
The Escherichia coli ribB riboswitch senses flavin mononucleotide within a defined transcriptional window
- Sébastien H. Eschbach1,
- Elsa D.M. Hien,
- Tithi Ghosh2,
- Anne-Marie Lamontagne3 and
- Daniel A. Lafontaine
- Department of Biology, Faculty of Science, RNA Group, Université de Sherbrooke, Sherbrooke, Quebec, Canada J1K 2R1
- Corresponding author: daniel.lafontaine{at}usherbrooke.ca
-
Handling editor: Eric Westhof
Abstract
Riboswitches are metabolite-binding RNA regulators that modulate gene expression at the levels of transcription and translation. One of the hallmarks of riboswitch regulation is that they undergo structural changes upon metabolite binding. While a lot of effort has been put to characterize how the metabolite is recognized by the riboswitch, there is still relatively little information regarding how ligand sensing is performed within a transcriptional context. Here, we study the ligand-dependent cotranscriptional folding of the FMN-sensing ribB riboswitch of Escherichia coli. Using RNase H assays to study nascent ribB riboswitch transcripts, DNA probes targeting the P1 and sequestering stems indicate that FMN binding leads to the protection of these regions from RNase H cleavage, consistent with the riboswitch inhibiting translation initiation when bound to FMN. Our results show that ligand sensing is strongly affected by the position of elongating RNA polymerase, which is defining an FMN-binding transcriptional window that is bordered in its 3′ extremity by a transcriptional pause site. Also, using successively overlapping DNA probes targeting a subdomain of the riboswitch, our data suggest the presence of a previously unsuspected helical region involving the 3′ strand of the P1 stem. Our results show that this helical region is conserved across bacterial species, thus suggesting that this predicted structure, the anti*-P1 stem, is involved in the FMN-free conformation of the ribB riboswitch. Overall, our study further demonstrates that intricate folding strategies may be used by riboswitches to perform metabolite sensing during the transcriptional process.
Keywords
INTRODUCTION
Riboregulation in bacteria encompasses a large variety of molecular regulators and signaling molecules, which are involved in the orchestration of regulatory mechanisms to ensure efficient cellular homeostasis (Chiaruttini and Guillier 2020; Chełkowska-Pauszek et al. 2021; Ng Kwan Lim et al. 2021; Schärfen and Neugebauer 2021; Evguenieva-Hackenberg 2022). Riboswitches are noncoding RNA regulators that are located in the 5′ untranslated regions (UTR) of mRNAs and that modulate gene expression by undergoing structural changes upon metabolite binding (Lotz and Suess 2018; Mahendran et al. 2022). These RNA elements are widely distributed across bacteria and represent >55 distinct ligand-binding motifs (Pavlova et al. 2019; Breaker 2022). While riboswitches modulate transcription attenuation (Lotz and Suess 2018; Turnbough 2019), they also control the initiation of translation (Breaker 2018) and mRNA decay (Caron et al. 2012; Takemoto et al. 2015). Interestingly, riboswitch regulation at the transcriptional and translational levels appears to be segregated in Gram-negative and Gram-positive bacteria, respectively, suggesting that the cellular context may be important for their regulatory mechanism (Arnvig 2019). Recent work has highlighted the versatility of riboswitches as molecular tools, which can be used as novel antibacterial targets (Abduljalil 2018; Panchal and Brenk 2021), synthetic molecular sensors (Sinumvayo et al. 2018; Chang et al. 2021; Tabuchi and Yokobayashi 2021; Hoetzel and Suess 2022), or modulators of biochemical pathways (Villa et al. 2018; Leistra et al. 2019; Kim et al. 2022). In addition, high-throughput and bioinformatics efforts suggest that several riboswitches are yet to be identified (Cole and Lupták 2019; Brewer et al. 2021), therefore providing strong optimism for the discovery of additional RNA-based regulation mechanisms involving important cellular metabolites (Breaker 2022). Lastly, several large noncoding RNAs have been recently uncovered (Harris and Breaker 2018), which could be particularly exciting in case such RNA elements exhibit similar regulating properties as riboswitches.
The flavin mononucleotide (FMN)-sensing riboswitch in Escherichia coli regulates the expression of ribB (Gelfand et al. 1999; Vitreschak et al. 2002; Winkler et al. 2002b), which is implicated in the biosynthesis of riboflavin (Richter et al. 1992). In the FMN-bound OFF state, crystal structures show that the aptamer domain folds into a compact structure that is organized around six helices (Fig. 1, P1–P6) and forms a binding site allowing high affinity and specificity for FMN (Serganov et al. 2009; Vicens et al. 2011). Interestingly, in the absence of ligand, the aptamer adopts a slightly different structure (Vicens et al. 2011), consistent with the riboswitch relying on a conformational selection folding mechanism to sense ligand. It is also predicted that the ON state structure contains the anti-P1 stem (Fig. 1), thereby allowing ribosomes to initiate ribB translation. In contrast to the FMN-sensing transcriptional riboswitch in Bacillus subtilis (Winkler et al. 2002b), ligand binding to the E. coli riboswitch inhibits the initiation of translation by sequestering the Shine–Dalgarno and AUG start codon sequences (Hollands et al. 2012; Pedrolli et al. 2015). Importantly, the E. coli FMN riboswitch was also shown to operate at the transcriptional level (Pedrolli et al. 2015), which relies on Rho transcription termination (Hollands et al. 2012; Bastet et al. 2017). In the context of Rho regulation, the current model proposes that the binding of FMN to the riboswitch aptamer domain leads to the exposition of Rho utilization (rut) sites located in the riboswitch sequence, and therefore to transcription termination within the expression platform (Fig. 1; Hollands et al. 2012; Bastet et al. 2017). Significant efforts have been put to characterize small antibacterial molecules targeting the FMN riboswitch (Lee et al. 2009; Ott et al. 2009; Long et al. 2010; Mansjö and Johansson 2011; Blount et al. 2015; Howe et al. 2015; Matern et al. 2016; Wang et al. 2017; Vicens et al. 2018; Motika et al. 2020), which make this riboswitch a prime candidate for the development of antibiotic compounds.
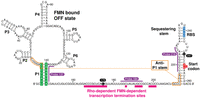
The sequence and secondary structure of the ribB riboswitch of E. coli. The nucleotide numbering is based on previous studies (Winkler et al. 2002b; Hollands et al. 2012). While the structure of the aptamer is depicted according to the consensus structure (Vitreschak et al. 2002; Winkler et al. 2002b), the organization of the expression platform is based on previous analyses (Hollands et al. 2012). The structure is depicted as when bound to FMN (OFF state). The aptamer domain consists of stems P1–P6, and the expression platform encompasses the sequestering stem. The P1 stem is highlighted in green. The anti-P1 stem that is forming in the FMN-free ON state is shown in orange. The ribosome-binding site (RBS) and AUG start codon are shown in blue and red, respectively. The pause sites 175 and 247 are indicated in black circles. The RNase H probes 137, 184, and 218 are shown in purple. Rho-dependent transcription termination sites are shown in pink (Hollands et al. 2012).
The transcriptionally regulating ribD FMN-sensing riboswitch in B. subtilis was previously shown to be kinetically driven (Wickiser et al. 2005b), i.e., that the riboswitch is not attaining thermodynamic equilibrium with FMN before RNA polymerase (RNAP) is committed either to stop or to elongate transcription. The presence of transcriptional pause sites within the riboswitch sequence allows more time for FMN sensing before RNAP reaches the terminator site, therefore creating a transcriptional window for ligand binding. Biochemical studies on the B. subtilis pbuE adenine-sensing riboswitch also revealed that ligand binding must be performed within a narrow transcriptional window for the riboswitch to modulate transcription (Wickiser et al. 2005a; Lemay et al. 2006), in agreement with such riboswitches regulating at the cotranscriptional level. Importantly, translationally regulating riboswitches also exhibit a transcriptional window for ligand binding. In the case of the E. coli thiC and tbpA riboswitches (Chauvier et al. 2017, 2021), which sense thiamin pyrophosphate (TPP), RNAP elongation downstream from the aptamer domain eventually leads to the formation of nascent RNA structures destabilizing the aptamer, thus preventing TPP binding. The destabilization of the aptamer domain during RNAP elongation effectively creates a transcriptional binding window, thereby requiring that TPP sensing is performed cotranscriptionally. Such a cotranscriptional binding mechanism is crucial for both thiC and tbpA riboswitches since transcription elongation is regulated through Rho transcription termination (Bastet et al. 2017; Chauvier et al. 2017). Interestingly, despite the E. coli ribB riboswitch primarily operating at the translational level, the presence of FMN downregulates the mRNA levels (Hollands et al. 2012; Pedrolli et al. 2015; Bastet et al. 2017), indicating that riboswitch FMN sensing is used to control both protein and mRNA levels. However, little is known whether FMN sensing is performed within a transcriptional window and what is the role of the nascent transcript in the sensing activity.
Here, we study the cotranscriptional FMN sensing of the E. coli ribB riboswitch. Our findings show that nascent ribB transcripts exhibit nanomolar affinities toward FMN. When assessing FMN binding on transcriptional complexes positioned at naturally occurring pause sites, we find that the affinity is modulated, in agreement with the formation of nascent structures affecting the sensing of FMN. Using RNase H assays, we find that transcriptional complexes at the 247 pause site exhibit small changes in cleavage accessibility, suggesting a lower capacity to bind FMN. By using several DNA probes covering the region 180–227 nt, our results reveal that the ribB riboswitch exhibits varying degrees of structural changes in the expression platform when elongation complexes (EC) are paused at the 247 pause site. Lastly, our results provide strong evidence suggesting the presence of a previously unidentified structure encompassing a portion of the ligand-free aptamer and the expression platform, therefore explaining the low binding affinity of the EC-247 complex. Together, our study sheds new insights about the folding of the ribB riboswitch and suggests that FMN sensing is also governed by the presence of a transcriptional window.
RESULTS
Nascent E. coli ribB transcripts undergo structural changes upon FMN sensing
To study how FMN sensing is achieved by ribB nascent transcripts, we used RNase H cleavage assays that have been previously employed for the structural characterization of nascent E. coli riboswitches (Ontiveros-Palacios et al. 2008; Perdrizet et al. 2012; Lussier et al. 2015; Bastet et al. 2017; Chauvier et al. 2017, 2021; Zeller et al. 2022). In these assays, we used a DNA template containing the E. coli lacUV5 promoter fused to the ribB riboswitch and the first 10 codons of the ribB coding region. Using this template, we performed in vitro transcription reactions using the E. coli RNAP as a function of varying ligand concentrations. The folding of nascent transcripts was assessed through RNase H cleavage activity using a DNA oligonucleotide designed to bind to the region 137–146 (Fig. 2A, probe 137). This region was selected due to its implication in the P1 stem that is expected to form upon FMN binding. In the absence of FMN, it is anticipated that nascent transcripts are hybridized to the probe 137 and cleaved by RNase H. In contrast, FMN binding to ribB transcripts should prevent cleavage through the stabilization of the P1 stem.
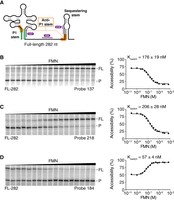
RNase H assays of FL ribB nascent riboswitch transcripts. (A) Schematic representing the ribB riboswitch in the FMN-bound OFF conformation. The P1 and anti-P1 stems are shown in green and orange, respectively. The RNase H probes 137, 184, and 218 are shown in purple. (B–D) RNase H probing assays performed on FL (282 nt) nascent ribB riboswitches. Reactions were done as a function of the FMN concentration using probes 137 (B), 218 (C), and 184 (D). Cleavage assays were performed using a range of 100 pM to 500 µM FMN. FL and cleaved products (P) are indicated on the right of the gels. For each reaction, the Kswitch value was determined by fitting the data to a two-state model. The experiments were performed in triplicate, and the most representative data are shown.
When nascent ribB transcripts were subjected to RNase H assays, a major product (P) was observed at low FMN concentrations (Fig. 2B, left). However, the use of higher concentrations of FMN reduced the fraction of cleaved products, which also led to higher levels of the full-length (FL) species. These results are consistent with the binding of FMN stabilizing the P1 stem, which prevents both the hybridization of probe 137 to ribB nascent transcripts and the cleavage by RNase H. Fitting analysis revealed a half FMN concentration (Kswitch) value of 176 ± 19 nM for the structural modulation (Fig. 2B, right). It was previously determined using mass spectrometry (LC-MS/MS) that the absolute concentration of FMN is ∼50 µM in exponentially growing E. coli with glucose as the carbon source (Bennett et al. 2009). Since this value is well above the Kswitch value of the ribB riboswitch (∼176 nM), it suggests that the riboswitch would always be bound to FMN in such conditions. However, the actual concentration of free FMN in bacteria could be significantly lower, thus allowing the ribB riboswitch to efficiently sense FMN.
According to the OFF-state secondary structure (Fig. 1), FMN binding to ribB transcripts leads to the inhibition of translation initiation by allowing the formation of the sequestering stem. To monitor this structural rearrangement, we next used a probe designed to target region 218–227 (probe 218) that is involved in the formation of the sequestering stem (Fig. 2A). Therefore, as observed for the probe 137 (Fig. 2B), it is anticipated that FMN sensing by ribB nascent transcripts inhibits the hybridization of probe 218, thus preventing RNase H cleavage. As expected, when performing RNase H assays with the probe 218 on ribB nascent transcripts, the amount of cleavage products was reduced upon increasing FMN concentration (Fig. 2C, left). Analysis of cleavage efficiencies provided a Kswitch value of 206 ± 28 nM (Fig. 2C, right). This value is similar to that obtained with the probe 137 (176 ± 19 nM), consistent with a direct correlation between FMN binding to the aptamer and formation of the sequestering stem (Fig. 2A).
To further study the binding of FMN by ribB transcripts, we next employed a probe targeting the region 184–193 nt that is located between the aptamer and the sequestering stem (Fig. 2A). This region was selected since there is still some uncertainty regarding how the transcript region located between the aptamer and the sequestering stem participates in the riboswitch regulatory function (Hollands et al. 2012; Pedrolli et al. 2015). Our RNase H assays showed that in contrast to data obtained with probes 137 and 218, the cleavage of nascent transcripts was increased at higher FMN concentrations (Fig. 2D, left). Furthermore, quantification of our data revealed that FMN induced a smaller change in RNase H cleavage (Fig. 2D, right). Fitting analysis revealed a smaller Kswitch value of 57 ± 4 nM (Fig. 2D, right), which is approximately threefold lower than what was obtained for probes 137 and 218. Such Kswitch variations have previously been observed for the adenosylcobalamin-sensing btuB riboswitch in E. coli (Lussier et al. 2015), most likely reflecting intrinsic variations either due to the local RNA structure or to the DNA oligonucleotides used for RNase H assays. Nevertheless, the information obtained with the probe 184 is important as it shows that this region becomes more accessible upon FMN binding. No cleavage of transcripts was observed in the absence of RNase H (Supplemental Fig. S1), consistent with the activity of RNase H.
FMN sensing occurs at transcriptional pause sites
An earlier study determined that the ribB riboswitch contains at least two transcriptional pause regions (Chauvier et al. 2017). The first pause region is located between the aptamer and the sequestering stem and is constituted of pause sites 175 and 181 (Fig. 1). The second region was determined to be nearby the AUG start codon (247–249 nt). Interestingly, the same study revealed that FMN binding to nascent ribB transcripts resulted in the pause half-life being decreased by ∼40% and increased by ∼20% for the upstream and downstream pause regions, respectively. To characterize the cotranscriptional sensing of ribB nascent transcripts at these transcriptional pause regions, we halted EC at defined positions using a biotin-streptavidin transcriptional roadblock (Frieda and Block 2012; Chauvier et al. 2017, 2021). To study both pause regions, we generated constructs allowing to stall RNAP at positions 175 and 247 (Fig. 1). Control experiments showed that EC were efficiently stalled by the biotin-streptavidin roadblocks (Supplemental Fig. S2), thus allowing us to study nascent transcripts at these positions.
We first monitored the binding of FMN on EC-175 complexes using probe 137. RNase H cleavage assays showed that EC-175 nascent transcripts exhibited a reactivity profile very similar (Fig. 3A) to that obtained with FL RNAs (Fig. 2B). Fitting analysis yielded a Kswitch value of 23 ± 3 nM for EC-175 complexes, suggesting that FMN is sensed ∼7.5-fold more efficiently when RNAP is located at this site. Importantly, the enhanced ability of ribB nascent transcripts to perform ligand sensing at pause sites is also shared by other riboswitches, such as thiC and tbpA (Chauvier et al. 2017; Chatterjee et al. 2021), suggesting that cotranscriptional sensing may be an inherent part of riboswitch regulatory activity.
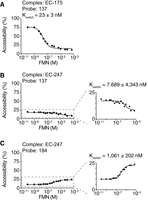
RNase H assays of transcription complexes located at positions 175 and 247. (A) Kswitch determination using the probe 137 for the elongation complex stalled at position 175 (EC-175). Cleavage assays were performed using a range of 100 pM to 500 µM FMN. The Kswitch value (23 ± 3 nM) was obtained using the probe 137. The experiments were performed in triplicate, and the most representative data are shown. (B,C) Kswitch determination using the probes 137 (B) and 184 (C) for the elongation complex stalled at position 247 (EC-247). Cleavage assays were performed using a range of 100 pM to 500 µM FMN. For each probe, a small change in cleavage efficiency was observed across the FMN concentrations, thus leading to a greater uncertainty of the determined Kswitch values. The experiments were performed in triplicate, and the most representative data are shown.
We next assessed the FMN sensing activity of transcription complexes at position 247. Using the probe 137, we found a very low RNase H activity across all tested FMN concentrations (Fig. 3B). The analysis of the small change in the extent of cleavage (∼10%) (Fig. 3B, right) allowed us to calculate a Kswitch value of 7689 ± 4343 nM (Fig. 3B), indicating poor FMN sensing activity at EC-247. Because this reactivity profile is markedly different from the one obtained in the context of the FL transcript and EC-175 (Figs. 2B and 3A), it suggests that ribB nascent transcripts adopt a different conformation in the context of EC-247. Further characterization of EC-247 nascent transcripts using the probe 184 revealed low RNase H cleavage efficiencies at all FMN concentrations (Fig. 3C), similarly to results obtained with probe 137 (Fig. 3B). The analysis of the reactive population (∼15%) yielded a Kswitch value of 1061 ± 202 nM, which is consistent with the poor FMN sensing activity of EC-247 nascent transcripts measured with the probe 137 (Fig. 3B).
Our data indicate that ribB nascent transcripts exhibit high and low FMN affinity at the first and second pause regions, respectively (Fig. 3A,B), in agreement with FMN sensing being performed within a transcriptional window located upstream of the second pause region. While the high FMN affinity of transcription complexes in the first region is mostly due to the aptamer structure being readily adopted by nascent transcripts, the low affinity observed in the second pause region is most probably because the presence of additional downstream sequence destabilizes the folding of the aptamer. Such a situation has previously been documented for the thiC and tbpA riboswitches, where the formation of the anti-P1 stem leads to the disruption of the aptamer (Chauvier et al. 2017, 2021). In the present case, it is entirely consistent that the formation of the anti-P1 stem in EC-247 nascent transcript inhibits the folding of the aptamer domain, therefore precluding the binding of FMN (Fig. 1). Such a mechanism favors the ligand sensing at the cotranscriptional level by EC within a transcriptional window located in the riboswitch expression platform.
RNase H probing of the FMN sensing transcriptional window
To gather additional information about the transcriptional window in which ribB nascent transcripts perform FMN sensing, we employed RNase H assays to probe the region 180–227 nt (Fig. 1). In these assays, we used a series of 10 nt DNA probes designed to cover the entire region and to be shifted by 1 nt between consecutive probes. A similar approach has previously been used to study the E. coli btuB and thiC riboswitches nascent transcripts (Perdrizet et al. 2012; Hien et al. 2024). An example of this strategy is shown in Figure 4A, where the probes targeting the subregion 180–198 nt are indicated at their corresponding targeted positions. RNase H assays performed within this region with and without FMN revealed large variations in cleavage where higher accessibilities were observed at the 3′ extremity (Fig. 4B). For instance, while the probe 180 showed accessibilities of ∼9% and ∼20% without and with FMN, respectively, the probe 189 yielded accessibilities of ∼80% and 91% in the same conditions (Fig. 4B).
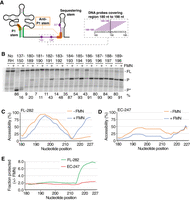
RNase H assays of the region 180–227 nt using FL transcripts and transcription complexes located at position 247. (A) Schematic representing the ribB riboswitch and the region 180–227 nt probed using RNase H assays (depicted in purple). The inset shows an example of DNA probes used to monitor the region 180–198 nt. For each RNA residue analyzed, overlapping DNA probes may be used to deduce an average RNase H accessibility. For example, the average of RNase H cleavage efficiency for position 189 is calculated using probes 180–189. (B) RNase H probing assays obtained on the FL ribB nascent transcripts without (−) and with (+) 100 µM FMN. The regions targeted by the DNA probes in each experiment are indicated at the top of the gel. The FL and cleaved products (P and P*) are indicated on the right of the gel. The length of the P* product obtained with the 137 probe is shorter than what was obtained with the other probes due to the targeted location on the transcript. Control experiments performed without RNase H (No RH) are shown on the left. RNase H assays were performed in the presence and absence of FMN using each probe. The most representative data are shown on the gel. (C,D) RNase H probing accessibility profiles for the ribB FL 282 nt transcript (C) or when in the EC-247 complex (D). The accessibility profile obtained without (−) and with (+) FMN is shown as a function of nucleotide position. For each position, the average of RNase H cleavage efficiency was calculated using the efficiencies obtained from all probes targeting the position. RNase H assays were performed in the presence and absence of FMN using each probe. The averages of overlapping probes are shown. (E) The fraction protected from RNase H cleavage by the binding of FMN as a function of nucleotide position in the context of FL transcripts (green) or in EC-247 (red). The fraction was obtained by performing a ratio of the averages of RNase H cleavage efficiencies obtained without and with FMN. RNase H assays were performed in the presence and absence of FMN using each probe. The averages of overlapping probes are shown.
Because DNA probes significantly overlap with each other, nucleotides of the analyzed region are targeted by multiple probes (except the first and last residues), therefore allowing to calculate the average of cleavage efficiencies obtained with different probes. For example, the average calculated for position 189 is acquired by combining the cleavage efficiencies obtained from probes 180–189 (Fig. 4A). Using this methodology, we calculated the average efficiency for each position of the 180–227 nt region that was probed both in the absence and presence of FMN (Fig. 4C). Without ligand, the profile of RNase H cleavage shows that the accessibility significantly increases from position 180 to position ∼195 (∼94%) (Fig. 4C) and gradually decreases to low levels at position 214 (∼13%). The downstream sequence (i.e., 215–227 nt) shows a large increase in accessibility, which suggests that the sequestering stem is not formed in this condition. In the presence of FMN, while a very similar pattern was obtained for the region 180–214 nt, a very low accessibility was detected for the 215–227 nt region (Fig. 4C), in agreement with the formation of the sequestering stem in the ligand-bound state (Fig. 1). Together, the matching profiles obtained −/+ FMN for the region 180–214 nt suggest that nascent transcripts exhibit a similar conformation in this region. However, the reduced accessibility of the downstream region in the presence of FMN is consistent with the formation of the sequestering stem that precludes translation initiation.
Next, to investigate the low FMN affinity exhibited by EC-247 nascent transcripts, we repeated RNase H assays to probe the region 180–227 nt in the absence and presence of FMN. Compared to the data obtained using the FL RNA (Fig. 4C), RNase H assays performed in the absence of FMN showed less variation across the entire region (<50% change) (Fig. 4D), suggesting a different global conformation in EC-247. When these assays were repeated with FMN, a similar profile was observed, but which exhibited an overall lower accessibility for the region 180–217 nt (Fig. 4D). The results also showed that in contrast to the FL RNA, the cleavage efficiencies were highly similar for the region 217–227 nt (Fig. 4D), indicating that the presence of FMN does not result in a detectable structural change for this region.
We compared the data obtained for the FL RNA and EC-247 by plotting the fraction protected in the presence of FMN across the region 180–227 nt (Fig. 4E). While no significant change was observed for nascent transcripts in EC-247, the fraction protected became more prominent in the region ∼215–227 nt for the FL RNA species. Our results are consistent with the idea that EC-247 nascent transcripts are refractory to bind to FMN, thus effectively constituting the 3′ boundary of the transcriptional sensing window.
A competing structure is involved in P1 stem formation
The variation observed for the accessibility profile of the FL RNA between positions 180–204 nt suggests that this region is structurally organized (Fig. 4C). Furthermore, the fact that nascent transcripts within EC-247 show very low levels of RNase H cleavage in this region (Fig. 4D) suggests that the structure is perhaps different or that it is stabilized in this context. We have previously observed such a case for the thiC riboswitch in which the anti-P1 stem is stabilized when RNAP reaches a specific pause site (Chauvier et al. 2017). To explore which structure(s) could be formed within this region, we examined the expression platform of FMN-sensing riboswitches in various bacterial species (Supplemental Fig. S3). Close examination of each riboswitch representative revealed that a previously unpredicted interaction could occur between positions 141–149 nt and 175–183 nt in the ON state (Fig. 5A, see inset). Because the 141–149 nt region is involved in the formation of the P1 stem, it suggests that the predicted interaction would be formed only in the absence of FMN and was therefore named the anti*-P1 stem.
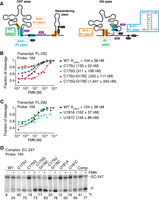
The ribB riboswitch contains an additional helical domain in the FMN-free ON state. (A) Schematics representing the ribB riboswitch in the FMN-bound OFF and FMN-free ON conformations. The regions involved in the predicted P1, anti*-P1, and anti-P1 stems are shown in green, blue, and orange, respectively. While the P1 stem is formed in the OFF state, the anti*-P1, and anti-P1 stems are formed in the ON state. The structure of anti*-P1 stem is shown on the right. The location of the RNase H probes and pause sites are indicated in purple and black, respectively. The line in gray in the ON state represents a section for which no attempt was made to draw the secondary structure. (B,C) Kswitch determination using the probe 184 for the FL transcripts. The mutants performed at positions C175/G176 (B) and U181 (C) were analyzed. Cleavage assays were performed using a range of 2 nM to 10 µM FMN. The fitting analyses were performed by normalizing the cleavage fraction to 1, and the Kswitch values are indicated for each mutant. The experiments were performed in duplicate, and the most representative data are shown. (D) RNase H probing assays obtained on EC-247 ribB nascent transcripts with the probe 184. Experiments were performed without (−) and with (+) 100 µM FMN. The WT and the mutants used are indicated at the top. The FL EC-247 transcripts (EC-247) and cleaved products (P) are indicated on the right of the gel. The experiments were performed in triplicate, and the most representative data are shown.
The inspection of the anti*-P1 stem shows that only two G-C base pairs are present, suggesting that they could be important for the stability of the structure. We first investigated the helical domain by introducing mutations to destabilize the formation of the G-C base pairs. The obtained mutants were assessed by determining the Kswitch values in the context of FL transcripts (FL-282) using the probe 184 (these experiments were performed using fluorescence-based assays, see Materials and Methods). The affinity reported for the WT construct by fluorescent assays (104 ± 38 nM) is similar to that obtained using radioactive transcription reactions (57 ± 4 nM) (Fig. 5B). When substituting the position C175 for an uracil (C175U) or a guanine (C175G), the FMN affinity was found to decrease by ∼1.25 and ∼3-fold, respectively (Fig. 5B; Supplemental Fig. S4). These results suggest that the C175U mutation does not affect riboswitch sensing, most probably since a G149:U175 base pair is formed in the anti*-P1 stem. However, given that the C175G mutant does not allow the formation of a base pair, a more disruptive effect on FMN sensing was obtained. We next introduced double mutations at positions C175 and G176 and monitored the effect on Kswitch values. When monitoring the C175U-G176C mutant, an affinity of 311 ± 108 nM (approximately threefold decrease) was obtained, suggesting that the riboswitch activity is perturbed (Fig. 5B). Importantly, the C175G-G176C mutant showed a strongly reduced affinity for FMN, where a Kswitch value of 1641 ± 345 nM was observed, which represents a ∼16-fold affinity decrease compared to the WT (Fig. 5B). We also investigated the importance of the residue U181 by introducing adenine and cytosine (U181A and U181C). For both mutants, relatively mild effects were observed on FMN sensing (162 ± 37 and 156 ± 86 nM, respectively) (Fig. 5C), suggesting that the identity of this residue is not crucial for the riboswitch sensing activity.
We next investigated the effects of mutations in the context of the EC-247 transcription complex. Unexpectedly, when performing RNase H assays using the C175U mutant with the probe 184, we obtained a higher cleavage efficiency in the absence of FMN (∼40%), which contrasts with that obtained with the WT construct (∼8%) (Fig. 5D). This result is consistent with the destabilization of the anti*-P1 stem allowing more efficient RNase H cleavage. Importantly, the addition of FMN resulted in a clear protection from RNase H cleavage, which showed an efficiency of ∼75% (Fig. 5D). When replacing C175 with a guanine (C175G), similar results were obtained albeit with a lower cleavage efficiency in the absence of FMN (∼19%) (Fig. 5D). When assessing the double mutant C175U:G176C, a similar effect to the single mutation constructs was obtained (Fig. 5D). However, the use of the mutant C175G:G176C led to high extents of cleavage both in the absence (∼77%) and presence of FMN (∼83%) (Fig. 5D). This result is consistent with the stronger effect of the mutation on the riboswitch affinity observed in the FL construct (Fig. 5B), consistent with a stronger destabilization of the anti*-P1 stem. Similarly to the G175 single mutant constructs, strong FMN-dependent protections were observed when using U181A and U181C mutants (Fig. 5D). Lastly, we replaced the complete region 175–183 nt for its Watson–Crick complementary counterpart (Comp mutant). When assessing the Comp mutant using RNase H assays, we found that the cleavage was efficient in the absence of FMN (∼41%) and significantly increased with FMN (∼77%) (Fig. 5D). Thus, our results suggest that the disruption of the anti*-P1 stem allows better RNase H cleavage efficiency using the probe 184 while still retaining FMN-binding activity.
To investigate further the role of the anti*-P1 stem on EC-247 nascent transcripts, we performed RNase H assays on the stem mutants using the probe 137 (Supplemental Fig. S5). As observed for the probe 184, we found that the cleavage efficiencies obtained in the absence of FMN were higher when mutating positions C175 and G176 than the position U181 (Supplemental Fig. S5). Our results also showed that higher RNase H activities were observed with FMN for all mutants when compared to the WT construct (Supplemental Fig. S5).
DISCUSSION
In this work, we suggest a model in which FMN binding to the riboswitch leads to the cotranscriptional formation of the P1 stem, which prevents the folding of the anti*-P1 and anti-P1 stems, thus consequently allowing the formation of the sequestering stem. Based on this model, it is expected that transcription of ribB nascent transcripts in the absence of FMN leads to the folding of the anti*-P1 stem prior to the anti-P1 stem (Fig. 5A), which should result in the ON structure of the aptamer region having both 5′ and 3′ extremities being involved in helical interactions. Because the anti*-P1 stem is predicted to form before the anti-P1 stem, it is plausible that the folding of the anti*-P1 stem enables a more efficient formation of the anti-P1 stem during transcription elongation. A similar competing structure was recently identified in the E. coli tbpA riboswitch (Berman et al. 2023), suggesting that such structural intermediates may be commonly used in riboswitch sensing to obtain biologically active structures. It is also expected that the additional structural motifs within the expression platform (i.e., region ∼150–210 nt) participate in either the ON or OFF ribB structures, or are involved in recruiting additional regulating factors such as Rho (Hollands et al. 2012; Bastet et al. 2017; Chauvier et al. 2017). For instance, in addition to regions of the aptamer recruiting Rho (Hollands et al. 2012), the FMN-dependent exposition of region 184–193 nt (detected by probe 184) suggests that this could also be involved in recruiting Rho in this region and terminate transcription. Furthermore, regions located further downstream in the coding region could also be involved to modulate the riboswitch structure, such as for the thiM riboswitch where the first eight codons are forming part of the sequestering loop (Winkler et al. 2002a; Bastet et al. 2017). Clearly, additional probing experiments will be required to fully understand how the structure of the expression platform is involved in using FMN sensing to regulate gene expression.
The assessment of ligand-binding activity of nascent transcripts at defined pause sites has revealed large differences both in RNA accessibility and FMN affinity. While the characterization of nascent RNA structure at EC-175 with the probe 137 showed >50% in variation of RNase H cleavage efficiency (Fig. 3A), only an ∼10% change was observed using the same probe in EC-247, clearly suggesting that the accessibility is reduced in the EC-247 transcriptional complex. A similarly low accessibility was monitored using the probe 184 (Fig. 3C), indicating that this region may be extensively structured in EC-247. Furthermore, the use of probe 137 allowed us to calculate Kswitch values of ∼23 nM and ∼7.6 µM for EC-175 and EC-247 complexes, respectively, suggesting that FMN sensing is decreased by a factor of ∼330-fold when EC reach the second pause region. This reduction in ligand affinity effectively restricts FMN sensing to be sensed cotranscriptionally before elongating the RNAP reach position ∼175. Such transcriptional windows for ligand binding have previously been observed for other riboswitches (Chauvier et al. 2017, 2021; Landgraf et al. 2022). Because transcription and translation are tightly coupled in E. coli (Proshkin et al. 2010), it suggests that in the event that ligand binding is not achieved when elongating RNAP are within the transcriptional window, it would presumably be difficult for FMN to be sensed posttranscriptionally by ribB nascent transcripts, which would be actively translated. In addition, if such a case was possible, it would suggest that FMN binding would only result in the inhibition of translation initiation, which would most probably lead to a lower level of regulatory activity. However, due to the relatively short half-life of ribB mRNA (∼2.6 min) (Bernstein et al. 2002), it suggests that such a genetic control is not likely the main determinant to regulate ribB expression. Nevertheless, this scenario could still constitute a viable regulatory mechanism for other riboswitches as previously demonstrated for the Vibrio vulnificus pbuE adenine-sensing riboswitch (Lemay et al. 2011).
Our results suggest that the ribB riboswitch ligand sensing is affected by both the positions of elongating complexes at different transcriptional pauses and the formation of the anti*-P1 stem. Our study of ribB riboswitch ligand sensing is mainly performed using RNase H assays, which is indirectly monitoring single-strand RNA regions using DNA oligonucleotides (Wong and Pan 2009; Chauvier and Lafontaine 2015). Because RNase H relies on the use of a DNA probe hybridizing to a specific region, the obtained results may potentially be biased due to the binding of the oligonucleotide competing with an RNA structure. Although such a case is more apparent when using long DNA probes (e.g., ∼20 nt), the use of 10-mer DNA molecules allows us to detect ligand-binding structural changes (Perdrizet et al. 2012; Lussier et al. 2015; Chauvier et al. 2017, 2021). Since the obtained Kswitch values are mostly in the nM and µM ranges, and are affected by the transcriptional conditions (Lussier et al. 2015; Chauvier et al. 2017, 2021), it suggests that RNase H assays are valuable to monitor cotranscriptional riboswitch folding. Furthermore, because E. coli riboswitches do not regulate intrinsic transcription terminators as B. subtilis riboswitches do, the ligand-binding activity of nascent E. coli riboswitches is not readily detectable using denaturing polyacrylamide gel electrophoresis. However, the use of a DNA oligonucleotide targeting a region of the RNA exhibiting structural changes upon ligand binding allows us to rely on RNase H cleavage activity to monitor riboswitch ligand sensing. Interestingly, Kswitch values were found to vary by approximately fourfold when using DNA probes targeting different regions of nascent btuB riboswitches (Lussier et al. 2015), which is similar to what we obtained here for the ribB riboswitch (Fig. 2). Such differences in Kswitch values could arise from multiple factors, such as the sequence of the DNA oligonucleotide and the RNA structural changes involved. Therefore, although it is not straightforward to compare Kswitch values obtained from different probes, the comparison of results obtained from a single probe is highly informative when employed to monitor ligand-binding affinity across different riboswitch mutants or transcription conditions affecting RNA folding.
The binding of metabolites to riboswitches was first studied, and is now widely employed, using the in-line probing technique that relies on the intrinsic stability of RNA molecules (Nahvi et al. 2002; Winkler et al. 2002a,b; Mandal et al. 2003). Compared to RNase H assays, in-line probing is a less intrusive technique that does not depend on the extrinsic use of additional reagents such as DNA probes. However, in-line probing data are mostly analyzed based on the ligand-binding riboswitch population and not on the total amount of transcripts, such as done using RNase H assays. This crucial difference in analyzing in-line probing may lead to overlooking the presence of multiple RNA populations exhibiting different metabolite-binding properties (Chauvier et al. 2021). Therefore, RNase H assays can constitute an important tool to analyze nascent RNA structures in specific conditions where it may complement in-line probing experiments.
Our study of the ribB riboswitch revealed that FMN sensing is performed at low nM concentrations (Fig. 2). It is informative to compare the Kswitch values with the intracellular FMN concentrations previously reported, which is ∼50 µM in exponentially growing cells (Bennett et al. 2009). Although this concentration is much higher than the Kswitch values (Fig. 2), it represents the total FMN concentration that could markedly differ from the free metabolite concentration. Furthermore, as previously observed for different riboswitches (Wickiser et al. 2005b; Lemay et al. 2011; Chauvier et al. 2017, 2021), the rate of transcription elongation is inversely proportional to ligand sensing when occurring within a transcriptional window. Since the NTP concentration in bacteria is higher than what we have used in our RNase H assays (Buckstein et al. 2008), it suggests that higher FMN concentrations would be required to bind ribB nascent transcript within the transcriptional window, thus allowing the riboswitch to efficiently sense FMN in E. coli.
RNase H data obtained with DNA probes 137 and 218, which respectively target the P1 and sequestering stems, are consistent with both helices being stabilized upon FMN binding by nascent ribB riboswitches (Fig. 2B,C). However, we obtained a different profile with the probe 184, where ligand binding is associated with an increase in RNase H cleavage (Fig. 2D). Based on our study, we postulated that the presence of the anti*-P1 stem, which involves residues 141–149 nt and 175–183 nt, is formed in the absence of FMN and interfere with the binding of probes 137 and 184 (Fig. 5). We measured RNase H activity in the context of FL transcripts for mutants of the first pause site (positions C175 and U181). Our results showed a protection of region 184 in the presence of FMN for all riboswitch mutants, except for the C175G-G176C construct (Fig. 5B,C; Supplemental Fig. S4). The increased effect of the C175G-G176C mutations could be due to the disruption of the pause site. Indeed, we noticed that the incoming nucleotide (+1) at the pause C175 is a guanine (G176) and the nucleotides at −11 and −10 positions are both guanines, consistent with the pausing consensus signal (Larson et al. 2014). Based on that, we hypothesized that the mutation effect on the FMN binding could also be due to the pausing consensus signal alteration that could impair RNA cotranscriptional folding and riboswitch sensing (Perdrizet et al. 2012; Saldi et al. 2021). Furthermore, since the RNAP pause (position −1) can occur on a uridine nucleotide in the context of a pausing consensus signal, we expected to observe a mild disruptive effect on ligand binding for C175U-G176C (Kswitch value is increased by ∼2.5-fold compared to the WT) (Fig. 5B). However, in the context of the C175G-G176C mutant, the pause site is predicted to be strongly disrupted, which is consistent with the much higher Kswitch obtained (∼16-fold increase) (Fig. 5B). Together, our results suggest that the disruptive effects observed for both double mutants could be due to either a reduced efficiency in transcriptional pausing or to the destabilization of the anti*-P1 stem. In our study, we did not seek to confirm the existence of the anti*-P1 stem using a mutagenesis approach in which the helical domain is obtained through “rescue experiments,” i.e., by using other compatible Watson–Crick base-pairing interactions. Indeed, since one of the strands participating in the anti*-P1 stem is also involved in the P1 stem, it makes it difficult to obtain a mutated riboswitch that contains an artificial anti*-P1 stem while still retaining FMN-sensing activity through P1 stem formation. Nevertheless, since the anti*-P1 stem is conserved across bacterial species, it supports its existence and its role in the riboswitch regulation mechanism.
In our study, we did not seek to confirm the existence of the anti*-P1 stem using a mutagenesis approach in which the helical domain is obtained through “rescue experiments,” i.e., by using other compatible Watson–Crick base-pairing interactions. Indeed, since one of the strands participating in the anti*-P1 stem is also involved in the P1 stem, it makes it difficult to obtain a mutated riboswitch that contains an artificial anti*-P1 stem while still retaining FMN-sensing activity through P1 stem formation. For the same reason, it is expected that the use of reporter gene assays would not be straightforward to obtain in vivo evidence for the formation of the anti*-P1 stem as gene expression of engineered mutants would not directly reflect their propensity to adopt the anti*-P1 stem. Nevertheless, since the anti*-P1 stem is conserved across bacterial species, it supports its existence and its role in the riboswitch regulation mechanism.
By performing extensive probing within the region 180–227 nt, RNase H assays revealed an accessibility profile displaying a certain level of structural complexity both in the absence and presence of FMN (Fig. 4C). While low RNA accessibilities were detected at the 5′ and 3′ of the regions probed, a high accessibility was detected at position ∼195 nt. The fact that a similar profile is obtained with and without FMN suggests that the accessibility of the RNA does not significantly change upon FMN binding. Importantly, a significant difference in accessibility is observed for positions 215–227 nt where low RNase H cleavage is detected with FMN, which is consistent with the formation of the sequestering stem upon FMN binding (Fig. 4C). The analysis of the 180–227 nt region when RNAP is stalled at the pause site 247 (EC-247) produced reactivity profiles that markedly differ from FL transcripts (Fig. 4E). The main difference lies in the lower efficiency of RNase H cleavage obtained without FMN (Fig. 4D). Interestingly, the efficiency was increased by ∼20% with FMN for the region 180–215 nt (Fig. 4D), which is an effect that is contrary to what was observed with the FL species (Fig. 4C). While it is difficult to speculate on the nature of the structural changes experienced by nascent transcripts in EC-247, previous studies have shown that ON state structures are favored at pause sites located 3′ of transcriptional windows (Chauvier et al. 2017, 2021). It is thus plausible that the ON state structure, which contains the anti*-P1 and anti-P1 stems, is adopted by most nascent transcripts in the context of EC-247. In the case of the FL transcripts (FL-282), the different RNase H profile is most probably a combination of several structures co-existing in the absence of FMN (ON and “OFF-like” states) (Fig. 4C). Clearly, together with our results, previous studies do not point to a clear picture concerning the structures involved in ribB riboswitch regulation (Hollands et al. 2012; Pedrolli et al. 2015), suggesting that more work is required to decipher the structures of nascent ribB riboswitches both in the absence and presence of FMN. We expect that such efforts will be crucial to understand the ribB riboswitch cotranscriptional sensing mechanism and how metabolite binding is converted into a regulatory control event.
In conclusion, our study has revealed key aspects of the sensing mechanism used by nascent ribB transcripts. While past studies have done extremely well to understand how the metabolite-riboswitch complex is achieved with such high degrees of specificity and affinity, it will be important to understand how the recognition event is performed within a transcriptional context and how the transcription machinery influences the riboswitch regulatory mechanism.
MATERIALS AND METHODS
DNA oligonucleotides and bacterial strains
DNA oligonucleotides were purchased from Integrated DNA Technologies. Transcription constructs were obtained by PCR reactions using E. coli genomic DNA as template. The strains used to construct the templates used for fluorescent transcription reactions are indicated in Supplemental Table S1. The PCR strategy and DNA oligonucleotides are listed in Supplemental Tables S2 and S3, respectively. Mutations performed in the ribB riboswitch were made using three PCR steps. PCR1 and PCR2 were performed with the genomic DNA, and PCR3 was done using products of the PCR1 and PCR2 reactions.
To prepare templates used for fluorescent transcription reactions, plasmids containing the WT and mutants were made. The ribB WT and mutant variants were produced using three fragments Gibson Assembly kits (pEASY-Uni Seamless Cloning and Assembly Kit from TransGen Biotech Co.). One fragment corresponding to the linear vector backbone (pUA66/pbpG-GFP) (Zaslaver et al. 2006) was amplified with primers Fwd RBS-GFP and Rev backbone-pUA66-lacUV5 overhang (see PCR strategy in Supplemental Table S2). The other fragments were used to make the riboswitch WT and mutant region (see PCR strategy in Supplemental Table S2). Templates used for transcription reactions were obtained by PCR amplification of the corresponding plasmids by targeting the region of the riboswitch WT or mutant sequence.
In vitro transcription assays
In vitro transcription reactions were performed essentially as previously described (Chauvier et al. 2017). The RNAP and sigma70 factor from E. coli were obtained from the Plateforme de purification des protéines (University of Sherbrooke). The purification procedures were done according to previous studies (Das et al. 1996; Svetlov and Artsimovitch 2015). Briefly, reactions were performed in 20 mM Tris-HCl pH 8.0, 20 MgCl2, 20 mM NaCl, 14 mM 2-mercapthoethanol, and 0.1 mM EDTA. The DNA template, sigma70 factor, and RNAP were incubated at 37°C for 5 min. The GCU initiator trinucleotide, ATP, UTP, and [α-32P] CTP were added and the reaction incubated at 37°C for 10 min, which allowed us to obtain EC-11 complexes. Samples were washed using G50 columns to remove unincorporated nucleotides. Reactions were allowed to resume by adding 100 µM NTP and 0.35 mg/mL heparin to ensure single-round conditions when using the FL template. A biotin-streptavidin roadblock was used to monitor FMN binding on EC (Chauvier et al. 2017, 2021). When analyzing the reaction in the absence and presence of FMN, a concentration of 100 µM FMN was used. Fluorescent in vitro transcription reactions were performed as described above but were initiated using Cy3-GUU, ATP, and UTP.
For Kswitch experiments, FMN was used at concentrations of 100, 1, 5, 10, 25, 50, 100, 250, 500 pM, 1, 2.5, 5, 10, 50, 100, and 500 µM. For Kswitch experiments performed on fluorescent transcription reactions, FMN was used at concentrations 2, 5, 10, 25, 50, 75, 100, 250, 500, 750 nM, 1, 2, 5, and 10 µM. Transcription reactions were incubated for 10 min at 37°C, which were then followed by RNase H probing assays.
RNase H probing assays
RNase H probing assays were performed by using an aliquot of the transcription reactions and by adding 20 µM of a DNA oligonucleotide for 5 min at 37°C (Chauvier et al. 2017). The RNase H cleavage reaction was initiated by adding a solution containing the transcription buffer (see above) with 0.12 U/µL of RNase H and was incubated for 5 min at 37°C. The reactions were stopped by adding at a 1:1 ratio a solution containing 95% formamide, 20 mM EDTA, and 0.4% SDS. The experiments were performed in triplicate, and the most representative data are shown. The reported standard deviations (SD) were obtained through fitting analysis, which was assumed to be approximated by the standard deviation of the points from the fitted curve (Flannery et al. 1992; Rist and Marino 2001). For the high-density probing (Fig. 4C,D,E), RNase H assays were performed in the presence and absence of FMN using each probe as a one replicate experiment, and the averages of overlapping probes were used to report the accessibility of the corresponding nucleotide.
For RNase H assays monitoring fluorescent transcription reactions, the transcription reactions were aliquoted with 2.5 µM of the DNA probe for 5 min at 37°C. A 10 µL aliquot of RNase H (0.03 U/µL) was added to the sample for 2 min at 37°C. Then, the reaction was stopped by adding a 1:1 ratio of a solution containing 95% formamide, 20 mM EDTA, and 0.4% SDS. The experiments were performed in duplicate, and the most representative data are shown.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Dr. Alain Lavigueur for discussion and critical reading of the manuscript. This work was supported by the Canadian Institutes of Health Research grant (CIHR) and Natural Sciences and Engineering Council of Canada (NSERC).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080074.124.
- Received April 23, 2024.
- Accepted September 24, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








