from 1 mM to 100 mM and ampicillin at concentrations ranging from 100 nM to 100 mM for 15 min at room temperature. The mixtures were resolved and analyzed as described above.
ACKNOWLEDGMENTS
We thank Dr. Mark White of the Sealy Center for Structure Biology at UTMB for help with X-ray data collection and helpful discussions. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). Beamline 8.2.2 of the Advanced Light Source, a DOE Office of Science User Facility under Contract No. DE-AC02-05CH11231, is supported in part by the ALS-ENABLE program funded by the National Institutes of Health, National Institute of General Medical Sciences, grant P30 GM124169-01. The authors acknowledge the Sealy Center for Structural Biology at the University of Texas Medical Branch at Galveston for providing research resources. This work was supported by NIH grants R01AI 187856 and U19AI171413 (to K.H.C.) and a Kempner predoctoral fellowship (to J.T.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079796.123.
- Received August 18, 2023.
- Accepted January 30, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Jerricho Tipo is the first author of this paper, “High-resolution RNA tertiary structures in Zika virus stem–loop A for the development of inhibitory small molecules.” Jerricho is an MD-PhD student studying at the University of Texas Medical Branch at Galveston and working in Kay Choi's lab at Indiana University. The laboratory aims to understand the role of noncoding viral RNAs in mediating viral life cycle events using structural and biochemical techniques.
What are the major results described in your paper and how do they impact this branch of the field?
Fragment-based drug discovery (FBDD) is a valuable method to generate small molecule inhibitors by identifying “fragment-sized” compounds that interact with a target of interest (often proteins) and enhancing the potency of hit fragments via linking, merging, or growing strategies. RNA molecules form intricate tertiary structures and act as important effector molecules as protein molecules do; however, their potential for FBDD has not been fully elucidated. Using X-ray crystallography, we isolated the structure of two RNA bulge substructures of the Zika virus stem–loop A, an RNA promoter structure for viral genome replication. By designing the RNA using complementary strands, we were able to visualize at least two sites of interest within the crystallographic asymmetric unit and found that these bulge regions are flexible and are stabilized by different interactions, despite having identical sequences. Furthermore, we tested the binding of small molecule fragments and show that they preferentially bind the two RNAs, thus offering the potential to exploit FBDD to develop small molecule inhibitors that can specifically target RNA molecules.
What led you to study RNA or this aspect of RNA science?
When I was an undergrad, how people determined the structures I saw in my textbooks was a big mystery to me. So, I entered my graduate studies wanting to learn “how to do structural biology.” Although I had no idea what system I wanted to study, I knew that I wanted to investigate the mechanisms of biomolecular interactions at the atomic level. In finding a laboratory to join, I met with Dr. Kay Choi, who works on understanding how structures within the RNA viral genome mediate essential processes in viral replication and infection. Admittedly, I previously considered RNAs to be useful only in protein translation, so in our first meeting, I was surprised by the importance of viral RNAs in mediating nontranslation events. The more we spoke, the more I realized that there is still much to be discovered within the RNA field, and the fact that I was left with multiple big question marks at the end of our meeting inspired me to join her laboratory.
I currently study RNA interactions using X-ray crystallography and biochemical techniques, and this has allowed me to develop a keen interest in RNA structures and their geometries. It amazes me how noncoding RNA structures play an important role in coordinating interaction with their protein-binding partners, almost as if they have a mind of their own. I want to continue studying RNA, specifically in understanding the chemistry and energetics driving specific RNA–RNA and RNA–protein interactions. Under Kay's guidance and the support of everyone in the laboratory, I've been able to grow as a junior scientist and find out what is interesting to me and how I can add to our understanding of RNA.
As Keerthi (a research scientist in our laboratory) told me when I first joined: “Why do I like RNA? It has four standard bases, but endless possibilities!”
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
I played the piano growing up. I remember I would get extremely frustrated with my piano teacher when she would emphasize the importance of developing a strong technique as a foundation before starting a new piece, so we would repeatedly crank out technique exercises. I remember I did not have the patience to painstakingly go through every note, and she would make sure to correct me when I misplayed them. And I remember that she would push me to keep trying at a section that I couldn't play, by helping me identify what my fingers wouldn't do. While the younger me would sometimes end these lessons in extreme frustration, these experiences instilled in me many skills that I fall back on today, such as developing a framework for learning new things, being detail-oriented, and remaining persistent. I often hear PI's saying that science is an art, and I am surprised at how often it takes a bit of creativity to troubleshoot experiments and a lot of inspiration to come up with new ideas to test. As a graduate student, I'm glad I have these foundational skills that I can continue to develop as I grow into the scientist that I would like to become in the future.
What are your subsequent near- or long-term career plans?
I am nearing the end of my research years, so my biggest short-term goal is to successfully defend my dissertation work, followed by finishing my remaining two years of medical school. I have only become more interested in RNA molecules and the many functions that they hold. Something I find particularly interesting is the oxymoronic nature of RNA to be highly flexible yet stably structured. My long-term career plans involve being able to further explore this facet of RNA molecules (such as with RNA scaffolds and manipulating RNA chemistry) and bringing them into the clinical setting as bioengineered therapeutics.








