
A circular split nanoluciferase reporter for validating and screening putative internal ribosomal entry site elements
- 1Department of Pharmacology, Weill Cornell Medicine, Cornell University, New York, New York 10065, USA
- 2Imperial College Centre for Synthetic Biology, Imperial College London, London SW7 2AZ, United Kingdom
- 3Department of Bioengineering, Imperial College London, London SW7 2AZ, United Kingdom
- Corresponding author: srj2003{at}med.cornell.edu
-
Handling editor: Fatima Gebauer
Abstract
Internal ribosomal entry sites (IRESs) recruit the ribosome to promote translation, typically in an m7G cap-independent manner. Although IRESs are well-documented in viral genomes, they have also been reported in mammalian transcriptomes, where they have been proposed to mediate cap-independent translation of mRNAs. However, subsequent studies have challenged the idea of these “cellular” IRESs. Current methods for screening and discovering IRES activity rely on a bicistronic reporter assay, which is prone to producing false positive signals if the putative IRES sequence has a cryptic promoter or cryptic splicing sites. Here, we report an assay for screening IRES activity using a genetically encoded circular RNA comprising a split nanoluciferase (nLuc) reporter. The circular split nLuc reporter is less susceptible to the various sources of false positives that adversely affect the bicistronic IRES reporter assay and provides a streamlined method for screening IRES activity. Using the circular split nLuc reporter, we find that nine reported cellular IRESs have minimal IRES activity. Overall, the circular split nLuc reporter offers a simplified approach for identifying and validating IRESs and exhibits reduced propensity for producing the types of false positives that can occur with the bicistronic reporter assay.
Keywords
INTRODUCTION
Cap-independent translation is an alternative mechanism of protein synthesis initiation that occurs independent of a 5′ cap structure on the messenger RNA (mRNA) (Jang et al. 1988; Pelletier and Sonenberg 1988). In cap-independent translation, the ribosome can be recruited to mRNA via an internal ribosomal entry site (IRES) to initiate protein synthesis. This mechanism allows for proteins to be synthesized (i) in circular mRNAs, which do not have an m7G cap; (ii) when multiple open reading frames (ORFs) are present in a long linear RNA; or (iii) when the cap-dependent translation machinery is compromised.
Several positive-stranded RNA viruses use IRESs to drive translation of viral proteins since their genomes lack a 5′ m7G cap (Jang et al. 1988; Pelletier and Sonenberg 1988; Belsham and Brangwyn 1990; Tsukiyama-Kohara et al. 1992). These viral IRESs tend to be large (400–800 nt) and have highly structured RNA sequences that can bind initiation factors to recruit the ribosome, or in some cases, directly recruit the ribosome (Kieft 2008).
Cap-independent translation is also thought to play a role in mRNA translation under stress conditions when global cap-dependent translation is suppressed (Spriggs et al. 2010). In these cases, translation is proposed to be mediated by cellular IRESs, which tend to be smaller and less structured than viral IRESs (Weingarten-Gabbay et al. 2016; Chen et al. 2021). Their mechanism of action is often difficult to determine, in part because it is difficult to distinguish the amount of translation that is cap-dependent and cap-independent since most cellular IRESs are in mRNAs that already contain a cap (Kozak 2003). Understanding the process of cap-independent translation is crucial for understanding viral translation and translation of cellular RNAs under cell stress conditions.
Cap-independent translation also serves as a valuable tool for therapeutics. IRESs are used in gene therapy vectors to achieve translation of two or more proteins from the same mRNA (Morgan et al. 1992; Okada et al. 1996, 1999; Urabe et al. 1997; Fan et al. 1998; Pizzato et al. 1998; Gardner et al. 2020). Furthermore, circular mRNA therapeutics rely on viral IRESs to achieve robust translation since circular RNA lacks a 5′ cap (Wesselhoeft et al. 2018; Chen et al. 2022; Qu et al. 2022). Screening new IRES sequences could therefore be useful for gene therapy and circular mRNA therapeutics.
The predominant method for screening IRES sequences is the bicistronic IRES reporter (Fig. 1; Jang et al. 1988; Jackson et al. 1990). The bicistronic reporter is an mRNA that contains two ORFs. The first ORF is often firefly luciferase, downstream from the 5′ m7G cap. The second ORF is often renilla luciferase located downstream from the first ORF. In this case, the renilla luciferase is preceded by a putative IRES sequence. Any translation of the renilla luciferase is thus inferred to be due to IRES activity. IRES activity can be measured by normalizing the renilla luciferase activity to the firefly luciferase activity using a standard dual-luciferase assay.
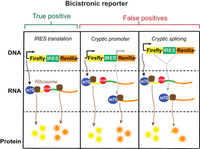
Schematic of potential outputs from bicistronic IRES reporter. The bicistronic reporter comprises a vector that makes an mRNA with a firefly luciferase (yellow) followed by a stop codon, followed by an IRES (green), followed by a renilla luciferase (orange). In the true positive scenario, cap-dependent translation drives translation of the firefly luciferase and the IRES drives translation of the renilla luciferase. A false positive result can occur when a cryptic promoter (gray) can create a new mRNA where the cap can drive translation of the renilla luciferase. A false positive result can also occur when cryptic splicing (gray) can splice out the IRES and stop codon and cause the renilla to be translated through a cap-dependent mechanism.
The bicistronic reporter, however, is known to be capable of producing false positive results. The two major causes of false positives are thought to be cryptic promoters within putative IRES sequences and cryptic splicing elements (Kozak 2003; Jackson 2013). Cryptic promoter activity can occur when a putative IRES sequence has a previously unrecognized promoter sequence. The cryptic promoter can induce the synthesis of new capped transcripts that comprise a portion of the IRES followed by the renilla luciferase ORF. Even small amounts of cryptic promoter activity can lead to synthesis of capped mRNA that is efficiently translated by conventional cap-dependent translation (Fig. 1).
Another source of false positives is cryptic splicing. This occurs when the putative IRES contains a cryptic splice site (Baranick et al. 2008). In this case, splicing skips over the stop codon of the first ORF and connects a part of the first ORF to the second ORF. Thus, cryptic splicing can lead to low levels of mRNA that encode some or all of the first ORF and all of the second ORF in a single continuous ORF (Fig. 1). In this case, both the firefly and the renilla would be translated in a cap-dependent manner.
Taken together, cryptic promoter activity and cryptic splicing can lead to false positive results from bicistronic reporters. Including additional controls that focus on detecting cryptic promoters and cryptic splicing by methods such as northern blotting can mitigate each of these false positives (Terenin et al. 2017). However, these alternative transcripts that produce false positives can be lowly abundant, which makes them difficult to detect. Therefore, a reporter assay that is not susceptible to false positives from cryptic splicing or cryptic promoters would streamline the process of screening for IRES activity.
Circular RNA reporters offer a promising alternative to bicistronic reporters for screening IRES activity. Since circular RNAs lack a 5′ m7G cap, these RNAs can be used to assess canonical IRESs that do not require 5′ ends or m7G caps. Recently, researchers performed a high-throughput screen to identify IRES sequences that function in the context of a circular RNA (Chen et al. 2021). The reporter comprised a split green fluorescent protein (GFP) that should produce fluorescence only when the RNA was circularized. However, the circular RNAs were produced using the back-splicing method, which has been shown to produce predominantly linear RNAs in some cases (Ho-Xuan et al. 2020; Jiang et al. 2021; Unti and Jaffrey 2024). Moreover, the intron homology that drives the back-splicing reaction can also cause a forward trans-splicing reaction to occur, which could result in false positives even when using a split GFP (Ho-Xuan et al. 2020; Jiang et al. 2021). We therefore wanted to create an IRES reporter using a high-efficiency, spliceosome-independent circularization method.
Here, we present a simple and accurate assay for screening IRES activity. This assay involves expressing a genetically encoded circular RNA using the Tornado expression system (Litke and Jaffrey 2019). The reporter is only produced if the RNA has circularized. The circular RNA lacks an m7G cap and is therefore only translated if the tested sequence has IRES function. Because of this, the circular split nLuc reporter does not produce false positives from cryptic promoters or cryptic splicing.
We show that the circular split nLuc reporter can accurately reproduce previously validated IRES activity. Furthermore, we use the circular split nLuc reporter to determine the activity of nine putative cellular IRESs. We show that the nine cellular IRESs we tested have negligible IRES activity compared to viral IRESs. Overall, the circular split nLuc reporter provides a more accurate method for screening IRES activity using a simple plasmid-based reporter system.
RESULTS
Circular split nLuc reporter does not produce the false positive results seen with the bicistronic reporter assay
Recently, we described a method to efficiently express circular mRNA in a mammalian cell (Unti and Jaffrey 2024) using the Tornado (Twister-optimized RNA for durable overexpression) system. The Tornado system involves expressing a linear RNA containing ribozymes at the 5′ and 3′ ends of the transcript. After the ribozymes undergo autocatalytic cleavage, the 5′ and 3′ ends are ligated by RtcB, an endogenous RNA ligase (Litke and Jaffrey 2019). This ligation produces the circular RNA.
To design our IRES reporter, we created a sequence that can only be translated into the nanoluciferase (nLuc) enzyme once the RNA is circularized (Fig. 2A). Thus, any linear precursor cannot produce signal.
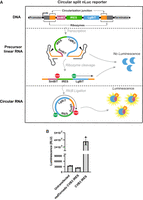
Schematic of the split nLuc Tornado translation system and the circular split nLuc reporter assay. (A) Construct design of the Tornado translation system using a split nLuc ORF. The circular split nLuc reporter comprises an SmBiT (pink), followed by an IRES (green), followed by an LgBiT (blue). When the RNA is transcribed, ribozyme (orange) cleavage and ligation will occur. The split nLuc protein will be translated by IRES translation. (B) Luminescence from circular split nLuc system is dependent on circularization. Luminescence from HEK293T cells transfected with plasmids encoding the circular split nLuc reporter with either a mutant ribozyme (mutTornado CVB3 IRES) or a functional ribozyme (CVB3 IRES). (CJ) Circularization junction.
The split nLuc reporter takes advantage of the NanoLuc Binary Technology (NanoBiT), comprising the Large BiT (LgBiT), and Small BiT (SmBiT) components (Dixon et al. 2016). These components have low affinity for each other and only produce luminescence when they are artificially brought together, for example, by two proteins or by a protein linker. The linear mRNA precursor was designed to contain three sequential components: (i) SmBiT followed by a stop codon; (ii) an IRES; and (iii) a start codon followed by the LgBiT sequence (Fig. 2A). The linker between the LgBiT and the SmBiT is the circularization junction and is designed to keep LgBiT and SmBiT in frame and lacks stop codons. Luminescence, therefore, only occurs when the mRNA is circularized and can be translated into a protein where the circularization junction tethers the SmBiT to the LgBiT. Our circular split nLuc reporter RNA is generated using a cytomegalovirus (CMV) RNA polymerase II promoter, which is similar to the promoter of many cellular mRNAs. Because the circular mRNA lacks a 5′m7G cap, the split nLuc system can therefore be used to screen putative cap-independent IRES elements.
We first wanted to determine whether the circular split nLuc reporter is dependent on circularization. To do this, we deleted the 3′ ribozyme sequence. This prevents formation of the 2′,3′-cyclic phosphate at the 3′ end of the RNA, which is needed for RtcB-mediated circularization (Tanaka et al. 2011). This construct is referred to as “mutTornado” (Fig. 2B). Cells transfected with the mutTornado plasmid produced only baseline levels of luminescence, similar to untransfected cells (Fig. 2B). However, cells transfected with the CVB3-translated circular split nLuc reporter with intact ribozymes produced substantial luminescence (Fig. 2B). These data show that the luminescence signal is dependent on circularization.
Unlike the bicistronic reporter assay, the split nLuc reporter assay theoretically would not produce false positive results from a cryptic promoter. If an IRES contains a cryptic promoter, the reporter plasmid would generate a linear mRNA that only contains the LgBiT (Fig. 3A). Thus, any transcript derived from a cryptic promoter should not affect the overall luminescence signal. To test this, we cloned a human PGK (hPGK) promoter into the IRES position of both the circular split nLuc reporter and the bicistronic reporter. As a negative control, we used a 42 nt random sequence with no known IRES activity (Fig. 3B). Notably, untransfected cells cannot be used as a negative control for the bicistronic reporter because the relative luminescence is calculated by dividing the level of renilla luminescence by the level of firefly luminescence.
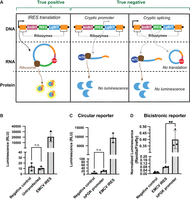
Circular split nLuc reporter does not produce false positive results. (A) Schematic of the circular split nLuc reporter for screening IRES activity. False positives from a cryptic promoter or cryptic splicing cannot occur since the mRNA will not be translated into the LgBiT fused to the SmBiT. (B) Negative control IRES does not produce background signal. Luminescence from HEK293T cells transfected with plasmids encoding the circular split nLuc reporter with either the EMCV IRES, negative control IRES, or no plasmid (untransfected). (C) hPGK promoter does not produce a false positive result. Luminescence from HEK293T cells transfected with plasmids encoding the circular split nLuc reporter with either the CVB3 IRES, negative control IRES, or hPGK promoter sequence in the IRES position. (D) hPGK promoter produces a false positive signal in the bicistronic reporter. HEK293T cells transfected with plasmids encoding the bicistronic reporter with either the 42 nt random sequence negative control, an hPGK promoter, or EMCV in the IRES position. (RLU) Relative luminescence units. Data are presented as mean values +/− one SD (n = 3 biological replicates). Significance was calculated using unpaired two-tailed Student's t-test. (****) P < 0.0001, (***) P < 0.001, (**) P < 0.01, (*) P < 0.05, (n.s.) P > 0.05.
We then compared the IRES activity of the hPGK to the negative control IRES and to a previously validated EMCV IRES (Wesselhoeft et al. 2018). We found that the hPGK promoter did not provide a statistically significant increase in luminescence compared to the negative control in the circular split nLuc reporter (Fig. 3C). However, the hPGK produced fivefold more signal than the EMCV IRES in the bicistronic reporter (Fig. 3D). This shows that the circular split nLuc reporter is not prone to the same false positive signals as the bicistronic reporter.
Similarly, the circular split nLuc reporter would theoretically not produce false positives from cryptic splicing. For example, if cryptic splicing removed the 5′ ribozyme, SmBiT, and IRES, no luminescence would occur since the RNA would not circularize or contain the SmBiT (Figs. 2A and 3A). Alternatively, if cryptic splicing removed just the IRES, the 5′ and 3′ ribozymes would still cleave, remove the m7G cap, and circularize. In this scenario, cap-dependent translation would not occur (Fig. 3A). Thus, the circular split nLuc reporter should not produce false positive results due to cryptic splicing.
To determine whether cryptic splicing would produce false positive signals, we tested the luminescence of the two theoretical products of cryptic splicing. The hPGK promoter cloned into the IRES position would create the end product of a cryptically spliced circular split nLuc reporter where the 5′ ribozyme, SmBiT, and IRES is removed (Fig. 3A). The hPGK promoter cloned into the IRES position did not produce luminescence (Fig. 3C). Furthermore, the 42 nt negative control cloned into the IRES position would create the end product of a cryptically spliced circular split nLuc reporter where the IRES is removed (Fig. 3A). The 42 nt negative control cloned into the IRES position also did not produce luminescence (Fig. 3B). Therefore, cryptic splicing should not lead to false positive results in the circular split nLuc reporter.
Circular split nLuc reporter can be used to determine IRES activity
To show that the circular split nLuc system can be used to screen IRES elements, we wanted to compare the IRES activity of known viral IRES in a bicistronic reporter and the circular split nLuc reporter. To do this, we cloned the hepatitis C virus (HCV) IRES and the encephalomyocarditis virus (EMCV) IRES into a bicistronic reporter and the circular split nLuc reporter. We used a 42 nt random sequence as a negative control. For the bicistronic reporter, we included an additional negative control comprised of a scrambled EMCV sequence after realizing the 42 nt random sequence had higher activity than several putative IRES sequences (Fig. 4A). We transfected cells and performed a luminescence assay 3 days later.
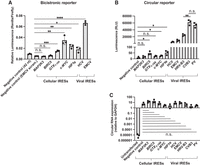
Circular split nLuc reporter accurately measures IRES activity. (A) Renilla luciferase normalized to firefly in HEK293T cells transfected with bicistronic reporters containing a putative IRES. Negative control is a scrambled EMCV sequence. (B) Luminescence from HEK293T cells expressing the circular split nLuc reporter with a putative IRES. (C) Circular RNA expression of the circular split nLuc reporter in HEK293T cells transfected with a putative IRES. Divergent primers that spanned the circularization junction were used for qPCR. RNA expression was normalized to GAPDH. The IRES does not affect circularization efficiency. (RLU) Relative luminescence units. Data are presented as mean values +/− one SD (n = 3 biological replicates). Significance was calculated using unpaired two-tailed Student's t-test. (****) P < 0.0001, (***) P < 0.001, (**) P < 0.01, (*) P < 0.05, (n.s.) P > 0.05.
In the case of the bicistronic reporter, we found that both the EMCV and HCV had higher relative luminescence than the negative control. Similarly, the EMCV and HCV IRES in the circular split nLuc reporter reflected the previously validated reports of IRES activity (Wesselhoeft et al. 2018). These results demonstrate that the circular split nLuc reporter and the bicistronic reporter can accurately detect EMCV and HCV activity.
However, the magnitude of difference between the negative control and the viral IRESs was significantly lower in the bicistronic reporter. In the circular split nLuc reporter, the HCV and EMCV IRES exhibited more than 250-fold and 650-fold higher IRES activity than the negative control, respectively (Fig. 4B). On the other hand, the HCV and EMCV IRES exhibited only threefold and 12-fold higher relative luminescence in the bicistronic reporter, respectively. This shows that the split nLuc system has much larger dynamic range for detecting viral IRES activity.
Previously described IRESs discovered through bicistronic reporters have low IRES activity in the circular split nLuc reporter
Several previous studies showed that cellular RNA sequences have IRES activity (Chappell et al. 2000; Stoneley et al. 2000a; Lang et al. 2002; Weingarten-Gabbay et al. 2016). The MAP2K3 and BIRC5 IRESs were discovered in a high-throughput screen of IRES activity in human and viral sequences (Weingarten-Gabbay et al. 2016). Both the MAP2K3 and BIRC5 IRESs were derived from the 3′ UTR of their respective mRNAs. The high-throughput screen used a bicistronic reporter and the MAP2K3 and BIRC5 IRESs have not been independently validated. The c-MYC IRES and the HIF1α IRES were initially discovered using a bicistronic reporter that showed they had increased IRES activity compared to the EMCV IRES (Stoneley et al. 2000a,b; Lang et al. 2002). Subsequent studies reported that the increased activity compared to the EMCV IRES was due to cryptic promoter activity and that both the c-MYC IRES and the HIF1α IRES had low activity compared to the EMCV IRES (Bert et al. 2006). The GTX1-166 IRES was discovered through a bicistronic reporter that showed it had increased IRES activity compared to the EMCV IRES in rodent cell lines (Chappell et al. 2000).
We therefore wanted to determine the IRES activity of these cellular IRESs in the circular reporter. We first inserted the cellular IRESs in a bicistronic reporter to recapitulate the previously reporter IRES activity in this assay system. We cloned the c-MYC IRES, MAP2K3 IRES, BIRC5 IRES, GTX1-166 IRES, and HIF1α IRES into the bicistronic reporter and measured the relative luminescence. We found that the MAP2K3 and BIRC5 IRESs exhibited no IRES activity. The c-MYC, GTX1-166, and HIF1α IRES, however, exhibited IRES activity comparable to the HCV IRES (Fig. 4A). These data confirm that the c-MYC, GTX1-166, and HIF1α IRES sequences exhibit IRES activity in the bicistronic reporter, comparable to the extensively validated HCV IRES.
Next, we tested the activity of the IRES using the circular split nLuc reporter. To do this, we cloned the c-MYC IRES, MAP2K3 IRES, BIRC5 IRES, GTX1-166 IRES, and HIF1α IRES into the circular split nLuc reporter and measured the luminescence. We found that the MAP2K3 IRES and the GTX1-166 IRES did not show any statistically significant increase in translational activity compared to the negative control. The c-MYC IRES, BIRC5 IRES, and HIF1α IRES showed higher activity than the negative control but substantially lower IRES activity than the EMCV and HCV IRESs in the circular split nLuc reporter (Fig. 4B).
Notably, the activity of these IRESs was <2% of the activity of the viral IRESs in the circular split nLuc reporter. These results validate the previous findings that show that the c-MYC IRES and the HIF1α IRES have low activity compared to viral IRESs (Bert et al. 2006). Furthermore, these results show that the GTX1-166 IRES has no IRES activity. It is possible that the GTX1-166 IRES (Chappell et al. 2000) was a false positive in the original study, but it should be noted that it may be a rodent-specific cellular IRES that does not produce signal in the human cell line we used. Interestingly, the BIRC5 IRES showed low but statistically significant IRES activity in the circular split nLuc reporter but not in the bicistronic reporter (Fig. 4A,B). This shows that the circular split nLuc reporter may have a greater sensitivity than the bicistronic reporter. Importantly, as a control, we tested the expression level of each construct and found that none of the putative cellular IRES sequences we tested changed the level of circular RNA expression (Fig. 4C).
It should be noted that the relatively low activity of the cellular IRESs that we found are unlikely to reflect differences in the promoters since the circular split nLuc reporter is generated from a Pol II reporter, like cellular mRNAs. However, cellular IRESs may require a 5′ end or poly(A) tail, which would not be present in the circular split nLuc reporter.
Circular split nLuc reporter can be used to screen viral IRESs
We next asked whether the circular split nLuc reporter can be used to screen viral IRESs for translational activity. To do this, we tested the translational activity of the coxsackievirus B3 (CVB3), poliovirus (PV), human rhinovirus B3 (HRV-B3), EMCV, and HCV IRESs, in the circular split nLuc reporter. We found that the PV and CVB3 IRES had the highest activity (Fig. 4B), which is similar to the findings of a previous report (Wesselhoeft et al. 2018). However, another previous study found that the HRV-B3 IRES had higher translational activity than the CVB3 IRES (Chen et al. 2022). Our results found that both the CVB3 and HRV-B3 have high translational activity, but the CVB3 showed approximately threefold higher translational activity than HRV-B3. Importantly, we showed decreased circular RNA expression was not the reason for HRV-B3 having a lower luminescence signal (Fig. 4C). Our study therefore provides further evidence that the CVB3 or PV IRES are thus far the viral IRESs with the highest translational activity in HEK293T cells.
Circular split nLuc reporter can be used in multiple cell types
Given that IRES activity can vary based on cell type and cell state, we wanted to determine whether the circular split nLuc reporter could be used in multiple cell types. We therefore tested several cellular and viral IRESs in HeLa and HepG2 cells. In HeLa cells, we found that the HIF1α and the BIRC5 IRESs showed low but statistically significant IRES activity. In HepG2 cells, we found that the BIRC5 IRES showed low but statistically significant IRES activity. Overall, we found that in both cell lines, the EMCV and CVB3 IRESs exhibited high translational activity, and the cellular IRESs exhibited low translational activity (Fig. 5A). These results resemble the results from HEK293T cells (Fig. 4B) and support the idea that the circular split nLuc reporter can be used to determine whether IRESs are cell-type specific.
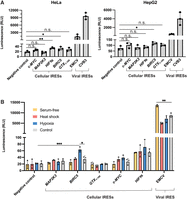
Circular split nLuc reporter can be used to test IRES activity in alternative cell types and states. (A) Luminescence from HeLa and HepG2 cells expressing the circular split nLuc reporter with a putative IRES. The overall trend in IRES activity of viral and cellular IRESs is similar in HepG2, HeLa, and HEK293T cells. (B) Luminescence from HEK293T cells transfected with the circular split nLuc reporter with a putative IRES. Cells were subjected to serum-free media (yellow), heat shock (salmon), hypoxia (blue), and no treatment control (gray). (RLU) Relative luminescence units. Data are presented as mean values +/− one SD (n = 3 biological replicates). Significance was calculated using unpaired two-tailed Student's t-test. (****) P < 0.0001, (***) P < 0.001, (**) P < 0.01, (*) P < 0.05, (n.s.) P > 0.05.
Screening IRES activity in cell stress conditions
We next wanted to use the circular split nLuc reporter to determine whether any of the cellular IRESs we tested are active under stressed conditions. We therefore tested the cellular IRESs under conditions of heat shock, starvation (serum-free media), and hypoxia (1% O2). Most cellular and viral IRESs were not affected by stressed conditions. The EMCV IRES showed an increase in activity under starvation conditions (Fig. 5B). This may reflect the phenomena where most cellular mRNAs are translationally repressed, thus enabling more ribosomes to bind to IRESs (Welnowska et al. 2011). Additionally, the BIRC5 IRES showed increased IRES activity under hypoxic conditions, reaching ∼1% the activity of viral IRESs (Fig. 5B). This highlights the use of the circular split nLuc reporter to determine whether cellular IRESs function in the context of cellular stress.
Circular split nLuc reporter can be used to screen circular RNA cellular IRESs
Recently, researchers performed a genome-wide screen of endogenous RNA sequences that can drive translation of a circular RNA (Chen et al. 2021). However, their screen used the back-splicing method that is known to produce linear RNA byproducts that can contribute to a false positive signal (Ho-Xuan et al. 2020; Jiang et al. 2021). We therefore wanted to test IRESs from this screen in an orthogonal circular RNA reporter. We cloned the IGFR2, TGFBR3, and CHD2 IRESs into the circular split nLuc reporter and compared their IRES activity to the EMCV IRES and the negative control. We found that the IGFR2 and the TGFBR3 IRESs had no activity but the CHD2 IRES had a statistically significantly higher translational output than the negative control. The CHD2 IRES is therefore capable of low-level translation from a circular RNA.
Another study has shown that a sequence from circZNF609 is capable of driving cap-independent translation (Legnini et al. 2017). We therefore wanted to test the circZNF609 IRES in the circular split nLuc reporter. We found that the circZNF609 IRES does not show any IRES activity (Fig. 6B). However, the study that originally described the circZNF609 IRES only tested its function in a bicistronic reporter. Importantly, they only found it had “IRES” activity when the bicistronic reporter was spliced. Furthermore, follow-up analysis has shown that the circZNF609 IRES is not capable of driving translation (Ho-Xuan et al. 2020). Our results therefore validate the previous report that shows that the putative IRES in circZNF609 is not capable of cap-independent translation.
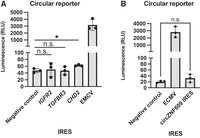
Circular split nLuc reporter can be used to test putative endogenous circular RNA IRESs. (A) Luminescence from HEK293T cells transfected with the circular split nLuc reporter with a putative circular RNA cellular IRES. (B) Luminescence from HEK293T cells transfected with the circular split nLuc reporter with the circZNF609 IRES, the EMCV IRES, or a 42 nt random sequence negative control. (RLU) Relative luminescence units. Data are presented as mean values +/− one SD (n = 3 biological replicates). Significance was calculated using unpaired two-tailed Student's t-test. (****) P < 0.0001, (***) P < 0.001, (**) P < 0.01, (*) P < 0.05, (n.s.) P > 0.05.
DISCUSSION
Here, we describe a simple and accurate method for screening IRES elements. This method is facilitated by the Tornado translation system, an efficient method for expressing circular mRNAs in mammalian cells, and a split nLuc protein encoded by the circular RNA. Because the reporter is a circular RNA, it lacks a 5′ cap, ensuring that any reporter signal derives from cap-independent mechanisms. Additionally, the reporter is a split nLuc that is only reconstituted by RNA circularization of the full-length precursor transcript. This ensures that any spurious transcript derived from a cryptic promoter or from cryptic splicing does not produce a signal because it cannot produce the circular RNA. Thus, the design of the circular split nLuc reporter ensures that it is less susceptible to the common mechanisms that lead to false positive results in the conventionally used bicistronic reporter. The circular split nLuc is therefore a substantially better alternative to the bicistronic reporter, which often produces false positive results due to cryptic promoters and cryptic splicing. While there have been substantial efforts made to test for the false positive results from a bicistronic reporter, these methods either require several rounds of experiments or testing a putative IRES using in vitro transcribed RNA. Researchers would benefit from having a one-step plasmid-based assay to detect IRES activity.
In this study, we first demonstrated the ability of the circular split nLuc reporter to bypass the major problem of the linear bicistronic reporter, which is false positive signals due to a cryptic promoter. We showed that insertion of an active promoter in place of an IRES in the circular split nLuc reporter produces no signal above background. We also tested a total of nine putative cellular IRESs. We found that the HIF1α, the CHD2, c-MYC, and BIRC5 IRES sequences exhibited statistically significant IRES activity compared to background, but far lower than EMCV. Importantly, the circular split nLuc reporter showed that the c-MYC IRES and the HIF1α had substantially lower IRES activity compared to EMCV. This agrees with a previous finding that the reason the c-MYC and HIF1α IRES were originally found to have greater activity than a viral IRES was due to cryptic promoter activity (Bert et al. 2006). Furthermore, this finding underscores the utility of the circular split nLuc reporter in avoiding false positive results.
Overall, we found that no cellular IRES we tested exhibited >2% of the IRES activity of EMCV. It is possible that this level of activity from cellular IRESs is sufficient to confer biologically meaningful levels of translation. However, our data suggest that viral IRESs should be used for therapeutic applications with circular mRNA since the IRES activity from cellular IRESs is low.
Lastly, we showed that the circular split nLuc reporter can be used to screen viral IRESs. The circular split nLuc reporter showed similar results of viral IRES activity to previous studies using in vitro transcribed and circularized mRNA (Wesselhoeft et al. 2018). The circular split nLuc reporter can therefore accurately predict IRES activity.
We found that the CVB3 IRES and the PV IRES are the viral IRESs that drive the highest level of cap-independent translation. A previous study suggested that the HRV-B3 IRES had higher translational activity than the CVB3 and PV IRES in a screen using an in vitro transcribed mRNA (Chen et al. 2022). It is worth noting that this study performed a direct comparison between the CVB3 IRES and HRV-B3 IRES in the same mRNA context, but the CVB3 IRES appeared to have anomalously low activity, as demonstrated by its much lower activity than the HCV IRES. Most studies show that the CVB3 IRES is much more active than the HCV IRES (Wesselhoeft et al. 2018). Thus, the superior performance of the HRV-B3 IRES may reflect a false negative result from the CVB3 IRES. However, the discrepancy between our study and the previous study could also be due to a difference in IRES activity when expressing the reporter from a plasmid-based system as opposed to directly transfecting an mRNA, as used in the above study.
Limitations
While the circular split nLuc reporter does not produce false positive results that we know of, there is a possibility that the circular split nLuc reporter could produce false negative results. False negative results can occur if the IRES somehow prevents ribozyme cleavage or ligation, perhaps by sequence interactions that prevent their folding. Our qRT-PCR data show that the IRESs we tested do not significantly change the level of circular RNA expression (Fig. 4C), so we think this mechanism is unlikely. However, this does not rule out the possibility that an IRES could interfere with the ability for the ribozymes to cleave or for the RNA to be ligated together. Thus, it is important to keep in mind that an IRES may be capable of driving high-level translation but is somehow not compatible with the Tornado translation system. Tests of circular RNA abundance should be used if this is of concern.
The circular split nLuc reporter only detects the activity of canonical IRESs, which are IRESs that function without an mRNA 5′ end or cap. However, cellular IRESs may depend on additional components such as the promoter identity, or the 5′ end of an mRNA. For example, cap-independent translational enhancers (CITEs), exhibit dependence on a 5′ end (Shatsky et al. 2018). Thus, just because a cellular IRES does not have a signal in the circular split nLuc reporter does not mean it lacks an important biological role in promoting translation when canonical cap-dependent translation is repressed.
Additionally, it is possible that some cellular IRESs function in specific cell types or specific cell states in which specific cellular factors are activated. Further analysis of these cellular IRESs can be done using the circular split nLuc reporter to determine whether putative cellular IRES are active in a specific cell state that was not tested in this study. Furthermore, we only tested nine putative cellular IRESs. It is possible that other cellular IRESs may exhibit higher translational activity. A high-throughput screen of endogenous sequences using the circular split nLuc reporter assay could be useful to determine whether any cellular IRESs exist that have activity levels similar to viral IRESs.
MATERIALS AND METHODS
Cloning IRES variants
IRES sequences were ordered as gene blocks (Integrated DNA Technologies or Twist) then cloned into the BsiWI, EcoRI restriction sites of the circular split nLuc plasmid (Addgene 212612).
Cell culture and transfection
HEK293T (ATCC CRL-11268), HepG2, and HeLa cells were cultured with ×1 DMEM (Thermo Fisher #11995-065) with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin under standard tissue culture conditions. Cells were cultured at 37 °C and 5% CO2 and passaged every 2–3 d. TrypLE Express (Thermo Fisher #12604013) was used to lift cells for passaging. Cells were plated at a density of 2 × 104 cells/cm2 20 h before transfection. Cells were transfected using a 1:1:1 ratio of RNAi plasmid to target plasmid to renilla plasmid. Cells were transfected using a 3:1 ratio of FuGENE (Promega #E5911) to DNA in OptiMEM I Reduced Serum Media (Thermo Fisher #31985). Cells were harvested 48 h after transfection.
qRT-PCR for split nLuc
Following RNA extraction or RNase R reactions, RNA was treated with DNase (Thermo Fisher #EN0521) according to the manufacturer's instructions. RNA was then directly used for cDNA synthesis with the Superscript III kit (Thermo Fisher #12574026). cDNA was diluted 1:10 added to an Eppendorf twin.tec 96 real-time PCR Plate (Eppendorf #0030132700) along with iQ Syber Green Supermix (Bio-Rad #1708880) and primers. Primers designed to amplify over the circularization junction and reference primers for GAPDH were chemically synthesized (Integrated DNA Technologies). qPCR was done using the Eppendorf Realplex qPCR machine. RNA quantification was done by using the 2−[ΔCt(target) − ΔCt(reference)] method.
Split nLuc expression analysis
Cells were harvested by directly lifting cells with ×1 phosphate buffered saline (PBS) (Thermo Fisher #10010031). For luminescence assays, cells were harvested 48 h after transfection unless otherwise stated. Media were aspirated off cells, then cells were resuspended in PBS. In total, 50 µL of cell suspension was transferred to a flat-bottomed white-walled 96-well plate (Corning). Nano-Glo Luciferase Assay System (Promega #N1110) reagent was prepared according to manufacturer's instructions. In total, 50 µL of Nano-Glo reagent was added to each well of cell suspensions. The plate was gently shaken, then luminescence detection was done using SpectraMax iD3 (Molecular Devices) machine with SoftMax Pro (v.7.1) software using the luminescence acquisition settings (end-point luminescence, 96-well standard opaque plate, integration time 1000 msec, 1 mm read height).
Dual-luciferase assay
Cells were harvested by directly lifting cells with ×1 PBS (Thermo Fisher #10010031). For luminescence assays, cells were harvested 72 h after transfection, unless otherwise stated. Media was aspirated off cells, then cells were resuspended in PBS. In total, 50 µL of cell suspension was transferred to a flat-bottomed white-walled 96-well plate (Corning). Dual-Glo Luciferase Assay System (Promega #E2920) reagent was prepared according to manufacturer's instructions. In total, 50 µL of Dual-Glo luciferase reagent was added to each well of cell suspensions. The plate was gently shaken, then luminescence detection was done using SpectraMax iD3 (Molecular Devices) machine with SoftMax Pro (v.7.1) software using the luminescence acquisition settings (end point luminescence, 96-well standard opaque plate, integration time 1000 msec, 1 mm read height).
Heat shock
HEK293T cells in a 12-well plate were placed in a 42°C incubator for 3 h, 69 h posttransfection and harvested immediately after for luminescence.
Hypoxia
HEK293T cells were placed in a 37°C, 5% CO2, 1% O2 chamber (InvivO2 300 Hypoxia Workstation) for 24 h, 48 h posttransfection and harvested immediately after for luminescence.
Serum-free treatment
The media HEK293T cells was changed to serum-free media (×1 DMEM [Thermo Fisher #11995-065] with 100 U/mL penicillin and 100 μg/mL streptomycin) for 24 h, 48 h posttransfection and harvested immediately after for luminescence.
Statistical analysis
For direct comparison of two groups, a paired two-tailed Student's t-test was performed using GraphPad Prism. P-values are shown in the figures.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health (NIH) grants R35NS111631, RM1HG011563, and S10 OD030335 (to S.R.J.) and a Moderna Global Fellowship (to M.J.U.). We thank Dr. Yueming Li and Dan Worroll for letting us use their hypoxia chamber.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.080008.124.
- Received February 26, 2024.
- Accepted July 14, 2024.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHORS

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Mildred Unti is the first author of this paper, “A circular split nanoluciferase reporter for validating and screening putative internal ribosomal entry site elements.” Mildred recently obtained a PhD, doing research in the Jaffrey lab at Weill Cornell. Mildred's research focused on creating a circular mRNA expression system and using it as a tool to screen internal ribosomal entry sites.
What are the major results described in your paper and how do they impact this branch of the field?
This paper describes a new method to screen internal ribosomal entry sites (IRESs). This method is not liable to false positive results from cryptic promoters or cryptic splicing—which makes it a great alternative to the bicistronic reporter assay.
What led you to study RNA or this aspect of RNA science?
I previously created a method for expressing large, protein-coding circular mRNAs. In doing so, we realized how useful this method could be for screening IRESs.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
As we were testing IRESs, we found that the activity of cellular IRESs was far lower than the activity of viral IRESs. We think this is primarily due to the lack of false positive signals in our assay.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
Taking a course on RNA biochemistry as an undergraduate at UCLA really spiked my interest in the world of RNA. During the course, I did a project on the EMCV IRES, and I was just in awe of how an RNA can create a structure that can recapitulate the entire translation initiation process of a 5′m7G cap.
If you were able to give one piece of advice to your younger self, what would that be?
Curiosity is a blessing, cherish it!
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My undergraduate advisor Dr. Kohn and my PhD advisor Dr. Jaffrey have both played a huge role in shaping me as a scientist.
What are your subsequent near- or long-term career plans?
I plan on working as a scientist at an exciting new startup.








