
LARP4 is an RNA-binding protein that binds nuclear-encoded mitochondrial mRNAs to promote mitochondrial function
- Benjamin M. Lewis1,2,3,4,
- Chae Yun Cho4,
- Hsuan-Lin Her1,2,3,5,
- Orel Mizrahi1,2,3,
- Tony Hunter4 and
- Gene W. Yeo1,2,3
- 1Department of Cellular and Molecular Medicine, University of California San Diego, La Jolla, California 92037, USA
- 2Institute for Genomic Medicine, University of California San Diego, La Jolla, California 92037, USA
- 3Stem Cell Program, University of California San Diego, La Jolla, California 92037, USA
- 4Molecular and Cell Biology Laboratory, Salk Institute for Biological Studies, La Jolla, California 92037, USA
- 5Bioinformatics and Systems Biology Graduate Program, University of California San Diego, La Jolla, California 92037, USA
- Corresponding authors: geneyeo{at}ucsd.edu, hunter{at}salk.edu
-
Handling editor: Fatima Gebauer
Abstract
Mitochondria-associated RNA-binding proteins (RBPs) have emerged as key contributors to mitochondrial biogenesis and homeostasis. With few examples known, we set out to identify RBPs that regulate nuclear-encoded mitochondrial mRNAs (NEMmRNAs). Our systematic analysis of RNA targets of 150 RBPs identified RBPs with a preference for binding NEMmRNAs, including LARP4, a La RBP family member. We show that LARP4's targets are particularly enriched in mRNAs that encode respiratory chain complex proteins (RCCPs) and mitochondrial ribosome proteins (MRPs) across multiple human cell lines. Through quantitative proteomics, we demonstrate that depletion of LARP4 leads to a significant reduction in RCCP and MRP protein levels. Furthermore, we show that LARP4 depletion reduces mitochondrial function, and that LARP4 re-expression rescues this phenotype. Our findings shed light on a novel function for LARP4 as an RBP that binds to and positively regulates NEMmRNAs to promote mitochondrial respiratory function.
Keywords
INTRODUCTION
A hallmark of eukaryotic cells is the presence of membrane-bound organelles, which facilitate the compartmentalization of biological processes. The proper targeting and localization of the protein components of these organelles is crucial for their function. Protein targeting to organelles is primarily facilitated by polypeptide sorting sequences contained within the proteins themselves. These cis-acting polypeptide sequences are recognized by other effector proteins that facilitate subcellular localization by various mechanisms (Bolender et al. 2008). An additional less explored pathway for protein targeting is mediated by the information contained within the mRNA sequence that encodes the protein. These cis-acting mRNA sequences are recognized by RNA-binding proteins (RBPs), which in turn influence the subcellular distribution and translational dynamics of their bound mRNA transcripts to promote the synthesis of proteins in the proximity of their functional locations (Béthune et al. 2019).
Efficient protein targeting is particularly important for mitochondrial proteins, many of which have biochemical or biological properties that are detrimental to the cell outside of the context of the mitochondrion (Yoo et al. 2005; Williams et al. 2014; Wrobel et al. 2015; Boos et al. 2020; Bykov et al. 2020). The classical pathway for protein targeting to the mitochondria is polypeptide mediated, facilitated by an N-terminal mitochondrial targeting sequence present on many but not all mitochondrial precursor proteins (Bolender et al. 2008). Additionally, several mRNA-mediated mitochondrial targeting pathways have been described in lower and higher eukaryotes (Béthune et al. 2019; Bykov et al. 2020). Many of the RBPs and interacting proteins involved in these mRNA-mediated pathways were first described in yeast or Drosophila (Fields et al. 1998; Eliyahu et al. 2010; Sen et al. 2015; Zhang et al. 2016) and were later found to have a conserved or similar function in human cells (Matsumoto et al. 2012; Gao et al. 2014; Gehrke et al. 2015; Schatton et al. 2017; Gabrovsek et al. 2020). Some factors involved in mitochondrial mRNA-dependent targeting described in yeast, however, do not have a human homolog with a conserved function (García-Rodríguez et al. 2007; Saint-Georges et al. 2008; Lesnik et al. 2014; Zabezhinsky et al. 2016). The extent to which human RBPs regulate expression of nuclear-encoded mitochondrial mRNAs (NEMmRNAs) is not fully understood.
With the goal of identifying human RBPs with a novel role in mitochondrial biology, we made use of the ENCODE collection of 223 enhanced cross-linking immunoprecipitation (eCLIP) data sets profiling 150 RBPs in K562 and HepG2 cell lines in a standardized workflow (Van Nostrand et al. 2020). Through a systematic computational analysis of these eCLIP data sets, several RBPs with RNA-target sets enriched for NEMmRNAs were identified. Of these NEMmRNA-enriched RBPs, LARP4, a La-related protein (LARP), was selected for further study, due to an enrichment for RNA-targets encoding respiratory chain complex proteins (RCCPs) and mitochondrial ribosome proteins (MRPs). LARP4 was first identified as one of the LARPs, each of which is a paralog to the conserved La protein, which functions as a processing chaperone for RNA transcripts produced by RNA polymerase III (Bousquet-Antonelli and Deragon 2009). The La protein and the LARPs each contain an evolutionarily related RBP domain called the La module. However, the La modules found on LARPs have diverged to bind RNA sequences distinct from their ancestral paralog. Additionally, many of the LARPs have gained additional protein domains to facilitate novel protein–protein interactions as well as new RNA interactions (Maraia et al. 2017). In an ancestral vertebrate, a gene duplication event occurred and gave rise to two paralogs of LARP4 (also referred to as LARP4A) and LARP4B (also referred to as LARP5) (Maraia et al. 2017; Coleman et al. 2023a). Upon initial characterization, LARP4 was shown to bind directly to poly(A) RNA as well as indirectly through protein–protein interactions with poly(A) binding proteins (PABPs) (Yang et al. 2011). Later, it was reported that the direct interaction with poly(A) RNA is mediated by the N-terminal region of LARP4 and not the expected La module (Cruz-Gallardo et al. 2019). Additionally, LARP4 was shown to interact with the 40S ribosomal protein RACK1 and assemble into translating polysomes (Yang et al. 2011). Later studies demonstrated that LARP4 plays a functional role in maintaining poly(A) tail (PAT) length, likely through competition with deadenylases for PABP binding sites (Mattijssen et al. 2017, 2020). Recently, LARP4 was found to colocalize with TOM20 at mitochondria as well as cofractionate with mitochondria in the MG63 and PC3 cancer cell lines (Coleman et al. 2023b). Additionally, LARP4 was reported to be recruited to the mitochondrial surface in HEK293 cells through an interaction with the PKA adaptor protein AKAP1, which also functions as an RBP (Gabrovsek et al. 2020). Without knowledge of the specific RNA targets of LARP4, those authors proposed LARP4 acted as a ribonucleoprotein (RNP) complex scaffolding factor which could facilitate the recruitment of associated translation factors to mitochondria to enhance local translation of mRNAs bound by AKAP1 at the mitochondrial surface (Gabrovsek et al. 2020).
In this study, we systematically determine the RNA targets of LARP4 to expand this model to support a role for LARP4 as a transcript-specific NEMmRNA recruitment factor. We identified and mapped the binding sites of LARP4 to 713 human mRNA targets, of which 186 (26%) are transcripts that encode mitochondrial proteins, with enrichment in proteins involved in oxidative phosphorylation (OXPHOS) (e.g., RCCPs) or mitochondrial translation (e.g., MRPs). Additionally, we showed that CRISPR-mediated LARP4 depletion results in reduced abundance of these two groups of proteins. Furthermore, LARP4-depleted cells show reduced OXPHOS capacity, and this phenotype is ameliorated by re-expression of LARP4. Our results indicate that LARP4 is an RBP that binds NEMmRNAs and promotes the expression of proteins essential to mitochondrial respiratory function.
RESULTS
LARP4 has a preference for binding nuclear-encoded mitochondrial mRNAs
The ENCODE consortium has made publicly available the RNA-binding profiles of hundreds of RBPs. This was done by performing the eCLIP assay for 150 RBPs in two human cell lines (HepG2 hepatocellular carcinoma and K562 erythroleukemia) for each RBP in duplicates (Van Nostrand et al. 2020). We analyzed these ENCODE eCLIP data sets for RBPs whose RNA-target sets were enriched for mRNAs encoding either proteins localized to the mitochondria (Mitocarta_2.0) or proteins specifically involved in oxidative phosphorylation (KEGG OXPHOS pathway) (Fig. 1A), and identified several RBPs with RNA-target sets substantially enriched for both: LARP4, DDX3X, RPS3, SUB1, PABPC4, YBX3, and several others (Fig. 1B,C). Among them, LARP4 showed the greatest enrichment and was chosen for more in-depth analysis. We performed an independent LARP4 eCLIP experiment in human HEK293 embryonic kidney cells (Fig. 1D) and discovered an even greater enrichment for RNA-targets encoding mitochondrial or OXPHOS proteins than observed in any of the other ENCODE eCLIP data sets available.
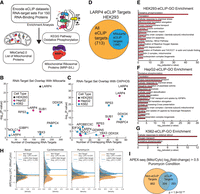
LARP4 interacts with NEMmRNAs. (A) Overview of enrichment analysis performed on eCLIP data sets for 150 RBPs in K562 and HepG2 cell lines. (B) Enrichment analysis for RNA-target set (eCLIP data) overlapping with mitochondrial genes. The significance of overlap (hypergeometric test) for each RBP data set is plotted on the y-axis and the number of overlapping targets is plotted on the x-axis. RBPs with significant overlap (P-value ≤ 0.05) and LARP4-K562 also shown (P-value = 0.06). (C) Enrichment analysis for RNA-target set (eCLIP data) overlapping with proteins in the KEGG OXPHOS pathway (HSA_00190). Only RBPs with statistically significant overlap (P-value ≤ 0.05, hypergeometric test) are shown. (D) Diagram showing the proportion of LARP4 eCLIP targets that encode mitochondrial proteins. (E–G) Metascape gene ontology (GO) analysis was performed on the three LARP4 eCLIP data sets from HEK293 (E), HepG2 (F), and K562 (G) cells. P-values are calculated by a hypergeometric test. (H,I) Comparison of mitochondrial enrichment scores from the APEX-seq atlas (Fazal et al. 2019) for LARP4 target and nontarget transcripts. eCLIP experiments were performed in independent biological duplicates for each cell line (N = 2). Gene-target lists of each eCLIP data set are defined as genes encoding an mRNA containing at least one peak that passes significance thresholds (−log10[P-value] ≥ 7, Fisher's exact test; log2[fold-change (IP/input)] ≥ 4). See also Supplemental Figure S1.
Overlap between the gene-target lists, defined as genes encoding an mRNA containing at least one significant LARP4 eCLIP peak, was greatest between the LARP4 eCLIP data set generated from HEK293 and HepG2 cells, which are both of epithelial origin, with the K562 cell data set generating fewer overall gene-targets (Supplemental Fig. S1A,B). GO analysis was performed on these eCLIP data sets, using similarly defined gene-target lists as a foreground and data set-specific background gene lists using the Metascape method (Fig. 1E–G; Zhou et al. 2019). OXPHOS (GO: 0006119) was the most enriched GO term for both the HEK293 and HepG2 cell data sets (Fig. 1E,F) and one of the top three terms for the analysis performed on the K562 eCLIP data set (Fig. 1G). The overlap between gene targets of each LARP4 eCLIP data set was even greater when only considering CLIP targets that are also mitochondrial mRNAs (Supplemental Fig. S1B).
The translation (RHSA:72766) GO term, which includes proteins that make up the cytosolic ribosome as well as the mitochondrial ribosome, was the second most enriched term in both the HEK293 and HepG2 cell eCLIP data sets. This enrichment for translational genes was also substantial when compared to other RBPs in the ENCODE data set collection (Supplemental Fig. S1E). Furthermore, mitochondrial translation elongation (RHSA-5389840), a term primarily composed of genes that encode MRPs, was also a top GO term of both the HEK293 and HepG2 cell eCLIP data sets (Fig. 1E,F). A separate analysis looking for overlap between the genes that encode LARP4's mRNA targets and the genes that encode the MRPs (GO: 0005761) confirms that MRPs are highly enriched (P-value = 6 × 10−11) in the HEK293 LARP4 eCLIP target set (Supplemental Fig. S1D). This enrichment for MRPs was also substantial when compared to other RBPs in the ENCODE data set collection (Supplemental Fig. S1F). A similar overlap analysis showed that genes encoding proteins that function in the OXPHOS pathway, that is, RCCPs, are also highly enriched (P-value = 5 × 10−37) in the LARP4 eCLIP target set (Supplemental Fig. S1C).
We conducted an analysis of the APEX-seq atlas to determine if LARP4 target mRNAs are enriched in the vicinity of the mitochondria. The APEX-seq atlas was generated using a method that utilizes proximity-labeling and sequencing to produce enrichment scores, which indicate the extent of mitochondrial localization for each transcript in HEK293 cells (Fazal et al. 2019). Along with basal conditions, the APEX-seq atlas includes conditions where proximity labeling was performed in the presence of either cycloheximide or puromycin which were used to accentuate mitochondrial targeting sequence-dependent (via ribosome stalling) and RNA-dependent (via ribosome–peptide–mRNA disassociation) modes of mRNA localization to the mitochondria, respectively. Our analysis revealed that, across all conditions studied in the APEX-seq atlas, the average mitochondrial enrichment for LARP4 targets was higher compared to nontargets. This difference was particularly evident in nonbasal conditions, such as the puromycin condition, which was used to highlight RNA-dependent modes of mRNA localization to the mitochondria (Fig. 1H). By setting a cutoff for mitochondrial enrichment at a log2(fold-change) > 0.5, we observed a highly significant overlap of LARP4 targets in the puromycin condition indicating many of the mRNAs which localize to mitochondria via RNA-dependent modes are also LARP4 targets (Fig. 1I). These findings highlight the potential involvement of LARP4 in regulating mitochondrial localization of specific mRNAs through RNA-dependent mechanisms.
How LARP4 achieves its RNA-target specificity is not fully understood. Using the eCLIP data sets, we performed k-mer analysis to identify possible linear sequence motifs in LARP4's RNA-target sets. While robust motifs were not identified, there was a weak degenerate C-rich motif present within the LARP4 peaks present in 3′ UTRs of target genes (Supplemental Fig. S1H). We also performed a similar k-mer analysis on the following subsets of LARP4 targets, nuclear-encoded mitochondrial genes, OXPHOS genes, and MRPs genes. These searches also did not identify any robust motifs. Metagene analysis of LARP4's eCLIP data shows that LARP4 coats many of its target mRNAs from CDS to 3′ UTR with the highest peak density centered around the stop codon and a small peak of density around the start codon; this pattern was consistent across subsets of LARP4 targets (Supplemental Fig. S1G), perhaps explaining why there is an absence of specific strong motifs. LARP4 contains both RNA-binding domains (La module and N-terminal region) and protein interaction domains that interact with other RBPs or RNA-associated proteins such as PABPs and RACK1 (Maraia et al. 2017; Cruz-Gallardo et al. 2019). It is possible that LARP4's RNA-target specificity is achieved through a cooperative mechanism involving both its RNA-binding domains and its protein interaction domains.
Together our data identify LARP4 as an RBP with a preference for NEMmRNAs. Of these NEMmRNA targets, LARP4 has a particular proclivity for binding mRNAs encoding RCCPs as well as MRPs.
Loss of LARP4 disrupts protein levels without affecting the mRNA abundance of mitochondrial targets
To study the functional consequences of LARP4 depletion, knockout (KO) cell lines were generated for HEK293 cells and the U2OS human osteosarcoma cell line. Genome editing was performed by transient expression of CRISPR/Cas9 and RNA guides targeting the first exon of LARP4 and the upstream promotor region. Disruption of LARP4 expression was validated by immunoblot analysis (Fig. 2A,B; quantified in Fig. 2D,E), and sequencing of genomic regions surrounding the CRISPR guide sites from the KO cell line subclones confirmed disruption of all LARP4 alleles (Supplemental Fig. S2A).
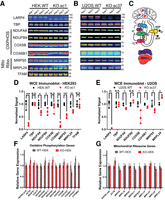
Loss of LARP4 disrupts protein levels without affecting the mRNA abundance of mitochondrial targets. (A,B) Immunoblot validation of LARP4 depletion by CRISPR/Cas9 and analysis of various OXPHOS proteins and MRPs in HEK293 KO and parental WT cells (A; N = 4) and in U2OS KO and parental WT cells (B; N = 3). (C) Diagram of the mitochondrial, oxidative phosphorylation proteins and MRPs. (D,E) Quantification of biological replicates shown in immunoblot panels. Band intensities are normalized by a quantitative total protein stain. (F,G) Analysis of mRNA abundance in the HEK293 KO/WT cells by qPCR using probes targeting mitochondrial ribosome subunit proteins (F) or OXPHOS proteins (G). LARP4 targets are denoted with an asterisk (N = 3). All averages shown are from independent biological replicates. Statistical significance of differences was assessed by two-tailed unpaired Student's t-tests (*) P ≤ 0.05, (**) P ≤ 0.001, (***) P ≤ 0.0001. (N.S.) Not significant. See also Supplemental Figure S2.
Because mRNAs encoding RCCPs and MRPs were particularly enriched in LARP4's RNA-target set, expression levels of representative proteins from these protein complexes (Fig. 2C) were characterized by immunoblot analysis. Based on the strong LARP4 binding to their encoding transcripts, NDUFA8, NDUFB9, COX5B, and COX6B1 were selected as representative RCCPs; MRPS5 and MRPL24 were selected as representative MRPs. The TATA-Box binding protein (TBP) was included as a non-LARP4 target loading control. Biological replicate samples of whole-cell extracts (WCEs) were collected from KO cell lines and their respective wild-type (WT) parental lines for analysis. Antibody signals were normalized with a quantitative total protein stain, and replicate averages were quantified. Samples from HEK293LARP4−/− KO cells showed a significant reduction in protein expression levels of multiple RCCPs (NDUFA8: KO/WT = 0.67, NDUFB9: KO/WT = 0.74, and COX6B1: KO/WT = 0.16) and multiple MRPs (MRPS5: KO/WT = 0.52 and MRPL24: KO/WT = 0.72). Parallel analysis of samples from U2OSLARP4−/− KO cells showed that protein levels were moderately reduced in about 60% of cases (NDUFA8: KO/WT = 0.83, COX5B: KO/WT = 0.48, COX6B1: KO/WT = 0.88, and MRPS5: KO/WT = 0.75) (Fig. 2A,B; quantified in Fig. 2D,E). The overall level of target protein disruption observed by immunoblot was less pronounced in the U2OS LARP4 KO line. We also depleted LARP4 in HEK293 cells by transduction with a lentivirus vector expressing a shRNA targeting LARP4 (shRNALARP4) or a nontargeting control shRNA (shRNANT). Depletion of LARP4 by this orthogonal method (shRNALARP4/shRNANT = 0.71) also resulted in a significant reduction in protein expression levels of OXPHOS proteins (NDUFA8: shRNALARP4/shRNANT = 0.71; NDUFB9: shRNALARP4/shRNANT = 0.85) and a representative MRP (MRPS5: shRNALARP4/shRNANT = 0.76) (Supplemental Fig. S2B,C). These data show that loss of LARP4 results in disruption of protein levels of selected proteins belonging to the RCCP and MRP gene groups that were enriched in LARP4's RNA-target set in the HEK293 cell line, and to a lesser extent the protein levels of some of these proteins were also disrupted in the U2OS cell line.
Given that LARP4 is thought to stabilize the mRNA levels of a subset of targets in certain contexts (Yang et al. 2011; Mattijssen et al. 2020), we sought to determine if the reduction in protein levels of RCCPs and MRPs observed in the HEK293LARP4−/− KO cells could be explained by a corresponding reduction in abundance of their encoding mRNAs. Analysis of mRNA abundance was performed by qPCR on a panel of mRNAs encoding RCCPs (Fig. 2F) as well as MRPs (Fig. 2G) on WT and HEK293LARP4−/− KO cells. None of the nine mRNAs encoding MRPs tested or the 13 mRNAs encoding RCCPs tested showed a significant change in abundance relative to WT HEK293 cells, indicating that the reduction in protein levels of RCCPs and MRPs observed in the HEK293LARP4−/− KO cells is not due to reduction in the abundance of the encoding mRNAs. These data show that the loss of LARP4 does not affect the levels of these target transcripts, suggesting that LARP4 is regulating the translation of these targets, possibly by promoting localized translation in the vicinity of the mitochondria.
To assess the effect of LARP4 depletion on mitochondrial gene expression, we measured relative mRNA abundance of a panel of genes encoded on the mitochondrial chromosome. We did not find any significant changes in the HEK293LARP4−/− KO cells relative to the WT HEK293 cells (Supplemental Fig. S2D). We also measured the relative mRNA abundance of panel genes regulated by the PGC-1α/NRF axis that controls TFAM expression and mitochondrial biogenesis. For the majority of these genes, we did not find any significant changes (Supplemental Fig. S2E). We also did not observe a significant change in TFAM protein expression by immunoblot (Fig. 2A). Together these data show that loss of LARP4 does not result in changes in mitochondrial gene expression levels or expression of TFAM.
Quantitative proteomic analysis of the LARP4 KO cell line reveals reduced abundance of mitochondrial ribosome proteins and OXPHOS proteins
To gain comprehensive insights into the proteomic consequences of LARP4 depletion and verify the reductions in protein abundance observed for RCCPs and MRPs by immunoblotting using an orthogonal method of measurement, quantitative tandem mass tagging (TMT) proteomics was performed on HEK293LARP4−/− KO and WT cells. To evaluate possible defects in protein localization to the mitochondria in the HEK293LARP4−/− KO cells, subcellular fractionation was performed in parallel. A rapid-magnetic mitochondrial enrichment strategy was used to generate two types of protein extracts for each genotype: a WCE and mitochondrial fraction extract (MITO) from the same cultures. Biological replicates (N = 4) of each type of protein extract were processed and analyzed by separate quantitative TMT proteomic experiments, to produce a WCE-TMT data set (Fig. 3A) and an MITO-TMT data set (Fig. 3B).
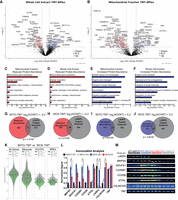
Quantitative proteomic analysis of the LARP4 KO cell line reveals reduced abundance of MRPs and OXPHOS proteins. (A) Volcano plot showing protein abundance (tag ratio of KO/WT) results from quantitative TMT analysis of WCEs (N = 4) prepared from HEK293LARP4−/− KO cells and WT cells. (B) Volcano plot showing protein abundance results from quantitative TMT analysis of mitochondrial enriched extracts (N = 4). (C,D) Metascape GO analysis of proteins with significant decreases in abundance identified by TMT analysis in mitochondrial enriched extracts (C) and WCEs (D). (E,F) Metascape GO analysis of proteins with significant increases in abundance identified by TMT analysis in mitochondrial enriched extracts (E) and WCEs (F). (G,H) Overlap of proteins with significant decreases in abundance identified by TMT analysis in mitochondrial enriched extracts (G) and WCEs (H). (I,J) Overlap of proteins with significant increases in abundance identified by TMT analysis in mitochondrial enriched extracts (I) and WCEs (J). (K) Violin plots comparing the changes in protein abundance of OXPHOS proteins and MRPs observed in the mitochondrial extract (green) and WCE (gray) proteomics experiments. Distributions shown are of average log2 (fold-change [KO/WT]) of proteins within each gene group. (L,M) Immunoblot analysis of WCEs (INPUT) and mitochondrial enriched extracts (MITO). Each lane was loaded with 10 μg of protein (M), and quantification of immunoblots from biological replicates (L). Significance thresholds for changes in protein abundance in TMT data are defined as P-value ≤ 0.05 and log2(fold-change [KO/WT]) ± 0.2. All averages shown are from independent biological replicates. Statistical significance of differences was assessed by two-tailed unpaired Student's t-tests (*) P ≤ 0.05. Statistical significance of enrichments was determined by the hypergeometric test.
To determine how gene groups that were overrepresented within LARP4's RNA-target set were affected by LARP4 depletion, GO analysis was performed using the Metascape method (Zhou et al. 2019). For each TMT data set (WCE or MITO), sets of proteins present in significantly increased (WCE: N = 203, MITO: N = 289) or decreased (WCE: N = 286, MITO: N = 494) abundance were defined (log2[fold-change] ± 0.2 and P-value < 0.05) and a separate GO analysis performed on each. For both the MITO and WCE-TMT data set, there was a significant overlap between the proteins with decreased abundance and the detected LARP4 targets (MITO: N = 71, P-value = 1.7 × 10−4, WCE: N = 50, P-value = 2.7 × 10−8) (Fig. 3G,H). The GO analysis of proteins with decreased abundance in HEK293LARP4−/− KO cells (Fig. 3C,D) revealed that many of the depleted proteins belonged to similar gene groups that were also overrepresented within LARP4's RNA-target set. In both proteomic data sets, two of the most enriched gene groups in the set of depleted proteins were proteins involved in mitochondrial gene expression and proteins involved in cellular respiration. The mitochondrial gene expression term had 104 proteins present in the set of depleted proteins, primarily MRPs, and the cellular respiration term had 60 proteins present almost entirely RCCPs (Fig. 3C,D). These gene groups were also enriched within LARP4's RNA-target set (Fig. 1E; Supplemental Fig. S1D,F), suggesting that LARP4 binding enhances protein expression of these target mRNAs.
The only mitochondrially encoded protein detected in the TMT-proteomics data set, MT-CO2, was depleted in the HEK293LARP4−/− KO cells (WCE: KO/WT = 0.86 and MITO: KO/WT = 0.76) to levels similar to many of the nuclear-encoded RCCPs, suggesting that either the depletion of MRPs results in decreased mitochondrial translation or other biological processes such as degradation or transcriptional buffering are occurring to bring the subunits of the respiratory complexes into stoichiometric balance. These same biological processes that keep the proportions of protein complex components in stoichiometric balance (Taggart et al. 2020) likely explain why the few non-LARP4 target RCCPs or MRPs present in the TMT-proteomics data set are depleted to similar levels as the rest of the members of their protein complexes.
Fewer proteins with significantly increased abundance in HEK293LARP4−/− cells were identified in both the WCE-TMT data set (N = 203) and MITO-TMT data set (N = 289), and fewer of these were also LARP4 targets (MITO: N = 34, WCE: N = 31) (Fig. 3I,J). The GO analysis of these proteins with increased abundance in HEK293LARP4−/− KO cells identified gene sets involved in biological processes that could conceivably compensate for reduced cellular respiration, for example, organic anion transport (WCE-TMT and MITO-TMT), glycolysis and gluconeogenesis (WCE-TMT), and transport of small molecules (WCE-TMT and MITO-TMT) (Fig. 3E,F). Furthermore, these biological processes were not enriched in the LARP4 RNA-target eCLIP data set (Fig. 1E). The increase in abundance of proteins with potential to compensate for reduced cellular respiration indicates the possibility that the HEK293LARP4−/− KO cells have undergone some type of genetic or metabolic compensation for the loss of LARP4 and associated reduction in abundance of proteins involved in cellular respiration. To validate some of the changes in protein abundance observed in the TMT experiments, quantification of protein abundance by immunoblot analysis of selected RCCPs and MRPs was also performed on similar WCE extracts and MITO extracts produced while optimizing the mitochondrial enrichment strategy. Although increased variability in these pilot experiments reduced the significance of some comparisons, significant differences were observed (MRPS5 and COX6B1) as well as similar downward trends (Fig. 3L,M).
Interestingly, when comparing the two TMT experiments, the reduction in protein abundance of RCCPs and MRPs was much more pronounced in the analysis of the mitochondrial enriched extracts compared to the analysis of the WCEs from the same cultures. For the OXPHOS proteins (RCCPs) the average fold change in TMT-quantified protein abundance was 0.72 in the mitochondrial extracts and 0.84 in the WCEs (Fig. 3K). A similar trend was observed for the MRPs with an average fold change of 0.73 in the mitochondrial extracts and an average fold change of 0.81 in the WCEs (Fig. 3K). Our results suggest that in the HEK293LARP4−/− KO cells targeting of these proteins to the mitochondria is impaired, supporting a model in which LARP4 promotes both the translation and subcellular targeting of RCCPs and MRPs to the mitochondria.
Loss of LARP4 reduces cell proliferation rates, levels of oxidized proteins, and translation rates
Given the essential role of mitochondria in cell proliferation (Birsoy et al. 2015; Sullivan et al. 2015) and the depletion of proteins essential to mitochondrial function observed in LARP4 KO cells, we hypothesized that cell proliferation would be reduced in LARP4 KO cells. Cell proliferation studies showed that the HEK293LARP4−/− KO cells had significantly reduced proliferation rates, averages from independent experiments (N = 3) showed significantly increased cell doubling times relative to WT cells, and this reduction in the rate of cell proliferation was completely rescued by re-expression of LARP4 (Fig. 4A). Additionally, compared to the parental U2OS osteosarcoma cancer line, the LARP4-depleted U2OSLARP4−/− KO cells showed a reduced rate of cell proliferation in two independent experiments (Fig. 4B). This is consistent with a recent study which showed that depletion of LARP4 in osteosarcoma cancer lines (MNNG/HOS; MG63) also reduced cell proliferation (Coleman et al. 2023b). These data indicate that LARP4 is required for normal rates of proliferation in HEK293 cells and that LARP4 depletion is also associated with reduced proliferation rates in the osteosarcoma cancer cell line U2OS.
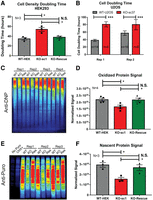
Loss of LARP4 reduces cell proliferation rates, levels of oxidized proteins, and protein translation rates. (A) Summary plot of average confluency doubling times for WT HEK293 cells, HEK293LARP4−/− KO cells, and HEK293LARP4−/− KO-Rescue cells from independent experiments. N = 3. (B) Plot of cell doubling times of WT U2OS cells and U2OSLARP4−/− KO cells, averages of technical replicates (n = 6–14) from each of two independent experiments are shown (Rep 1 and Rep 2). (C,D) Analysis of oxidized protein abundance by the OxyBlot assay (C) and quantification of normalized oxidized protein signal averages (N = 4) for each cell line, each replicate represents an independent experiment (D). (E,F) Analysis of translation rates by the puromycin incorporation assay (E) and quantification of normalized puromycin incorporation signal averages (N = 3) for each cell line, each replicate represents an independent experiment (F). Statistical significance of differences was assessed by two-tailed unpaired Student's t-tests (*) P ≤ 0.05, (***) P ≤ 0.0001. (N.S.) Not significant. See also Supplemental Figure S3.
Because mitochondria are a major source of reactive oxygen species (Balaban et al. 2005), which are a cause of protein oxidation, levels of oxidized proteins were measured, using a derivatization and immunoblot approach (OxyBlot Kit). This analysis found that oxidized proteins were present in significantly lower abundance in HEK293LARP4−/− KO cells compared to WT cells, and that this phenotype was completely rescued by re-expression of LARP4 (Fig. 4C,D; Supplemental Fig. S3A). These data indicate that LARP4 function indirectly promotes the production of oxidized proteins, likely through enhanced mitochondrial respiration rates.
LARP4 has previously been shown to associate with translational machinery (Yang et al. 2011) and our data above indicates enrichment of mRNAs encoding proteins involved in translation in its RNA-target set (Fig. 1E,F; Supplemental Fig. S1E). Since protein translation is an energy-intensive process and LARP4 targets and regulates the expression of proteins essential to the energy-producing capacity of mitochondria, we assayed translation rates. Using the puromycin incorporation assay, we found that translation rates in HEK293LARP4−/− KO cells were substantially reduced relative to WT cells; re-expression of LARP4 returned translation rates to levels similar to those of WT cells (Fig. 4E,F; Supplemental Fig. S3B). These data indicate that LARP4 is required for normal translation rates in HEK293 cells.
LARP4 promotes oxidative phosphorylation function
As proteins essential for cellular respiration (RCCPs and MRPs) were significantly depleted in the HEK293LARP4−/− KO cells, the functional impact on OXPHOS function and capacity was investigated. The Seahorse Extracellular Flux Analyzer was used to measure oxygen consumption rates (OCRs) (a parameter directly related to the OXPHOS function). The analyzer was configured to run the Mito-stress-test (MST) assay, which assesses basal respiration and maximal respiration, measured in terms of OCRs.
In every MST Seahorse assay performed, the HEK293LARP4−/− KO cells exhibited significantly reduced basal respiration and maximal respiration compared to WT cells (Fig. 5A,B; Supplemental Fig. S4A–D). Similarly, the U2OSLARP4−/− KO cells exhibited deficiencies in OXPHOS function as measured by the MST Seahorse assay (Fig. 5C,D; Supplemental Fig. S4E–H). Furthermore, we demonstrate that rescue of LARP4 depletion by stable re-expression of LARP4 at somewhat elevated levels in HEK293LARP4−/− KO cells significantly improved the OXPHOS deficiency in the LARP4-deficient cells although not completely to WT levels (Fig. 5A,B; Supplemental Fig. S4A–D). It is likely that during continued culture the LARP4 KO cells undergo compensatory metabolic changes following genetic disruption of LARP4 and these changes prevent a complete restoration of OXPHOS rates upon LARP4 re-expression. Evidence of compensatory metabolic change was observed in the analysis of the HEK293LARP4−/− KO cells by TMT-proteomics, for example, up-regulation of proteins involved in glycolysis and gluconeogenesis (Fig. 3E,F). These results from HEK293 cells demonstrate that loss of LARP4 reduces OXPHOS rates, indicating that depletion of RCCPs and MRPs observed in LARP4 KO cells by TMT-proteomics and immunoblot analysis has a functional impact on the biological processes carried out by these protein complexes.
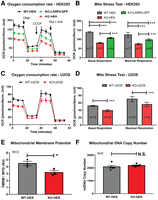
LARP4 promotes OXPHOS function. (A,B) Seahorse extracellular flux analysis of OCRs from WT HEK293 cells, HEK293LARP4−/− KO cells, and HEK293LARP4−/− KO-LARP4-GFP Rescue cells. Basal and maximal respiration rates (B) are calculated from the changes in OCR (A) in response to inhibitor addition. Averages shown are from technical replicates (N = 7 or 8, as indicated). (C,D) Seahorse extracellular flux analysis of OCRs from WT U2OS cells and U2OSLARP4−/− KO cells. Averages shown are from technical replicates (n = 12). (E) Analysis of mitochondrial membrane potential (MMP) by flow cytometry of WT HEK293 cells and HEK293LARP4−/− KO cells stained with the potential dependent dye tetramethylrhodamine–methyl-ester–perchlorate (TMRM) and the potential independent dye MitoTracker Green. Averages shown are from three independent experiments (N = 3). (F) Analysis of mitochondrial mass by proxy using qPCR measurements of mitochondrial DNA copy number from WT HEK293 cells and HEK293LARP4−/− KO cell samples. Averages shown are from samples from five independent experiments (N = 5). Statistical significance of differences was assessed by two-tailed unpaired Student's t-tests (*) P ≤ 0.05, (***) P ≤ 0.0001. (N.S.) Not significant. See also Supplemental Figure S4.
LARP4 depletion causes mitochondrial membrane potential disruption
The effect of reduced OXPHOS rates on the MMP was also explored in LARP4-deficient cells. Flow cytometry analysis on the HEK293LARP4−/− KO cells stained with both TMRM (MMP dependent) and MitoTracker Green (mitochondrial mass dependent, MMP independent) showed a significantly reduced MMP as measured by the ratio of TMRE staining and MitoTracker Green staining (Fig. 5E). Analysis of mtDNA levels was also performed as a measure of mitochondrial mass. This analysis found no significant difference in mtDNA levels between the HEK293LARP4−/− KO cells and WT cells (Fig. 5F), indicating that significant changes in mitochondrial mass in HEK293LARP4−/− KO cells are unlikely to account for any phenotypic changes.
LARP4 promotes mitochondria-associated translation
Because some of the protein products of the RNA targets of LARP4 were more depleted in the proteomic analysis of mitochondrial extracts compared to the analysis of WCEs from LARP4-depleted cells (Fig. 3K), we hypothesized that reduced mitochondria-associated translation results in impaired protein targeting in HEK293LARP4−/− KO cells. To assess the effect of LARP4 depletion on mitochondria-associated translation, the puromycin incorporation assay was performed on mitochondrial extracts. Cells were treated transiently with puromycin before the preparation of mitochondrial extracts, and puromycin incorporation was measured by immunoblot (Fig. 6A,B). These experiments showed that mitochondria-associated translation as measured by puromycin incorporation was significantly reduced in the HEK293LARP4−/− KO cells relative to the WT cells and that re-expression of LARP4 ameliorated this phenotype (Fig. 6A,B). Another prediction of this model is that impaired protein targeting will result in increased mislocalization of some LARP4 target mitochondrial proteins. To test this prediction, we used immunofluorescence staining analysis and high-resolution confocal imaging to measure the percent cytosolic signal (nonmitochondrial cell signal) of the nuclear-encoded mitochondrial OXPHOS protein COX7A2 in HEK293 LARP4 KO and WT cells. This protein is encoded by a LARP4 target mRNA and was also more depleted in the MITO-TMT (KO/WT = 0.64) compared to the WCE-TMT (KO/WT = 0.86). This analysis revealed a significantly greater nonmitochondrial COX7A2 signal present in HEK293LARP4−/− KO cells (KO: 76% vs WT: 57% and KO: 66% vs WT: 47%) in two independent experiments (Fig. 6C,D) indicating COX7A2 mislocalization. Together these observations support a model in which LARP4 promotes mitochondria-associated translation to enhance protein targeting of some of its NEMmRNA targets. This provides mechanistic insights into how LARP4 promotes OXPHOS function.
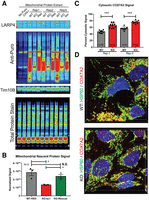
LARP4 promotes mitochondria-associated translation. (A,B) Analysis of mitochondria-associated translation rates by isolating mitochondria from puromycin-treated cells and measuring puromycin incorporation. (A) Puromycin immunoblots used for mitochondrial puromycin incorporation assay. (B) Quantification of average normalized mitochondria-associated puromycin signal from three independent biological replicates (N = 3). Controls include immunoblots for LARP4 to show depletion and TIM10B to show mitochondrial enrichment as well as a quantitative total protein stain for normalization. (C,D) Analysis of protein localization of COX7A2 (LARP4 target) by immunofluorescence staining with the mitochondrial protein HSP60 used as a mitochondrial marker and low threshold mask for cell area for image analysis. (C) Average percent cytosolic COX7A2 signal was quantified from nine fields of view (n = 9) of the HEK293LARP4−/− KO cells and WT cells from two independent biological replicates (Rep-1 and Rep-2). (D) Representative images from each condition and replicate are shown. Statistical significance of differences was assessed by two-tailed unpaired Student's t-tests (*) P ≤ 0.05, (***) P ≤ 0.0001. (N.S.) Not significant.
DISCUSSION
Mitochondrial gene expression presents several unique biological challenges that must be overcome to ensure mitochondrial protein homeostasis and function. The overwhelming majority of mitochondrial proteins are encoded by mRNAs transcribed from nuclear genes, all of which must be translated by cytosolic ribosomes and imported into the mitochondria by various partially redundant pathways (Bykov et al. 2020). These pathways include polypeptide-dependent pathways, facilitated by the N-terminal mitochondrial targeting sequence present on many but not all mitochondrial proteins (Bolender et al. 2008), as well as mRNA-dependent targeting pathways, which are facilitated by RBPs that influence the abundance, spatial distribution, and translational activity of their RNA targets (Béthune et al. 2019).
Efficient targeting of proteins to the mitochondria is particularly important for certain classes of proteins. These include proteins whose ectopic expression in the cytosol has potentially deleterious effects for the cell. This has been demonstrated for both RCCPs and MRPs (Yoo et al. 2005; Williams et al. 2014; Wrobel et al. 2015; Rawat et al. 2019; Boos et al. 2020; Bykov et al. 2020). Additionally, for proteins that assemble into large multiprotein complexes, such as RCCPs and MRPs (Fig. 2C), spatial and temporal coordination of translation also has the potential to promote cotranslational complex assembly, a process that has been demonstrated to be beneficial in several contexts (Keil et al. 2012; Kamenova et al. 2019; Schwarz and Beck 2019).
Post-transcriptional regulation of NEMmRNAs including localization to the mitochondrial membrane and localized translation is a fundamentally conserved biological process from yeast to metazoans; however, the protein factors (RBPs) and the mRNA sequences involved differ. A more complete characterization of the RBPs and their RNA interactions involved in this process in the context of human cells will facilitate greater insight into the mechanisms by which these RBPs promote efficient expression of mitochondrial proteins, potentially leading to a better understanding of the plethora of diseases where mitochondrial dysfunction is present.
In this report, we elucidated a novel function of LARP4 by which it binds functionally important NEMmRNAs to help maintain mitochondrial protein homeostasis and facilitate proper OXPHOS function. Our results provide direct evidence that LARP4 binds mRNAs encoding RCCPs and MRPs, and implicate LARP4 in positively regulating the expression of these proteins. Furthermore, we demonstrated that LARP4 is required for normal OXPHOS rates. Additionally, we show that LARP4 is required for normal rates of proliferation in HEK293 cells as well as the osteosarcoma cancer cell line U2OS, adding to the body of literature describing the role of LARP4 in cell proliferation (Coleman et al. 2023a).
We found that the RBP LARP4 is required for normal translation rates, consistent with previous reports linking LARP4 to polysome stability (Yang et al. 2011). Later studies demonstrated LARP4 to have a functional role in maintaining PAT length across the transcriptome via competition with deadenylases (Mattijssen et al. 2017, 2020). Interestingly, LARP4-mediated PAT length maintenance was observed across the transcriptome, except for a subset of NEMmRNAs where LARP4 did not appear to play a significant role in maintaining PAT length (Mattijssen et al. 2020). A potential explanation for this, made possible by our results, is a model in which direct RNA-targets of LARP4, such as NEMmRNAs, are bound by LARP4 in a way that does not reduce deadenylase activity, whereas indirect LARP4 RNA-targets interact with LARP4 in a way that leads to reduced deadenylase activity, possibly through competing with deadenylases for protein–protein interactions with PABPs.
In two recent studies, LARP4 was found to colocalize with TOM20 at mitochondria in HEK293 cells as well as in the MG63 and PC3 cancer cell lines (Gabrovsek et al. 2020; Coleman et al. 2023b). In HEK293 cells the recruitment of LARP4 to the surface of mitochondria was shown to be mediated by the protein kinase A adaptor protein AKAP1, where LARP4 acts as a general translation factor to promote the local translation of AKAP1-associated mRNAs (Gabrovsek et al. 2020). Our results extend beyond this study by showing that LARP4 functions not only as an RNP complex scaffolding factor involved in enhancing mitochondria-associated translation of AKAP1-associated mRNAs but also as an RBP with its own specific NEMmRNA targets that are translated in a LARP4-dependent manner. Additionally, using a similar assay as Gabrovsek et al. (2020), we show that LARP4 depletion reduces mitochondria-associated translation, phenocopying the effect of AKAP1 depletion on mitochondria-associated translation. The mitochondria-associated puromycin translation assay we used only gives information about global mitochondria-associated translation and does not provide information about the translation of individual gene products. Further technical advances enabling the measurement of localized translation of specific gene products in mammalian cells (i.e., mammalian proximity Ribo-seq) would be useful in investigating if the translation of LARP4 mRNA targets is specifically promoted in the vicinity of the mitochondria.
In their proteomic screen for AKAP1 interacting partners, Gabrovsek et al. (2020) also found PABPC1 and PABPC4, two of the PABP paralogs, as highly enriched AKAP1 interactors. Interestingly, in our analysis of the ENCODE eCLIP data sets, we found that PABPC4 also has an RNA-target set highly enriched for NEMmRNAs. Future studies should investigate what role PABPC4 plays in mitochondrial gene expression and if AKAP1, LARP4, and PABPC4 function cooperatively.
The results presented in this study provide strong support for a model in which LARP4 binds select mitochondrial mRNAs and positively influences the abundance of their protein products through post-transcriptional processes maintained by LARP4. Because we did not observe significant changes in mRNA abundance in the LARP4 target genes studied, it is unlikely that LARP4 functions as a selective regulator of transcript stability in the context of these mRNAs (if transcriptional rates for those mRNAs are similar in the WT and KO cells). However, because mRNA abundance is a product of both stability and transcription rate, it remains a possibility that changes in the transcriptional rate of LARP4 target mRNAs could be masking the role of LARP4 in regulating target mRNA stability. Given that recruitment of LARP4 to the cytoplasmic surface of the mitochondria has been previously demonstrated (Gabrovsek et al. 2020; Coleman et al. 2023b) and we demonstrated that mitochondria-associated translation was reduced upon LARP4 depletion, LARP4 may play a direct role in the recruitment of a subset of its target mRNAs to the vicinity of the mitochondria to enhance their local translation and mitochondrial protein targeting. Additionally, both our proteomic analysis and immunofluorescence staining analysis of LARP4-depleted cells demonstrated nonmitochondrial accumulation of protein products encoded by mitochondrial RNA targets of LARP4, implicating LARP4 in the process of mitochondrial protein targeting. Together these results are consistent with LARP4 promoting OXPHOS function by enhancing mitochondria-associated translation to facilitate efficient protein targeting to the mitochondria.
MATERIALS AND METHODS
DNA constructs
The coding region of full-length LARP4 was amplified from pFLAG-CMV2 plasmid containing a LARP4 expression cassette (gift from Richard Maraia) and cloned into a Gateway entry plasmid by Gibson assembly. Subsequently, either LARP4-GFP or LARP4-WT lentiviral expression plasmids were produced via Gateway cloning. The lentiviral expression plasmids used a CAG promotor to express either LARP4 or LARP4-GFP and contained a puromycin expression cassette to enable selection of transduced cells. The LARP-GFP lentiviral vector was used for the Seahorse assay rescue experiments; all other rescue experiments used the LARP4-WT lentiviral vector. CRISPR plasmids were cloned according to Zhang lab protocols (Ran et al. 2013).
Cell culture and manipulations
All cell lines were maintained in DMEM (Corning, 10-013-CV) supplemented with 10% fetal bovine serum at 37°C with 10% CO2. Cultures were passaged with Accutase (Innovative Cell Technologies, AT104) every 3 d. HEK293 and U2OS cell lines were obtained from ATCC and cultured for 30 passages or less. Periodic testing for mycoplasma was performed. All transfections were performed using Lipofectamine 2000 (Thermo Fisher Scientific, 11668030) according to the manufacturer's instructions. All transductions were performed using filtered cell culture media from cells transfected with lentiviral production plasmids.
CRISPR knockout cell line generation
LARP4 knockout cell lines were generated with CRISPR/Cas9 using a two-guide transfection strategy. Cas9 and sgRNA guides were introduced into cells by transfection of plasmids expressing Cas9 and sgRNA guides. Forty-eight hours after transfection, cells were dissociated and sorted into individual wells by flow cytometry to generate subclone cultures. Recovered clones were first screened by PCR using primers flanking the two adjacent CRISPR target sites. Clones showing evidence of a deletion in the target region were selected for validation by immunoblot analysis. Sequences of CRISPR guide RNAs and PCR primers used are listed in Supplemental Table 1. For the KO clones used in this study, the genomic region flanking the gRNA target site was amplified and cloned into a cloning vector and plasmids from 10 individual transformants for each clone sent for sequencing.
Incucyte cell proliferation assays
For analysis of cell proliferation rates, the Zoom Incucyte live cell imaging device was used to make image-based cell density measurements of cell cultures at regular intervals. The manufacturer's image analysis software was used to calculate either percent well confluency (HEK293) or direct cell counts (U2OS) at each time point. These cell density measurements were used to calculate density doubling times (HEK293) or cell doubling times (U2OS) for individual wells of the cell culture plate. For experiments with HEK293 cells, 6000 cells were plated into wells of 96-well plates, 24 h prior to transfer into the Incucyte device, and growth measurements were recorded for 4–5 d. Reported confluency doubling times represent averages of three biological replicates, each of which represents an average of technical replicates, which consisted of eight individual wells. For experiments with U2OS cells, cells were first transduced with a lentivirus expressing a nuclear red fluorescent protein to directly track cell counts. For experiments with U2OS cells, two biological replicates were performed and cell doubling times reported are averages of technical replicates.
Immunoblotting
For standard immunoblot analysis, lysates were prepared in RIPA lysis buffer (Thermo Fisher Scientific, 89900) supplemented with EDTA-free complete protease inhibitors (Sigma 5892791001). Protein concentrations were determined by the BCA assay (Thermo Fisher, 23227). Samples concentrations were normalized and prepared for PAGE in a reducing loading buffer; depending on the target protein, 10–30 µg of protein per sample were resolved through acrylamide gels. Proteins were then transferred onto PVDF membranes (Millipore, IPFL00010) via the wet-transfer procedure. Total transferred protein was then quantified using the Revert fluorescent total protein stain (LI-COR, 926-11011) according to the manufacturer's instructions. Membranes were then blocked via 1-h incubation in blocking buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 5% w/v bovine serum album). Primary antibody incubations were carried out for 2 h at room temperature in blocking buffer supplemented with 0.1% Tween-20. Secondary antibody incubations were carried out for 1 h at room temperature in blocking buffer supplemented with 0.1% Tween-20 and 0.01% SDS. The following primary antibodies were used at 1:1000 dilution unless stated otherwise: LARP4, gift from R. Maraia; NDUFA8, Abcam, ab184952, dilution: 1:20,000; NDUFB9, Abcam, ab200198, dilution: 1:5000; COX5B, Abcam, ab180136, dilution: 1:10,000; COX6B1, Abcam, ab131277; MRPS5, Abcam, ab96291; MRPL24, Santa Cruz Biotechnology, sc-393857, dilution: 1:100; NDUFA1, Abcam, ab176563; ATP5L, Invitrogen, PA5-60783; Calnexin, Santa Cruz Biotechnology, sc-11397, dilution: 1:100; TBP, Cell Signaling, 44059; TIMM10B, Invitrogen, PA597081; COX7A2, Proteintech, 18122-1-AP, dilution 1:100; anti-puromycin, Millipore, MABE343, dilution: 1:20,000; and anti-DNP, Millipore, S7150, dilution: 1:150. The following secondary antibodies were used at a 1:20,000 dilution: goat anti-rabbit DyLight 800, Invitrogen, SA535571; goat anti-mouse DyLight 800, Invitrogen, SA535521; goat anti-rabbit Alexa Fluor 680, Invitrogen, A-21109; and goat anti-mouse Alexa Fluor 680, Invitrogen, A-21058. After each antibody incubation, membranes were washed three times in wash buffer (150 mM NaCl, 50 mM Tris, pH 7.5, 0.1% Tween-20) for 5 min. Stained membranes were rinsed in TBS (150 mM NaCl, 50 mM Tris, pH 7.5) and visualized using the Odyssey imaging system. The LI-COR imaging software was used to quantify both the total transferred protein per lane and the signal intensity of the target band. All reported immunoblot signal quantifications are the target band signal normalized by the total protein signal of the lane containing the target band. Antibodies and other key reagents are listed in Supplemental Table 2.
OxyBlot assay
For analysis of oxidized protein abundance using the OxyBlot assay kit (Millipore, S7150), lysates were prepared in RIPA lysis buffer supplemented with 50 μM DTT. Protein concentrations between samples were normalized and oxidized proteins were then derivatized with DNP according to the manufacturer's instructions. Immunoblotting was then performed and quantified as previously described, using the anti-DNP primary antibody provided with an OxyBlot assay kit (Millipore, S7150).
Puromycin incorporation assay
For analysis of translation rates by the puromycin incorporation assay (Schmidt et al. 2009), cells in culture were incubated in the presence of puromycin (10 μg/mL) for 10 min prior to lysate collection in RIPA buffer. Immunoblotting was then performed and quantified as previously described using an anti-puromycin primary antibody (Millipore, MABE343). Negative control sample cell cultures were processed similarly, with the exception that cells were either incubated with unmodified media, or puromycin labeling media supplemented with cycloheximide at a final concentration of 100 μg/mL.
RT-qPCR analysis
For analysis of mRNA abundance, total RNA extracts were collected from cell cultures containing approximately 500,000 cells by directly applying TRIzol reagent (Thermo Fisher Scientific, 15596026). Total RNA was isolated using the Direct-zol column purification kit (Zymo, R2050). Reverse transcriptase reactions were carried out using the SuperScript III Reverse Transcriptase kit (Invitrogen, 18080044), according to the manufacturer's instructions. A qPCR analysis was then performed on the resulting cDNA with Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, 4368577) using primer pairs listed in Supplemental Table 1. For each sample type, three biological replicates (independent collection days) were assayed, with at least two technical replicates (replicate qPCR reactions) performed for each biological replicate.
mtDNA abundance analysis
For analysis of mtDNA abundance, approximately 500,000 cells were collected and pelleted. Cell pellets were then resuspended in 50 mM NaOH and incubated at 95°C for 1 h. Samples were then neutralized with 1 M Tris, pH 8.0 using one-tenth the volume of lysis buffer used. DNA concentration was then estimated by spectrophotometry and qPCR performed using 3 ng of DNA per 10 μL reaction of Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, 4368577). For each sample, two reactions were prepared using primers targeting either the nuclear DNA (β2-microglobulin) or primers targeting the mitochondrial DNA (mitochondrially encoded tRNA leucine 1). The ratio of Ct values from the two primer sets was used to estimate the ratio of mitochondrial DNA to nuclear DNA. For each sample type, at least three biological replicates (independent collection days) were assayed, with at least two technical replicates (replicate qPCR reactions) performed for each biological replicate.
Magnetic isolation of mitochondria
Magnetic isolation of mitochondria was performed using the Militenyi Biotec Human Mitochondria Isolation Kit, according to the manufacturer's instructions with additional modifications and the following experiment-specific details. HEK293 cells were seeded at a density of 15 × 106 per 15 cm plate, 24 h before collection. Cells were harvested using DPBS and a rubber policeman, ∼10% of the cells collected were pelleted and set aside to be used as matched WCE samples, and the remaining 90% were pelleted and resuspended in 1 mL of the kit's lysis buffer, which was supplemented with protease inhibitors (Sigma, 5892791001). A 26-gauge needle and 1 mL syringe were used to homogenize the cell lysates. After optimization, the following protocol was found to be effective for 85%–95% cell disruption: five strokes of the entire homogenate volume, ice for 60 sec then repeat three more times for a total of 20 stroke repetitions. The final pellet for the mitochondria extracts and the WCEs were resuspended in RIPA buffer supplemented with protease inhibitors (Sigma, 5892791001). To remove the antibody-conjugated magnetic nanobeads prior analysis by quantitative TMT mass spectrometry, lysed mitochondria extracts were passed over fresh LS columns from the isolation kit that had been equilibrated with RIPA buffer.
TMT mass spectrometry and analysis
Samples were precipitated by methanol/chloroform and redissolved in 8 M urea/100 mM TEAB, pH 8.5. Proteins were reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich) and alkylated with 10 mM chloroacetamide (Sigma-Aldrich). Proteins were digested overnight at 37°C in 2 M urea/100 mM TEAB, pH 8.5, with trypsin (Promega). The digested peptides were labeled with 10-plex TMT (Thermo Fisher Scientific, 90309), and pooled samples were fractionated by basic reversed-phase chromatography (Thermo Fisher Scientific, 84868).
The TMT-labeled lysate samples were analyzed on a Fusion Lumos mass spectrometer (Thermo Fisher Scientific). Samples were injected directly onto a 25 cm, 100 μm ID column packed with BEH 1.7 μm C18 resin (Waters). Samples were separated at a flow rate of 300 nL/min on an Easy-nLC 1200 column (Thermo Fisher Scientific). Buffers A and B were 0.1% formic acid in water and 90% acetonitrile, respectively. A gradient of 1%–25% B over 180 min, an increase to 40% B over 30 min, an increase to 100% B over another 20 min, and held at 90% B for 10 min was used for a 240 min total run time.
Peptides were eluted directly from the tip of the column and nanosprayed directly into the mass spectrometer by application of 2.8 kV voltage at the back of the column. The Lumos was operated in a data-dependent mode. Full MS1 scans were collected in the Orbitrap at 120k resolution. The cycle time was set to 3 sec, and within this 3 sec, the most abundant ions per scan were selected for CID MS/MS in the ion trap. MS3 analysis with multinotch isolation (SPS3) was utilized for detection of TMT reporter ions at 60k resolution (McAlister et al. 2014). Monoisotopic precursor selection was enabled and dynamic exclusion was used with exclusion duration of 10 sec.
Protein and peptide identification were done with Integrated Proteomics Pipeline—IP2 (Integrated Proteomics Applications). Tandem mass spectra were extracted from raw files using RawConverter (He et al. 2015) and searched with ProLuCID (Xu et al. 2015) against the Uniprot human database. The search space included all fully tryptic and half-tryptic peptide candidates. Carbamidomethylation on cysteine and TMT on lysine and peptide N-term were considered as static modifications. Data were searched with 50 ppm precursor ion tolerance and 600 ppm fragment ion tolerance. Identified proteins were filtered using DTASelect (Tabb et al. 2002) and utilizing a target-decoy database search strategy to control the false discovery rate to 1% at the protein level (Peng et al. 2003). Quantitative analysis of TMT was done with Census (Park et al. 2014) filtering reporter ions with 20 ppm mass tolerance and 0.6 isobaric purity filter.
Protein total intensity on each channel was normalized by the sum of all proteins in the same channel. These normalized intensity values for each replicate (N = 4) were averaged together by group (WT or KO) and used to calculate the average normalized protein abundance for each protein. These values were used to calculate KO/WT ratios for each protein and determine sets of proteins present in increased or decreased abundance for GO analysis. Common keratin contaminates were removed manually. Supplemental Table 3 lists the complete set of proteins passing the thresholds of log2(KO/WT) ± 0.2 and P-value < 0.05 as well as the set of background proteins (all detected proteins in each TMT experiment) used for the GO analysis of the proteins with increased and decreased protein abundance in the HEK293 TMT data.
Extracellular flux analysis
OCRs and proton efflux rates were measured using the XF-96 extracellular flux analyzer (Agilent) and XF-96 FluxPaks (Agilent). The MTS assay was carried out according to the manufacturer's instructions with additional modifications and the following experiment-specific details. Approximately 24 h prior to assay, cells were seeded onto XF-96 cell culture plates and at a density of 25,000 or 16,000 cells per well for HEK293 cells or U2OS cells, respectively. For experiments with HEK293 cells, the following final concentrations of small molecules were applied: oligomycin (1.5 μM), CCCP (1.0 μM), rotenone (0.5 μM), and antimycin A (0.5 μM). For experiments with U2OS cells, the following final concentrations of small molecules were applied: oligomycin (1.0 μM), CCCP (2.0 μM), rotenone (0.5 μM), and antimycin A (0.5 μM). The following small molecules required for the MTS assay were ordered individually, and 10 mM stocks were prepared and stored at −20°C: CCCP, Sigma C2920; oligomycin, Sigma O4876; and antimycin A, Sigma A8674. The assay media used was XF DMEM base media (Agilent 103575-100) supplemented with 10 mM glucose, 1 mM pyruvate, and 2 mM glutamine. For MTS assay well normalization, DAPI (Thermo Fisher Scientific, D3571) was added to the final small molecule injection solution such that the final well concentration was 1 μg/mL. After completion of flux measurements, the cell culture plates were transferred to the Celigo plate cytometer (Cyntellect), DAPI-stained nuclei imaged, and direct cell counts for each well made using the Celigo cytometer software. For analysis of extracellular flux measurements, the Wave Desktop program (Agilent) was used.
Flow cytometry
For flow cytometry measurements, cells were seeded 24 h prior to collection at a density of 250,000 cells per well of a 12-well plate. Cells were harvested with Accutase, pelleted, and resuspended in flow buffer (DPBS, 2% FBS, 1 μg/mL DNase I, 1 μg/mL DAPI) prior to flow analysis. Cell suspensions were analyzed with an LSR II flow cytometer (BD Biosciences), and data were analyzed with FACSDiva software (BD Biosciences). For analysis of MMP, cells were costained with the MMP-dependent dye, TMRM (Thermo Fisher Scientific, T668), and the mitochondrial mass-dependent dye, MitoTracker Green (MTG) (Thermo Fisher Scientific, M7514). MMP measurements were then normalized by the mitochondrial mass-dependent dye measurements. For flow cytometry staining, media was replaced with staining media containing TMRM (50 nM) and MTG (50 nM), and cultures were incubated at 37°C for 25 min prior to harvest. To validate the membrane potential of TMRM dye, control samples were created by adding either a depolarizing agent (CCCP, 30 μM) or a hyperpolarizing agent (oligomycin, 10 μM) to the staining media.
eCLIP library preparation and analysis
For analysis of RNA-targets bound by LARP4 in HEK293 cells, eCLIP experiments were carried out in biological duplicates as previously described (Van Nostrand et al. 2017), with the following experiment-specific details. UV-crosslinked LARP4 was immunoprecipitated with anti-LARP4 rabbit polyclonal antibody (Bethyl A303-900A). Libraries were sequenced as 75 bp single-end reads on an Illumina HiSeq4000 instrument to approximately 20 million reads. Reads were processed through the previously described eCLIP0v0.40 pipeline (Van Nostrand et al. 2017). Input normalized peaks were further filtered by the irreproducible discovery rate (IDR) and merged using merge_peaks-v0.05 ([IDR] https://github.com/YeoLab/merge_peaks).
To find specific pathway genes, we combined annotations from the MitoCarta database with Gencode annotations v19 to filter IDR peaks in specific gene groups. Nuclear-encoded mitochondrial genes are defined as all genes in the MitoCarta database that are not encoded on chrM. Mitochondrial-encoded ribosomes are defined as any gene name within MitoCarta that contains RPS/RPL. OXPHOS is defined with the pathway annotation in MitoCarta. The filtered IDR peaks are subsequently fed into the MetaPlotR pipeline and generated the metagene plots. For motif analysis, we define a set of strong binding sites with −log10(P-value) > 7 (Fisher's exact test), log2(fold-change [IP/input]) > 4. Along with the gene groups defined above, we ran HOMER motif analysis using our in house-pipeline (https://github.com/YeoLab/clip_analysis_legacy) for each set of IDR peaks. Briefly, peaks were stratified by regions (CDS, UTR, introns), and then HOMER was used de novo to find motifs with GC-matched, region-matched background sequences. Supplemental Table 4 lists significantly enriched eCLIP peaks.
Gene ontology analysis
For all GO analyses, the Metascape GO method and server were used (Zhou et al. 2019). From each data set, two gene sets were generated, a set of foreground genes and a background gene set. For analysis of LARP4 eCLIP data sets (HEK293, HepG2, and K562), the set of foreground genes (genes that encode RNA targets of LARP4) was generated from the annotated bed file of the merged IDR peaks. Genes with an eCLIP peak passing the following filters were included in each set of foreground genes: log2(fold-change [CLIP-IP/Input]) > 4 and −log10(P-value) > 7 (Fisher's exact test). For background gene sets, the matched eCLIP input data sets were used; all genes with at least five reads in any exonic region were included in each background set. For analysis of the TMT mass spectrometry data sets, foreground gene sets were generated from proteins present in either increased abundance or decreased abundance, and background gene sets were defined by all proteins detected in each mass spectrometry data set (mitochondrial extracts or WCEs). Proteins with significantly increased abundance were defined as having a log2(fold-change [KO/WT]) > 0.2 and P-value < 0.05 (two-tailed unpaired Student's t-test). Proteins with significantly decreased abundance were defined as having a log2(fold-change [KO/WT]) < 0.2 and P-value < 0.05 (two-tailed unpaired Student's t-test).
Statistical analyses
Average values were tested for statistical differences using two-tailed unpaired Student's t-tests. Averages of biological replicates were plotted as mean values ± standard error and replicate number denoted with a capital N. Averages of technical replicates were plotted as mean values ± standard deviation and replicate number denoted with a lowercase n. The statistical significance of differences in averages are indicated by (*) P ≤ 0.05, (**) P ≤ 0.001, (***) P ≤ 0.0001, (****) P ≤ 0.00001. Statistical significance of enrichments was determined by the hypergeometric test.
Reagents, resources, and data
All key reagents, resources, and data are listed in Supplemental Tables 1–4. eCLIP sequencing data has been deposited to GEO under accession number GSE193134. The TMT mass spectrometry data has been deposited to the MassIVE data repository at MSV000093340.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
COMPETING INTEREST STATEMENT
G.W.Y. is a cofounder, a member of the Board of Directors, a scientific advisor, an equity holder, and a paid consultant for Locanabio and Eclipse BioInnovations. G.W.Y. is a visiting professor at the National University of Singapore. The interest(s) of G.W.Y. have been reviewed and approved by the University of California San Diego in accordance with its conflict-of-interest policies. The authors declare no other competing financial interests.
ACKNOWLEDGMENTS
We thank Dr. Eric Van Nostrand for initial guidance on the project. B.M.L. and this work were supported by a National Institutes of Health (NIH) T32 Cell and Molecular Genetics Training Program (GM007240), as well as grants and awards from the NIH to T.H. (CA080100, CA082683, and CA242443). This work was supported by grants from the NIH to G.W.Y. (HG009889 and HG004659). G.W.Y. is supported by an Allen Distinguished Investigator Award, a Paul G. Allen Frontiers Group advised grant of the Paul G. Allen Foundation. T.H. is a Frank and Else Schilling American Cancer Society Professor and holds the Renato Dulbecco Chair in Cancer Research. O.M. was supported by a Gruss-Lipper postdoctoral fellowship. This work was supported by the Mass Spectrometry Core of the Salk Institute with funding from NIH-NCI CCSG: P30 CA014195 and the Helmsley Center for Genomic Medicine. We thank J. Diedrich and A. Pinto for proteomics technical support. We thank Dr. Sandy Mattijssen and Dr. Richard J. Maraia for providing a LARP4 antibody.
Author contributions: Conceptualization, B.M.L., G.W.Y., and T.H.; investigation, B.M.L. and O.M.; vector cloning, B.M.L. and C.Y.C.; formal analysis and visualization, B.M.L.; metagene analysis, B.M.L. and H.L.H.; writing—original draft, B.M.L.; writing—review and editing, B.M.L., G.W.Y., and T.H.; funding acquisition, G.W.Y. and T.H.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079799.123.
-
Freely available online through the RNA Open Access option.
- Received August 11, 2023.
- Accepted November 25, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International license), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Ben Lewis is the first author of this paper, “LARP4 is an RNA-binding protein that binds nuclear-encoded mitochondrial mRNAs to promote mitochondrial function.” Ben was a graduate student of the UCSD Biology Department PhD program. His thesis research focus was on RNA-binding proteins and mitochondrial homeostasis, under the supervision of Gene Yeo and Tony Hunter. His laboratory work was primarily done in Tony Hunter's lab in the Molecular and Cellular Biology Lab (MCBL) at the Salk Institute.
What are the major results described in your paper and how do they impact this branch of the field?
In this study, through a systematic analysis of eCLIP data sets for 150 RNA-binding proteins (RBPs) from the ENCODE collection, we have identified several RBPs with RNA-target sets enriched for mRNAs encoding mitochondrial proteins, including the LARP4. Our in-depth characterization of LARP4's RNA-target set reveals an enrichment for mRNAs encoding respiratory chain complex proteins (RCCPs) and mitochondrial ribosome proteins (MRPs). Furthermore, our functional studies demonstrate that LARP4 depletion disrupts the expression of these two groups of proteins. Finally, we established that LARP4 is required to maintain normal rates of proliferation, translation, and respiratory (OXPHOS) function. These results support a novel function of LARP4 by which it binds functionally important nuclear-encoded mitochondrial mRNAs to help maintain mitochondrial protein homeostasis and facilitate proper respiratory function.
What led you to study RNA or this aspect of RNA science?
Since high school, I have been fascinated by mitochondria, molecular biology, and neurodegeneration. Later in graduate school, I was drawn to study RNA-binding proteins because they are implicated in many neurodegenerative diseases, and there was a lot of opportunity to study interesting molecular biology using new sequencing-based tools and computational approaches, which was exciting. By looking for RNA-binding proteins involved in mitochondrial biology, I was able to bring together the biology that interested me the most.
If you were able to give one piece of advice to your younger self, what would that be?
A piece of advice I would give to my younger self entering graduate school would be to make more time to attend social events with your fellow PhD students. The samples in the freezer aren't going anywhere, but social opportunities become increasingly scarce as time goes on.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
Both of my PhD thesis advisors, Tony Hunter and Gene Yeo, served as exceptional role models who led by example, profoundly influencing my approach to science. I'd like to highlight a few of their attributes that have left a lasting impact on me. Tony Hunter possesses a remarkable ability to pose insightful questions that address critical aspects of a project, and he does so with tact and kindness. His example has reinforced my belief in the importance of asking critical questions in a constructive and considerate way. Gene excels at integrating computational and experimental methods. As a laboratory leader, he drives performance by setting high standards and encouraging trainees to collaborate on projects and learn from each other. My experience in the Yeo lab taught me the value of an interdisciplinary approach to science and the value of disseminating your specific expertise to your teammates or collaborators.
What are your subsequent near- or long-term career plans?
After completing my PhD, I transitioned into an industry scientist position in the regenerative medicine space. In my new role, I primarily carry out multiomics data analysis and integration, as well as lead a pipeline development project. The transition from academia to industry has been a rewarding experience, providing me with the opportunity to apply my training to the development of therapies and collaborate with professionals who share a passion for advancing regenerative medicine.








