
Recoding UAG to selenocysteine in Saccharomyces cerevisiae
- Kyle S. Hoffman1,5,
- Christina Z. Chung1,6,
- Takahito Mukai1,7,
- Natalie Krahn1,
- Han-Kai Jiang1,
- Nileeka Balasuriya2,
- Patrick O'Donoghue2,3 and
- Dieter Söll1,4
- 1Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut 06511, USA
- 2Department of Biochemistry, The University of Western Ontario, London, Ontario N6A 3K7, Canada
- 3Department of Chemistry, The University of Western Ontario, London, Ontario N6A 3K7, Canada
- 4Department of Chemistry, Yale University, New Haven, Connecticut 06511, USA
- Corresponding author: dieter.soll{at}yale.edu
Abstract
Unique chemical and physical properties are introduced by inserting selenocysteine (Sec) at specific sites within proteins. Recombinant and facile production of eukaryotic selenoproteins would benefit from a yeast expression system; however, the selenoprotein biosynthetic pathway was lost in the evolution of the kingdom Fungi as it diverged from its eukaryotic relatives. Based on our previous development of efficient selenoprotein production in bacteria, we designed a novel Sec biosynthesis pathway in Saccharomyces cerevisiae using Aeromonas salmonicida translation components. S. cerevisiae tRNASer was mutated to resemble A. salmonicida tRNASec to allow recognition by S. cerevisiae seryl-tRNA synthetase as well as A. salmonicida selenocysteine synthase (SelA) and selenophosphate synthetase (SelD). Expression of these Sec pathway components was then combined with metabolic engineering of yeast to enable the production of active methionine sulfate reductase enzyme containing genetically encoded Sec. Our report is the first demonstration that yeast is capable of selenoprotein production by site-specific incorporation of Sec.
Keywords
INTRODUCTION
The twenty-first genetically encoded amino acid, selenocysteine (Sec), is a valuable component for recombinant protein engineering. Diselenide bonds are more stable than disulfides and are resistant to irreversible oxidation. Sec is a stronger nucleophile than cysteine (Cys), providing improved reactivity when substituted in the active site of certain enzymes (Reich and Hondal 2016). Nature has replaced Cys codons with Sec in the catalytic site of oxidoreductase enzymes where the enhanced nucleophilic and reducing properties are exploited to reduce reactive oxygen species and prevent oxidative damage in cells (Shchedrina et al. 2007; Metanis and Hilvert 2014; Reich and Hondal 2016).
Unlike the majority of proteinogenic amino acids, Sec is biosynthesized in a tRNA-dependent manner and has no dedicated aminoacyl-tRNA synthetase. Sec biosynthesis begins when tRNASec is initially charged with serine (Ser) by seryl-tRNA synthetase (SerRS). In eukaryotes and archaea, the seryl moiety of Ser-tRNASec is next phosphorylated by O-phosphoseryl-tRNASec (Sep-tRNASec) kinase (PSTK) (Carlson et al. 2004). Sep-tRNASec is rapidly converted to Sec-tRNASec by Sep-tRNA:Sec-tRNA synthase (SepSecS) (Yuan et al. 2006; Xu et al. 2007; Palioura et al. 2009). In bacteria, however, Ser-tRNASec is directly converted to Sec-tRNASec by selenocysteine synthase (SelA) (Forchhammer et al. 1991). The transfer of selenium by SepSecS or SelA originates from selenophosphate, which is produced from selenide by selenophosphate synthetase 2 (SPS2) in eukaryotes (Guimaraes et al. 1996), or by selenophosphate synthetase (SelD) in bacteria (Fig. 1A; Veres et al. 1992).
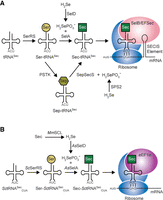
Natural and synthetic Sec translation systems. (A) Natural Sec translation in all domains of life initiates with serylation of tRNASec by SerRS. Conversion to Sec occurs in a single step in bacteria (via SelA) or two steps in archaea and eukaryotes (Ser to Sep by PSTK and Sep to Sec by SepSecS). Selenophosphate (H2SePO3−) is generated from SelD (in bacteria) or SPS2 (in eukaryotes) to support the conversion to Sec. Sec-tRNASec then recodes a UGA codon upstream of a stem–loop RNA structure (SECIS element) by a specialized elongation factor (SelB or EFSec). (B) Synthetic Sec translation system in S. cerevisiae (Sc) initiates with an engineered SctRNASec to recode UAG codons. Initial serylation occurs with endogenous ScSerRS before conversion to Sec by A. salmonicida (As) SelA. Selenophosphate is provided through breakdown of free Sec to selenide by M. musculus (Mm) SCL and then selenide to selenophosphate by AsSelD. Sec-SctRNASec is then recognized by eukaryotic EF1α and installs Sec at UAG codons.
Normal insertion of Sec during translation is specified by recoding designated UGA codons. Recoding of UGA from a termination codon to Sec requires specialized translation factors and a stem–loop element in the selenoprotein messenger RNA (mRNA). A selenocysteine-specific elongation factor called EFSec (SelB in bacteria) preferentially binds Sec-tRNASec over Ser-tRNASec and is recruited to the ribosome by a stem–loop RNA structure known as the Sec insertion sequence (SECIS) element. The SECIS element is positioned directly downstream from the UGA codon in bacteria and in the 3′ untranslated region in eukaryotic selenoprotein mRNAs (for review, see Squires and Berry 2008).
Selenoprotein biosynthetic pathways have widely been lost in evolution of the fungal kingdom, except for nine species recently discovered that have selenoprotein genes along with the biochemical pathway components required for Sec-tRNASec biosynthesis (Mariotti et al. 2019). Despite Saccharomyces cerevisiae lacking Sec utilizing traits, including a tRNASec, selenium can accumulate intracellularly at high concentrations, largely in the form of selenomethionine (SeMet) (Ponce de Leon et al. 2002). Selenium can also infiltrate the trans-sulfuration and Cys biosynthesis pathways in S. cerevisiae (Lazard et al. 2015), which leads to the production of free Sec. In fact, free Sec is misincorporated at Cys codons during translation and causes protein aggregation (Lazard et al. 2017). In mammals, free Sec is hydrolyzed by selenocysteine lyase to dehydroalanine and selenide (Esaki et al. 1982), providing the necessary form of selenium for biosynthesis of Sec-tRNASec and specific insertion of Sec at UGA codons. With this knowledge and minimal metabolic engineering, we aimed to construct a Sec translation system in S. cerevisiae for production of selenoproteins.
We (Bröcker et al. 2014; Miller et al. 2015) and others (Thyer et al. 2015) have engineered bacteria for efficient selenoprotein production (Mukai et al. 2018; Thyer et al. 2018). Building on these findings, we introduced part of the bacterial pathway into yeast for selenoprotein production (Fig. 1B). Our approach addresses a critical biotechnology gap, since not all selenoproteins can be efficiently synthesized in bacteria (Miller et al. 2015), and eukaryotic selenoproteins produced in bacteria may lack important post-translational modifications. To date, photocaged Sec has been incorporated in proteins in yeast; however, uncaging requires isolation of the recombinant protein from cell lysate and exposure to UV light (Rakauskaitė et al. 2015), which cannot be used for studies in live cells. Direct incorporation of Sec and expression of selenoproteins in S. cerevisiae would enable systems biology studies of interaction networks when implemented in yeast two-hybrid screens or in synthetic genetic arrays. Here, we engineered a novel yeast translation system dedicated to Sec incorporation. We demonstrated site-directed Sec incorporation using a Sec-dependent genetic reporter in live cells and by mass spectrometry (MS) analysis. Lastly, we produce an active methionine sulfate reductase enzyme (MsrB1) containing Sec to demonstrate an application of the Sec translation system to produce recombinant human selenoproteins in yeast.
RESULTS
Engineering translation components for selenocysteine insertion in Saccharomyces cerevisiae
Naturally, tRNASec is a poor substrate for SerRS relative to tRNASer, resulting in a low level of Sec-tRNASec within the cell (Wang et al. 2015). This is likely due to the low number of Sec incorporations required for normal selenoprotein expression. For improving the yield of recombinant selenoprotein production, it is necessary to have high cellular concentrations of Sec-tRNASec, to decode an overexpressed selenoprotein mRNA and outcompete the release factor for Sec insertion at a stop codon. Considering this, and the fact that the anticodon of tRNASer is not an identity element for aminoacylation by SerRS (Lenhard et al. 1999), we used the major S. cerevisiae tRNASer (SctRNASer) isoacceptor, tRNA-Ser-AGA-1-1 (Chan and Lowe 2016) to engineer a UAG-specific suppressor tRNA for Sec incorporation. The design of this yeast tRNASec molecule was inspired by our previous development of a UAG-specific bacterial tRNA for efficient selenoprotein production (Fig. 1B; Mukai et al. 2018; Prabhakar et al. 2022). Yet eukaryotic tRNASec species have a 13 bp acceptor domain (acceptor stem and T-stem combined), a distinguishing feature from canonical tRNAs that contain a shorter (12 bp) acceptor domain, required for UGA recognition by Sec-specific proteins (Krahn et al. 2020). Therefore, we used Aeromonas salmonicida SelA as it recognizes its cognate tRNASec that has a 12 bp acceptor domain and is known to enable high efficiency selenoprotein production in bacteria with other 12 bp acceptor domain tRNASec species (Mukai et al. 2016, 2018). A. salmonicida SelD was also taken from the bacterial system and codon optimized for expression in yeast. Further metabolic engineering was also needed to supply selenide for SelD at the initial step of Sec-tRNASec biosynthesis. Knowing that S. cerevisiae can accumulate free Sec when growth medium is supplemented with SeMet, we chose to express Mus musculus selenocysteine lyase (MmSCL) for hydrolysis of free Sec to dehydroalanine and selenide (Mihara et al. 2000). As an indication that the enzyme functions in S. cerevisiae, expression of MmSCL improved growth in medium containing toxic levels of SeMet (Supplemental Fig. S1), conditions known to cause accumulation of free Sec that induce protein aggregation and slow growth (Lazard et al. 2017).
Suppression of amber codon with tRNASec variants
Three tRNASec variants were constructed and tested for Sec incorporation in a reporter protein using SctRNASer as a scaffold (Fig. 2A). First, a CUA anticodon was installed for recoding amber (Am) (UAG) codons. Second, to facilitate recognition by AsSelA for Ser to Sec conversion, three additional base pairs in the D-arm were inserted to resemble AstRNASec. Third, to maintain the D-loop and T-loop tertiary interactions a G59U mutation was inserted. Finally, in efforts to sequentially change the tRNA core to resemble AstRNASec, the core G–C positions were swapped (G9C and C48G). This set of mutations resulted in the first variant, SctRNASecCUA-1. The second variant (SctRNASecCUA-2) was designed based on SctRNASecCUA-1. To continue sequentially changing the tRNA core to resemble AstRNASec, an additional change, U8A, was installed. Furthermore, from previous studies in Escherichia coli, we knew that stabilizing the D-arm can promote Ser to Sec conversion and that the native AstRNASec sequence is not efficient in this pathway (Mukai et al. 2018). Therefore, we made an additional mutation in the D-arm, U14C, to increase its stability. The third variant (SctRNASecCUA-3) built on SctRNASecCUA-2 with one additional change, C9U. This finished the sequential replacement of the tRNA core nucleotides to AstRNASec (Fig. 2A; outlined). To test nonsense suppression efficiency of the first tRNASec variant (SctRNASecCUA-1), we used two different reporters to compare it to an E. coli tRNATyr/TyrRS suppression system previously characterized in S. cerevisiae (Edwards and Schimmel 1990). First, we measured fluorescence resulting from suppression of an Am codon at position 2 in superfolder GFP (sfGFP). EctRNATyr/EcTyrRS was observed to have roughly 1.5-fold higher fluorescence (P < 0.001, n = 12) compared to the background levels. SctRNASecCUA-1, however, surpassed that with a fivefold increase in fluorescence (P < 0.0001, n = 12) (Fig. 2B) functioning as a more efficient suppressor than EctRNATyr/EcTyrRS (P < 0.0001, n = 12) (Edwards and Schimmel 1990).
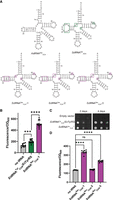
Design of tRNAs for Sec translation in S. cerevisiae. (A) Secondary structure representation of SctRNASec designed from replacing regions of SctRNASer with features from AstRNASec (outlined in purple). As the enzymes used in this study originated from a small group of Aeromonas species which irregularly utilize the ACA anticodon (and UGU codon pair) and thrive at a wide temperature range (Pfeiffer et al. 2018), the AstRNASec shown has an AGA anticodon. (B) sfGFP-S2Am readthrough assay found SctRNASecCUA-1/AsSelA (purple) to be a strong suppressor compared to an E. coli tRNATyrCUA/TyrRS pair (green) (P < 0.0001, n = 12). (C) Gal4-R110Am assay showed similar cellular growth of S. cerevisiae in the absence of uracil for SctRNASecCUA-1 compared to an E. coli tRNATyrCUA/TyrRS pair. (D) sfGFP-S2Am readthrough assay compared all SctRNASecCUA variants with AsSelA, highlighting the highest suppression levels with SctRNASecCUA-1.
The second reporter used Gal4 with an R110Am mutation coupled to URA3 transcription (Chin et al. 2003). Amber codon suppression in gal4-R110Am is required for growth on medium lacking uracil, as a strain lacking an Am suppressor tRNA does not grow (Fig. 2C; empty vector control). SctRNASecCUA-1 is shown to suppress gal4-R110Am, growing similarly to a strain expressing the EctRNATyr/EcTyrRS pair (Fig. 2C). The findings agree with our GFP fluorescence data and demonstrate that SctRNASecCUA-1 is an efficient UAG suppressor but also suggest that codon context or the reporter method could influence the efficiency of suppression. Given that our sfGFP-S2Am reporter had a wider dynamic range for analyzing suppression, we proceeded to compare all three SctRNASec variants in this manner. Compared to background (no tRNA), SctRNASecCUA-1 had the highest GFP fluorescence, followed by SctRNASecCUA-3. SctRNASecCUA-2 lead to GFP fluorescence that was not statistically different compared to cells expressing no additional tRNA (Fig. 2D). These assays specifically investigate suppression efficiency regardless of the amino acid inserted. Given that the Sec pathway we used may also misincorporate Ser (Chung et al. 2022), we required a reporter capable of distinguishing between Ser and Sec.
Development of a selective reporter for Sec incorporation
Since sfGFP-S2Am and gal4-R110Am reporters are not specific for Sec, we designed a Gal4 reporter that would be functional with Sec incorporation but not Ser (Fig. 3A). In E. coli, Cys residues in the iron–sulfur cluster of formate dehydrogenase can be replaced with Sec while still maintaining enzyme activity, while insertion of Ser abolishes activity (Mukai et al. 2018). Thus, we hypothesized that Cys residues in the Gal4 DNA binding domain could be replaced by Sec for coordinating the zinc ion. Mutating Cys11 or Cys21 to Ser in Gal4 resulted in loss of growth on medium lacking uracil but containing the selenium sources SeMet and sodium selenite, even in the presence of the SctRNASec variants (Fig. 3B). This indicates that insertion of Ser by Ser-tRNASec will not lead to proper zinc-coordination of Gal4 to promote transcription of URA3 required for cell growth in the absence of uracil. To increase the stringency of our reporter, we engineered both Cys positions (11 and 21) in the Gal4 DNA binding domain to encode for Ser. We verified that the Ser in both Cys positions cannot support growth presumably due to the inability to coordinate zinc (Fig. 3B).
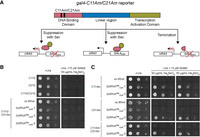
Sec-specific Gal4 reporter. (A) Design of Gal4 reporter with up to two Cys coded for by an Am codon (C11Am and/or C21Am) in the DNA binding domain. If Ser is inserted in either of those positions, an inactive Gal4 will be expressed which is unable to bind to the Gal4 upstream activation sequence (UAS) to activate transcription of URA3. The same is true if suppression does not occur and Gal4 is truncated. URA3 promotes S. cerevisiae growth in the absence of uracil. If Sec is inserted, then the DNA binding domain of Gal4 is active to bind to the GAL4UAS and activate URA3 transcription. (B) S. cerevisiae growth is dependent on the presence of a Cys (or Sec) at positions 11 and 21 to produce active Gal4. Mutation of either of those positions to Ser (C11S or C21S) does not promote cell growth. (C) Testing individual (C11Am or C21Am) or dual (C11Am/C21Am) Sec insertion found that SctRNASecCUA-3 promotes cell growth better than SctRNASecCUA-1.
We next tested suppression of gal4-C11Am, gal4-C21Am, and gal4-C11Am/C21Am with SctRNASecCUA-1 and SctRNASecCUA-3. We confirmed that both gal4 alleles are not suppressed in the absence of a suppressor tRNA (Fig 3C; no tRNA). However, expression of either SctRNASecCUA-1 or SctRNASecCUA-3 results in gal4-C11Am suppression and uracil prototrophy. Sec insertion at this position appears to be equivalent for both tRNAs (Fig. 3C), yet varies when we look at a different position, gal4-C21Am. At position 21, only SctRNASecCUA-3 was found to insert Sec, and it follows that the double insertion gal4-C11Am/C21Am was also only possible by this variant (Fig. 3C). This suggests that SctRNASecCUA-3 is robust with regards to sequence variation surrounding the Am codon, and it is more efficient at Sec insertion. These results also indicate that Sec can substitute zinc-coordinating Cys residues in Gal4. As SctRNASecCUA-3 is most efficient at inserting Sec, we performed all further experiments with this tRNA and renamed it to SctRNASU to denote a S. cerevisiae tRNASer that functions as a tRNASec molecule (U is the one letter code for Sec).
Production and characterization of a recombinant human selenoprotein in yeast
To demonstrate the ability of the system to produce a selenoprotein, human MsrB1 was expressed and purified from S. cerevisiae cells containing AsSelA, AsSelD, MmSCL, and SctRNASU. The UGA codon at position 95 encoding for Sec was replaced with UAG for recognition by SctRNASU. The enzyme was able to reduce l-methionine-sulfoxide (l-Met-SO) to l-methionine (lMet), confirming production of an active selenoprotein (Fig. 4A). To validate Sec insertion, purified MsrB1 was analyzed by liquid chromatography–tandem mass spectrometry (LC–MS/MS). The enzyme was reduced, alkylated, and digested with trypsin prior to LC–MS/MS analysis. All raw data were searched against a database containing the MsrB1 sequence appended to the S. cerevisiae proteome and a 1% false discovery rate filter was applied. We identified tryptic peptides containing either Ser or carbamidomethylated Sec at position 95 (Supplemental Fig. S2) in MsrB1 for all three biological replicates (representative LC–MS/MS spectra shown in Fig. 4B). Peptides of similar length were analyzed for their presence of Ser or Sec. Abundance values for each set of peptides were compared and averaged. From this, we estimated that Sec-containing MsrB1 accounts for about 20% of the total MsrB1 yield (Fig. 4C). The MS/MS data are consistent with independent elemental analysis by ICP-MS of the MsrB1 to determine Sec occupancy. ICP-MS determined that the mass fraction of the measured amount of selenium in MsrB1 made up on average 16 ± 1% of the expected mass of selenium if Sec incorporation efficiency was 100% (Supplemental Table S1). Taken together, these results validate and confirm Sec incorporation and estimate that approximately 1/5 of the purified MsrB1 protein contains Sec.
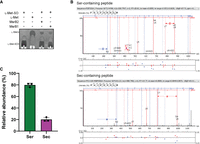
Production of selenoprotein MsrB1 in S. cerevisiae. (A) Purified MsrB1 from S. cerevisiae shows conversion of l-Met-SO to l-Met which is comparable to an MsrB2 control. (B) Representative LC–MS/MS spectra show the presence of both Ser and Sec peptides of MsrB1. (C) Quantitative analysis of three separate spectra show ∼20% of MsrB1 contains Sec.
DISCUSSION
The Sec-decoding trait is found among organisms representing all three domains of life (Romero et al. 2005; Su et al. 2009), yet selenoprotein biosynthesis is notably absent in higher plants and most Fungi except for a few species among the early branching fungal phyla, including the Zoopagomycota and the Chytridiomycetes (Mariotti et al. 2019). Phylogenetic analysis suggested that the Sec trait was lost early in the evolution of the Dikarya, a fungal subkingdom that includes S. cerevisiae. Thus, S. cerevisiae is separated from the Sec-decoding trait by a deep evolutionary divide.
In this study, for the first time, we bridged the evolutionary gap to generate an S. cerevisiae strain that genetically encodes 21 amino acids, including Sec. Several independent components of selenoprotein biosynthesis were required to expand the genetic code of yeast with Sec. Transplanting a eukaryotic or fungal Sec-biosynthetic system, such as the complete Sec system found in Bifiguratus adelaidae (Mariotti et al. 2019), would represent a fascinating experiment. Such an approach, however, would produce a yeast strain that encodes Sec at UGA codons in the appropriate sequence context of a required SECIS element. Since we desired to develop a Sec system in yeast to enable general site-directed Sec insertion at an Am codon and independent of the SECIS element, we imported Sec genes from A. salmonicida. Because we had shown that the A. salmonicida Sec-biosynthetic machinery was highly efficient in engineering a Sec incorporation system that works well without the SECIS element in bacteria (Mukai et al. 2018), we reasoned that the same components may also function in yeast following development of a new “yeast” tRNA. Thus, we engineered a yeast tRNASer mutant that could be recognized by both yeast SerRS for initial serylation and A. salmonicida SelA for conversion to Sec. To generate a complete Sec-biosynthetic pathway in yeast, we also imported A. salmonicida selD, and expressed murine Sec lyase to provide a cellular selenide source for SelD.
We designed a novel and functional Cys/Sec-specific reporter to detect Sec incorporation in proteins produced in yeast. In characterizing the reporter, we found that suppression of up to two Am codons in place of essential Cys residues in Gal4 suggests that Sec-SctRNASU was produced at high enough levels to incorporate two Sec residues into the same peptide, while effectively competing with the endogenous translation release factor and peptide chain termination. Although our approach has clearly demonstrated selenoprotein synthesis in yeast, the next steps in our future work will involve improving the fidelity of the Sec translation system. Translation fidelity is an on-going challenge in the field of genetic code expansion (George et al. 2016; Gan and Fan 2017), which we and others have improved by increasing tRNA copy number (Aerni et al. 2015), evolving more efficient tRNA or aminoacyl-tRNA synthetase variants (O'Donoghue et al. 2013), or by increasing the cellular concentration of the noncanonical amino acid (Lacoursiere et al. 2020).
In addition to these findings, our work also suggests that eukaryotic elongation factor (eEF1α) binds Sec-SctRNASU and delivers it to the ribosome in a noncanonical manner that does not require an mRNA structural element (SECIS) or a specialized elongation factor for Sec (SelB or EFSec). Thus, our approach enables flexible and site-directed Sec incorporation in yeast. Because the selenoprotein translation system does not require a SECIS element in the mRNA, the approach can be used to place a Sec residue at any desired location in a recombinant protein or in any endogenous yeast protein by introducing Am codon mutations at specific sites in the genome. Our work will enable the production of many kinds of recombinant and engineered selenoproteins in yeast using Aeromonas pathway components that also enabled efficient selenoprotein production in bacteria (Mukai et al. 2018). Producing selenoproteins in S. cerevisiae will facilitate protein engineering and systems biology studies of enzyme function and protein–protein interactions. Expanding the yeast genetic code with Sec will allow new approaches to cost-effectively produce medically or industrially relevant selenoproteins that require or are improved by synthesis in a eukaryotic system.
MATERIALS AND METHODS
Strains and growth conditions
All yeast strains used in this study were derived from either BY4741 (Winzeler and Davis 1997) or MaV203 (listed in Supplemental Table S2; Vidal et al. 1996a,b). Unless otherwise indicated, yeast strains were grown in yeast peptone (YP) medium containing 2% glucose, or in minimal medium (MM) supplemented with required amino acids. All cultures were grown at 30°C. KHY1 was created by disrupting the HIS3 allele in MaV203 with a KanMX integration cassette (described in DNA molecules). The KanMX cassette was transformed into MaV203 and plated onto YPD plates containing 200 µg/mL G418 (Sigma-Aldrich). Histidine auxotrophic transformants were validated by patching onto MM plates lacking histidine, and integration of the KanMX cassette was confirmed by PCR.
DNA molecules
Mus musculus selenocysteine lyase, A. salmonicida SelA (AsSelA), and A. salmonicida SelD (AsSelD) were expressed from the ADH1 constitutive promoter (700 bp) and terminated by the RPL41B terminator. A construct containing ADH1, MmSCL, and the RPL41B terminator (sequence listed in Supplemental Material) was synthesized (IDT) and cloned into pRS425 to generate pKH24 (plasmids listed in Supplemental Table S3). AsSelA and AsSelD were codon optimized (sequences listed in Supplemental Material), synthesized (IDT), and cloned into NcoI/NheI digested KH24 by infusion, thereby replacing MmSCL and creating pKH29 and pKH30, respectively. To generate pKH39, ADH1-MmSCL-RPL41B was PCR amplified from the KH24 template using ASR-F and ASR-R (oligonucleotides listed in Supplemental Table S3) and cloned by infusion into PciI cut pRS424. AsSelA was PCR amplified using primers adh1-F and rpl41b-R and KH29 as templates and cloned by infusion into BamHI/XhoI digested pRS424 and KH39 to create pKH31 and pKH47, respectively. The sfGFP reporter plasmid pKH75 was created by amplifying the ADH1-sfGFPS2Am-RPS20 (sequence in Supplemental Material) and cloning into pRS426.
GAL4, from −15 to +2946 of the translational start, was PCR amplified from S. cerevisiae genomic DNA in two fragments using primers gal4a with gal4b and gal4c with gal4d. Full-length ADH1 promoter (1400 bp) was amplified using primers fADH1-F and fADH1-R. The three PCR productions were cloned by infusion into BamHI cut pRS424 to produce pKH58. The R110Am mutation was introduced by QuikChange using pKH58 as template to produce pKH60. The gal4-C11S, gal4-C21S, gal4-C11Am, and gal4-C21Am alleles were made by QuikChange (Supplemental Table S4; primer names indicate mutations) using pKH58 as template, generating pKH79, pKH80, pKH81, and pKH82, respectively. Each of the gal4 alleles were moved into pRS423 as a BamHI fragments to create pKH85, pKH99, pKH100, and pKH101. pKH85 was amplified with primers C11Am-F and C11Am-R to create pKH95. Similarly, pKH97 was made by amplifying the pKH79 template using primers C21S-F and C21S-R, and pKH98 by amplifying pKH95 with C11S-F and C11S-R primers. pKH102 contains the gal4-C11S/C21S allele subcloned from pKH97 into pRS423 as a BamHI fragment.
tS(AGA)D2, including −500 bp and +300 bp of the tRNA sequence, was PCR amplified from S. cerevisiae genomic DNA using primers tSD2-F and tSD2-R, and cloned by infusion into BamHI digested pRS425 to create pKH66. The anticodon was changed to CUA by two-step PCR using primers tScua-F with tSD2-R and tScua-R with tSD2-F. The two PCR products were mixed at a 1:1 molar ratio, denatured at 95°C, annealed at 55°C, and extended for 5 min at 72°C to produce full-length tS(CUA)D2. The full-length product was PCR amplified using primers tSD2-F and tSD2-R and cloned into BamHI cut pRS425 by infusion to generate pKH71. pKH74 was made by inverse PCR using primers tSec-F and tSec-R with the pKH71 template, followed by infusion. tU(CUA)1 was subcloned into KH30 as a BamHI fragment to create pKH77. tU(CUA)2 and tU(CUA)3 were introduced by inverse PCR, amplifying the pKH77 template using primers tSec2-F with tSec-R and tSec3-F with tSec-R, respectively. Infusion cloning of the PCR products generated pRS425-AsSelD/tU(CUA)2 (pKH84) and pRS425-AsSelD/tU(CUA)3 (pKH86).
Human MsrB1(95TAG) was ordered as a DNA fragment from Twist BioScience and cloned into KH75 (pRS426-ADH1-sfGFP-S2Am-RPS20) by replacing sfGFP-S2Am. The plasmid, including the C-terminal His6-tag, was amplified using primers 4936_op_F and 4936_op_R, and MsrB1 was amplified using primers MsrB1_PRS_F and MsrB1_PRS_R. The two PCR products were assembled using Gibson Assembly (NEBuilder HiFi DNA Assembly). An N-terminal Flag-tag was inserted by amplifying the plasmid using primers FlagMsr_F and FlagMsr_R. Both primers contain a BamHI cut site on the 5′-end for ligation. The PCR product was subjected to DpnI-mediated digestion at 37°C for 1 h and gel purified. The purified product was digested using BamHI-HF (NEB) at 37°C for 1 h, gel purified, and ligated with T4 DNA Ligase (NEB) at 37°C for 1 h to generate pRS426-ADH1p-Flag-hMsrB1(95TAG)-RPS20.
To disrupt the HIS3 allele in MaV203, a KanMX disruption cassette was made by three sequential PCRs. KanMX was PCR amplified using primers U2 and D2. This product was used as a template for PCR using primers his3-F and his3-R. Primers his3-P and his3-T were used for PCR to extend the HIS3 homologous sequences flanking KanMX to 96 bp. Integration of the KanMX marker was validated by PCR using primers kanMX-F and lys2-R.
Spot plating and growth assays
For testing SeMet resistance of strains expressing MmSCL, BY4741 containing either KH39 or pRS424 was grown in MM lacking tryptophan, then diluted to an optical density (OD600 nm) of 0.01 in the same medium but containing 10 µM SeMet. Cultures were grown in a 96-well clear-bottom plate (Corning Inc.) at 30°C for 48 h in a BioTek Synergy HT plate reader.
Suppression of gal4-C11S/C21Am (pKH98), gal4-C11Am (pKH101), gal4-C21Am (pKH85), and gal4-C11Am/C21Am (pKH95) was analyzed in yeast strain KHY1 containing pKH47, and either pKH77, pKH84, pKH86, or pRS425 (no tRNA control). Each strain was grown in MM lacking leucine, tryptophan, and histidine to stationary phase, then diluted into YPD medium contain 10 µM SeMet and 30 µg/mL sodium selenite and grown for 5 h. Cells were spotted onto MM plates depleted of leucine, tryptophan, histidine, and uracil, but containing 10 µM SeMet and 30 µg/mL sodium selenite in 10-fold serial dilutions. Each spot plate was grown at 30°C for 72 h. As controls for Sec-specific gal4 suppression, the KHY1 strain containing pKH47, pRS425, and either gal4-C11S (pKH99), gal4-C21S (pKH100), or gal4-C11S/C21S (pKH102) was grown and spotted as described above.
Analysis of sfGFP fluorescence
KHY1 strains containing KH75 (sfGFP-S2Am), KH47 (MmSCL/SelA), or KH39 (MmSCL), and either pRS425, KH77 [tU(CUA)1], KH84 [tU(CUA)2], or KH86 [tU(CUA)3] were grown to stationary phase in MM lacking histidine, tryptophan, and leucine, then diluted 1:1000 in the same medium and grown in a 96-well clear-bottom black-walled plate (Corning Inc.) for 48 h. Cell density and fluorescence were measured every hour at 600 and 528 nm, respectively. sfGFP fluorescence was normalized to cell density (OD528 nm/OD600 nm) for each strain and was compared at mid-logarithmic growth phase.
Expression and purification of MsrB1
MaV203 containing pRS426-ADH1p-Flag-hMsrB1(95TAG)-RPS20, KH86 [pRS425-SelD/tU(CUA)3], and KH47 (MmSCL/SelA) were grown overnight at 30°C in 2.5 mL of MM lacking uracil, leucine, and tryptophan. The next day, the 2.5 mL preculture was diluted into 180 mL of the same MM and grown at 30°C overnight. The following day, 20 mL from the preculture was diluted into 980 mL MM (1:50 dilution) containing 15 µM SeMet and 50 µg/mL sodium selenite and grown at 30°C overnight. A total of 8 L was grown. Cells were harvested and the pellet was weighed (17.4 g total). The pellet was resuspended in 45 mL Y-PER Yeast Protein Extraction Reagent (Thermo Fisher Scientific) and left on a rotator at room temperature for 20 min. The lysate was centrifuged at 14,000g for 10 min at 4°C and the supernatant was loaded onto a HisPur Cobalt Resin (Thermo Fisher Scientific) preequilibrated with Y-PER. The column was washed with 50 mM sodium phosphate, 300 mM sodium chloride, and 10 mM imidazole (pH 7.5), and the bound protein was eluted with 50 mM sodium phosphate, 300 mM sodium chloride, and 250 mM imidazole (pH 7.5) into one tube. The elution was concentrated and buffer exchanged to Y-PER to 1 mL using Amicon Ultra-4, 3K MWCO (Millipore). The sample was moved to a microcentrifuge tube containing 150 µL Pierce Anti-DYKDDDDK Affinity Resin (Thermo Fisher Scientific) preequilibrated with Y-PER. The sample was left on a rotator at room temperature for 20 min and moved to a capped gravity column. Once the beads settled, the cap was removed, and the flowthrough was collected. The beads were washed according to the manufacturer's instructions and eluted using the acid elution protocol. The elution was concentrated and buffer exchanged to 50 mM sodium phosphate and 100 mM sodium chloride (pH 7.5) to 45 µL using Amicon Ultra-0.5 mL, 3K MWCO (Millipore Sigma). The sample was immediately used for the activity assay and a sample was analyzed via SDS-PAGE.
MsrB1 methionine reductase activity assay
The reductase activity assay was based on methods previously described (Kim and Gladyshev 2004). For the reduction of l-Met-SO to lMet, 50 µL reactions were prepared containing (1) 8.5 mg/mL l-Met-SO, (2) 8.5 mg/mL l-Met, (3) 8.5 mg/mL l-Met-SO and 5 µg MsrB2 (Cytoskeleton Inc.), or (4) 8.5 mg/mL l-Met-SO and 5 µg MsrB1. All reactions included 20 mM dithiothreitol (DTT) and 25 mM sodium phosphate (pH 7.5). The reactions were incubated at 37°C for 2 h, and 6 µL were spotted in 1 µL increments onto a TLC PEI Cellulose F plate (Millipore Sigma) and left to dry. The plate had been preactivated with water and dried prior to spotting. The plate was run in a TLC chamber with n-butanol:acetic acid:water (60:15:25) until the solvent front was near the top of the plate. The plate was removed from the chamber and left to dry. Visualization of the amino acids was achieved by pouring ninhydrin stain on the plate (0.02 g ninhydrin, 10 mL ethanol, 50 µL acetic acid). The plate was left to dry and the spots to develop. Images were taken with the ChemiDoc MP (Bio-Rad).
Mass spectrometry analysis
Samples were then prepared for LC–MS/MS analysis at Bioinformatics Solutions Inc. Briefly, samples were reduced with 10 mM DTT (Sigma-Aldrich) alkylated with 20 mM iodoacetamide (Sigma-Aldrich), acetone precipitated and digested overnight with MS grade trypsin (Promega). Digested samples were lyophilized. Lyophilized samples were resuspended in 0.2% trifluoroacetic acid (TFA) and desalted using a C18 spin column.
C18 desalted samples were resuspended in 12 µL buffer A (0.1% TFA in water). Six microliters of each sample was injected into the timsTOF Pro (Bruker Daltronics) by nanoflow liquid chromatography using a Bruker NanoElute chromatography system (Bruker Daltronics). Liquid chromatography was preformed using a constant flow of 300 µL/min and a 15 cm reversed-phased column with a 75 µm inner diameter filled with Reprosil C18 (PepSEP). Mobile phase A was 0.1% formic acid and mobile phase B was 99.9% acetonitrile, 0.1% formic acid. Peptide separation was carried out over 30 min as follows; linearly 2% A to 35% B over 30 min with an increase to 95% B over 30 sec and held constant for 2.62 min to clean the column. Column equilibration was done prior to automatic sample loading at a temperature of 50°C.
The timsTOF Pro was outfitted with a CaptiveSpray source (Bruker Daltronics), operated in PASEF mode. Trapped ion mobility separation was achieved by using an accumulation time of 100 msec in the first TIMS region and ramps of the TIMS region from 0.85 to 1.30 V.s/cm2, with each ramp lasting 100 msec. MS and MS/MS scans were limited to 100–1700 m/z, and a polygon filter was applied to the m/z and ion mobility dimensions to select for multiple charged ions most likely to be peptide precursors. Collision energy was applied as a function of ion mobility with a linear regression using the following parameter settings: 0.85 V.s/cm2 → 27 eV, 1.30 V.s/cm2 → 45 eV. TIMS voltage was calibrated using ions from the Agilent Tune Mix (m/z 622, 922, 1222). Active exclusion of MS/MS scans was enabled at a setting of 0.40 min. Quadrupole isolation was set to 2 m/z for m/z less than 700, and 3.0 m/z for ions with an m/z greater than 800. A linear regression calculation was done automatically for ions in between m/z 700 and 800. All MS experiments were completed at the MS laboratory of Bioinformatics Solutions Inc.
MS Raw Files were processed using PEAKS XPro (v10.6, Bioinformatics Solutions Inc.). The data were searched against a custom database containing the MsrB1 sequence in conjunction with the S. cerevisiae proteome (UniProt reviewed database). Precursor ion mass error tolerance was set to 15 ppm and fragment ion mass error tolerance was set to 0.05 Da. The search was performed with a fixed modification of carbamidomethylation (57.02 Da) on cysteine residues. Variable modifications of deamidation (0.98 Da) on asparagine and glutamine, as well as oxidation (15.99 Da) on methionine, were also specified. To search for the presence of Sec in MsrB1, Ser was included at position 95 in the MsrB1 sequence database, and carbamidomethylated Sec was identified by the mass shift of +120.94 Da from Ser. The false discovery rate was set to 1% for the database search and only modifications supported by an AScore of 20 were considered. MS Raw files and Search files were deposited into the PRIDE Repository.
Inductively coupled plasma mass spectrometry (ICP-MS) analysis of MsrB1
Purified MsrB1 protein samples (27 µM) were subjected to ICP-MS analysis to detect the amount of Se incorporated in MsrB1. Protein samples were analyzed in triplicate, each containing 13 µg of protein in 100 µL of buffer (50 mM sodium phosphate, 100 mM sodium chloride, and 10% glycerol, pH 7.4), using an Agilent 7700 series ICP-MS coupled to an Agilent 1260 Infinity high-performance liquid chromatography (HPLC) system at the Biotron Facility at the University of Western Ontario.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
H-K.J. held a graduate student fellowship from the Taiwan Academic Talents Overseas Advancement Program from the Ministry of Science and Technology (MOST 110-2917-I-007-006), and C.Z.C. held a postdoctoral fellowship from the Natural Sciences and Engineering Research Council of Canada (NSERC). This work was supported by grants from the National Institute of General Medical Sciences (R35GM122560, R35GM122560-05S1 to D.S.), the DOE Office of Basic Energy Sciences (DE-FG02-98ER20311 to D.S.), the Natural Sciences and Engineering Research Council of Canada (04282 to P.O.), the Canada Research Chairs (232341 to P.O.), and the Canadian Institutes of Health Research (165985 to P.O.).
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079658.123.
-
Freely available online through the RNA Open Access option.
- Received March 10, 2023.
- Accepted May 16, 2023.
This article, published in RNA, is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.
REFERENCES
MEET THE FIRST AUTHOR

Meet the First Author(s) is an editorial feature within RNA, in which the first author(s) of research-based papers in each issue have the opportunity to introduce themselves and their work to readers of RNA and the RNA research community. Kyle Hoffman is the first author of this paper, “Recoding UAG to selenocysteine in Saccharomyces cerevisiae.” Kyle did this work as a postdoctoral fellow with Dr. Dieter Söll at Yale University, after which he has taken on the role of Applications Manager at Bioinformatics Solutions Inc., where mass spectrometry–based proteomics research and services are provided to academic and industry sectors.
What are the major results described in your paper and how do they impact this branch of the field?
For the first time, we directly incorporated selenocysteine site-specifically into yeast proteins. This involved engineering yeast tRNA-Ser so that it is recognized by selenocysteine biosynthesis pathway components for conversion of Ser-tRNA to Sec-tRNA. This demonstrates that yeast is capable of selenoprotein production, despite widespread loss of the selenocysteine biosynthesis pathway from the fungal kingdom. Lastly, we produce an active methionine sulfate reductase enzyme (MsrB1) containing Sec to demonstrate an application of the Sec translation system to produce recombinant human selenoproteins in yeast.
What led you to study RNA or this aspect of RNA science?
In my PhD, under the supervision of Dr. Chris Brandl, I discovered a suppressor tRNA in yeast that inserts alanine at proline codons, causing mistranslation of the genetic code. How a single mutation in a tRNA could alter the genetic code was exciting and fascinating. This led me into the field of genetic code expansion to further explore how the genetic code could be manipulated to incorporate noncanonical amino acids.
During the course of these experiments, were there any surprising results or particular difficulties that altered your thinking and subsequent focus?
I was quite skeptical going into this project, since there was literature suggesting the yeast ribosome could not accommodate selenocysteine. When I observed growth that was dependent on selenocysteine insertion in Gal4, I knew I needed to continue pursuing this project and validate my observations with mass spectrometry data.
What are some of the landmark moments that provoked your interest in science or your development as a scientist?
In general, the “aha” moments and excitement when you discover something that could really impact the field continues to provoke my interest in science. In my current position, I collaborate with researchers who are performing antibody discovery or immunopeptidomics for cancer vaccine development. Helping other researchers discover and develop novel therapeutics that can impact human health continues to drive my interest in science.
If you were able to give one piece of advice to your younger self, what would that be?
Don't give up on anything that is worth pursuing. Perseverance will pay off.
Are there specific individuals or groups who have influenced your philosophy or approach to science?
My supervisors throughout graduate school and my postdoctoral work, Drs. Jim Karagiannis, Chris Brandl, and Dieter Söll, greatly influenced my approach to science. They taught me to take responsibility and independence over my own projects but also to foster relationships with colleagues and collaborators to improve the quality and impact of research and publications. From each of them, I learned that nothing comes easy, but hard work and critical thinking will help to overcome many challenges.
What are your subsequent near- or long-term career plans?
My long-term career plan is to continue developing my skills as Applications Manager at Bioinformatics Solutions Inc. and further expand our mass spectrometry laboratory services to offer research solutions that fit within protein therapeutics and cancer vaccine development workflows.








