
Structural and dynamic effects of pseudouridine modifications on noncanonical interactions in RNA
- Jennifer Vögele1,
- Elke Duchardt-Ferner1,
- Holger Kruse2,
- Zhengyue Zhang2,3,4,
- Jiri Sponer2,
- Miroslav Krepl2 and
- Jens Wöhnert1
- 1Institute of Molecular Biosciences and Center for Biomolecular Magnetic Resonance (BMRZ), Goethe-University Frankfurt, 60438 Frankfurt, Germany
- 2Institute of Biophysics of the Czech Academy of Sciences, 612 65 Brno, Czech Republic
- 3CEITEC−Central European Institute of Technology, Masaryk University, 625 00 Brno, Czech Republic
- 4National Centre for Biomolecular Research, Faculty of Science, Masaryk University, 625 00 Brno, Czech Republic
- Corresponding authors: krepl{at}seznam.cz, woehnert{at}bio.uni-frankfurt.de
Abstract
Pseudouridine is the most frequently naturally occurring RNA modification, found in all classes of biologically functional RNAs. Compared to uridine, pseudouridine contains an additional hydrogen bond donor group and is therefore widely regarded as a structure stabilizing modification. However, the effects of pseudouridine modifications on the structure and dynamics of RNAs have so far only been investigated in a limited number of different structural contexts. Here, we introduced pseudouridine modifications into the U-turn motif and the adjacent U:U closing base pair of the neomycin-sensing riboswitch (NSR)—an extensively characterized model system for RNA structure, ligand binding, and dynamics. We show that the effects of replacing specific uridines with pseudouridines on RNA dynamics crucially depend on the exact location of the replacement site and can range from destabilizing to locally or even globally stabilizing. By using a combination of NMR spectroscopy, MD simulations and QM calculations, we rationalize the observed effects on a structural and dynamical level. Our results will help to better understand and predict the consequences of pseudouridine modifications on the structure and function of biologically important RNAs.
Keywords
INTRODUCTION
Pseudouridine (5-ribosyl-uridine, ψ) was the first modified nucleotide discovered in natural RNA (Cohn and Volkin 1951; Davis and Allen 1957) and is the most abundant RNA modification in nature. It is present in most tRNAs and ribosomal RNAs throughout all kingdoms of life (Björk et al. 1987; Ofengand and Bakin 1997) and has also been found in many other classes of functional RNAs such as spliceosomal snRNAs (Reddy and Busch 1988), the U3 snoRNA involved in ribosome biogenesis (Greenwood et al. 1996), telomerase RNA (Schwartz et al. 2014), viral RNAs (Becker et al. 1998), and long noncoding RNAs (Carlile et al. 2014). Recently, pseudouridine was also identified as an abundant epitranscriptomic mark in eukaryotic mRNAs with a putative role in the regulation of gene expression (Carlile et al. 2014; Lovejoy et al. 2014; Schwartz et al. 2014).
Pseudouridine differs from uridine with regard to the linkage of the uracil base with the ribose sugar. In pseudouridine the N-glycosidic bond between the N1 atom of uracil and the C1′ atom of the ribose is replaced with a carbon-carbon bond between the C5 atom of uracil and the C1′ of the ribose. Accordingly, pseudouridine features an additional hydrogen bond donor group—the H1N1 imino group—while the arrangement of hydrogen bond donor and acceptor groups on the Watson–Crick edge of the base is the same for both uridine and pseudouridine (Fig. 1A). The C5–C1′ carbon-carbon bond connecting the base and the ribose in pseudouridine is slightly longer (1.50 Å) than the corresponding N1-C1′ N-glycosidic bond (1.48 Å) in uridine (Rohrer and Sundaralingam 1970; Sundaralingam 1975). The bond angle involving the C6, C5, and C4 carbon atoms in pseudouridine is slightly smaller (118°) than the corresponding bond angle involving C6, N1, and C2 in uridine (122°) (Neumann et al. 1980). Together, these differences in bond lengths and bond angles lead to a preference for a syn conformation of the base in pseudouridine and an anti conformation in uridine (Nanda et al. 1974; Neumann et al. 1980). The ribose in both pseudouridine and uridine nucleosides and nucleotides is in a conformational equilibrium between the C3′ endo and the C2′ endo conformation with very similar populations (Neumann et al. 1980). However, when pseudouridine is incorporated into small single stranded RNAs, it shows a stronger preference for a C3′ endo ribose conformation, the anti conformation of the base and enhanced stacking interactions with the neighboring bases in comparison to uridine (Davis 1995; Davis et al. 1998). Pioneering structural studies by the Steitz group comparing the X-ray structures of unmodified and fully modified glutaminyl-tRNA bound to glutaminyl-tRNA synthetase (Arnez and Steitz 1994) showed that the presence of the additional H1N1 imino group as a hydrogen bond donor in pseudouridine creates a highly occupied hydration site, where a water molecule is complexed by the H1N1 imino group and the 5′-phosphate group of the pseudouridine as well as the 5′-phosphate group of the preceding nucleotide. This leads to an additional stabilization of the RNA structure in comparison to the unmodified RNA. Similar water binding sites were observed in the X-ray structures of other tRNAs as well as in structures of other functional RNAs such as the U2 snRNA branch point helix (Kim et al. 1974; Suddath et al. 1974; Moras et al. 1980; Lin and Kielkopf 2008). The presence of the highly occupied pseudouridine-dependent water binding site in the branch point helix was also confirmed in solution in a pioneering NMR study (Newby and Greenbaum 2002a,b). Taken together, the stabilizing effects of pseudouridine incorporation observed for single stranded RNAs and the presence of additional highly occupied hydration sites due to the H1N1 imino group in pseudouridine containing structured RNAs gave rise to the widely held notion that in general, pseudouridine incorporation in RNA has a structure-stabilizing effect (Carlile et al. 2014; Schwartz et al. 2014; Borchardt et al. 2020). While this has been borne out in thermodynamic and structural studies in other systems (Davis 1995; Durant and Davis 1999; Sumita et al. 2005; Kierzek et al. 2014), there is at least one report of a pseudouridine modification with a destabilizing effect located in the loop region of an RNA hairpin structure corresponding to helix 63 of the 23S rRNA from E. coli ribosomes (Meroueh et al. 2000), thus challenging the generality of this concept. However, no structural rationalization for this observation was provided.
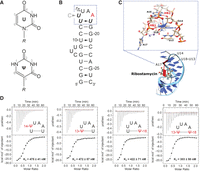
Pseudouridine modifications of the neomycin-sensing riboswitch (NSR). (A) Comparison of the chemical structures of uridine (top) and pseudouridine (bottom) and their atomic numbering schema. (R) Ribose. (B) Secondary structure of the WT NSR and nucleotide numbering. The apical loop comprising the U-turn motif (U14, U15, A16) and the closing U13:U18 base pair is highlighted by a blue frame. A17—the only nucleotide of the apical loop in contact with the ligand—is colored in red. Nucleotides that were replaced with pseudouridine in the course of this study are highlighted in bold italic. The position of the U14C mutation is also indicated. (C) Close-up of the three-dimensional structure of the apical loop of the NSR in the complex with ribostamycin. The signature interactions HB1—the hydrogen bond between the U14 imino group and the A17 5′-phosphate group, HB2—the hydrogen bond between the U14 2′-hydroxyl group and the N7 nitrogen of A16 and the lone-pair-π interaction between the Rp oxygen atom of the 5′-phosphate group of A16 and the base of U14 are highlighted. (D) Ribostamycin affinities of the four pseudouridine containing WT NSR variants U14ψ (far left), U13ψ (middle left), U18ψ (middle right) and U13ψ_U18ψ (far right). Shown are the respective ITC thermograms (top) and the resulting binding isotherms (bottom). The respective values of the dissociation constants (KD) which are the averages of three independent experiments are given as insets.
So far, investigations into the effects of pseudouridine modifications on RNA structure and stability are limited to a few model systems including tRNAs (Davis and Poulter 1991; Arnez and Steitz 1994) and tRNA fragments (Durant and Davis 1999; Yarian et al. 1999), ribosomal and spliceosomal RNA fragments (Meroueh et al. 2000; Newby and Greenbaum 2001; Sumita et al. 2005) and model duplexes with canonical and noncanonical pseudouridine containing base pairs (Kierzek et al. 2014; Deb et al. 2019). Here, we introduce the synthetic neomycin-sensing riboswitch (NSR) bound to ribostamycin as a new model system to characterize the structural and dynamic effects of pseudouridine modifications in functional RNAs (Fig. 1B).
The NSR is the smallest known biologically active riboswitch element and consists of only 27 nt (Weigand et al. 2008; Weigand et al. 2011). Upon ligand binding it is able to suppress translation initiation in Saccharomyces cerevisiae when it is incorporated into the 5′-UTR region of mRNAs. Its ligand bound structure has been solved to high resolution by NMR spectroscopy in solution (Duchardt-Ferner et al. 2010, 2016). Upon binding to ribostamycin—a neomycin analog—the riboswitch adopts a compact conformation with a ligand binding pocket in the major groove where almost all functional groups of the ligand are recognized via hydrogen bonding and electrostatic interactions to RNA bases and backbone phosphate groups. Of particular interest for the work presented here is the apical hexaloop of the NSR with a 5′-U13U14U15A16A17U18-3′ sequence which, in the presence of the ligand, folds into a U-turn structure with a U13:U18 closing base pair (Fig. 1C). However, only the base of nucleotide A17 which is a NSR-specific insertion between the U-turn motif and the U:U closing base pair is in contact with the ligand while all other nucleotides of the apical loop are not (Duchardt-Ferner et al. 2016). In agreement with the general structural consensus for this motif (Moore 1999), the U-turn structure is stabilized by two signature H-bonds. The first one involves the H3N3 imino group of U14 and the 5′-phophate group of A17 (henceforth denoted as HB1) and the second one involves the 2′-OH group of U14 and the N7 atom of the A16 base (henceforth denoted as HB2) (Fig. 1C). Furthermore, there is an anion-π interaction between the nonbridging Rp oxygen atom (OP2) of the A16 5′-phosphate group and the base of U14 as well as a potassium ion complexed by the U13, U14, and U18 O4 base oxygen atoms and the A17 phosphate group (Fig. 1C; Krepl et al. 2018). Interestingly, U14 can be replaced by C14 which, in this particular structural context, is protonated at the N3 nitrogen atom even at pH values above 8 and forms a highly stable C14+–A17 phosphate hydrogen bond (Gottstein-Schmidtke et al. 2014).
The structure and dynamics of the NSR in the ligand-bound state have been extensively characterized by NMR (Duchardt-Ferner et al. 2010, 2016). Furthermore, the system has been established as a well-behaved model system for molecular dynamics (MD) simulations, and its signature interactions were described in great detail using quantum chemical (QM) calculations (Krepl et al. 2018; Chyży et al. 2021; Zhang et al. 2021). Notably, uridine residues are involved in almost all important noncovalent interactions stabilizing its apical loop but do not interact with the ligand. The U-turn in general is a very common structural motif which is frequently found in all types of functional RNAs (Quigley and Rich 1976; Pley et al. 1994; Huang et al. 1996; Stallings and Moore 1997; Puglisi and Puglisi 1998; Gutell et al. 2000; Liu et al. 2007; Akiyama et al. 2016) and is also a common site for pseudouridine modifications. Therefore, we used the NSR bound to ribostamycin as a model system for exploring the effects of pseudouridine replacements in the context of its intricate, but well-characterized interaction network. We replaced U13 and U18 of the U:U closing base pair and U14 of the U-turn individually by pseudouridine in both the NSR-WT and the U14C-mutant and investigated the effects of these replacements on the structure, stability of its signature interactions, potassium binding and hydration with dedicated NMR-experiments and interpreted our results in the framework of extensive MD simulations and supplementary QM calculations. Interestingly, our results showed that the effects of uridine replacements by pseudouridine are highly dependent on the structural context. Depending on its structural environment pseudouridine can be either destabilizing, lead to a local stabilization or even induce a more global stabilization of the entire apical loop. Thus, our results challenge the oversimplified notion that pseudouridine in general is a stability-enhancing modification and suggest that it is necessary to take the precise structural context into account when predicting the effects and functional consequences of pseudouridine modifications in functional RNAs.
RESULTS
Pseudouridine replacements do not interfere with ligand binding and the global fold of the NSR ribostamycin complex
According to ITC experiments the three single pseudouridine replacement variants U13ψ, U14ψ, and U18ψ in the WT NSR context bound ribostamycin with dissociation constants (KD) of ∼0.5 µM; very similar to the value previously reported for the WT NSR (∼0.3 µM) under the same conditions (Fig. 1D; Duchardt-Ferner et al. 2016). Likewise, the KD of the variant with simultaneous U13ψ and U18ψ replacements (∼0.3 µM) is also very close to the one for the WT NSR (Fig. 1D). The ribostamycin KDs of the U13ψ and U18ψ replacement variants in the context of the U14C NSR are also similar to the KD of the unmodified U14C NSR (Supplemental Fig. S1). Therefore, the uridine replacements with pseudouridine did not interfere with ligand binding.
The well-resolved imino proton regions of the WT and U14C NSR 1D-1H-NMR spectra bound to ribostamycin (Fig. 2A, top; Supplemental Fig. S2A, top) feature signals for the imino protons of U13, U14/C14+, and U18. Their chemical shifts correspond to their hydrogen bonding status in the asymmetric U13:U18 base pair with two direct hydrogen bonds between the two H3N3 imino groups and their C2 and C4 carbonyl groups, respectively, and the hydrogen bonding interaction between the U14/C14+ H3N3 imino groups and the A17 phosphate group (HB1). The imino proton spectra of the pseudouridine variants bound to ribostamycin are similar to the spectra of the corresponding unmodified RNAs (Fig. 2A; Supplemental Fig. S2A) confirming that the fold of these RNAs is not altered by the pseudouridine replacements. However, significant chemical shift differences are observed for the signals of the imino protons at the replacement sites as well as for adjacent nucleotides, indicating local structural and dynamical effects of the pseudouridine substitutions as described in detail below. Furthermore, the imino proton spectra of all pseudouridine variants feature additional signals for the H1N1 imino groups of the pseudouridines with widely divergent chemical shifts. Sequential assignments for all imino protons in all variants were confirmed in 2D-1H,1H-NOESY and if necessary 1H,13C-HSQC-experiments for aromatic CH-groups at natural abundance (Supplemental Figs. S3–S5). Pseudouridine H1 and H3 imino protons were distinguished from each other by the presence of a strong NOE between the H1 imino proton and the H6 proton of the same base (Supplemental Figs. S3–S5). Smaller chemical shift differences far away from the apical loop region (G1, G2, and U26) likely reflect the chemical differences between the unmodified RNAs produced by in vitro transcription and hammerhead ribozyme cleavage featuring a triphosphate group at their 5′-end and a 2′–3′ cyclic phosphate group at their 3′-end and the chemically synthesized pseudouridine variant RNAs featuring hydroxyl groups at both their 5′- and 3′-end. Further analysis of NOESY spectra showed that in all variants the pseudouridines adopted an anti conformation and used their Watson–Crick faces with the H3N3 imino group for intramolecular hydrogen bonding interactions.
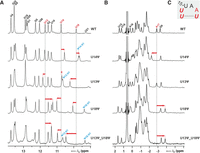
Effects of individual pseudouridine replacements on chemical shifts in the WT NSR bound to ribostamycin. Comparison of the imino proton 1H (A) and 31P (B) 1D NMR-spectra of the WT-NSR (top), the U14ψ variant (second row from top), the U13ψ variant (middle), the U18ψ variant (second row from bottom) and the U13ψ/U18ψ double replacement variant (bottom). The relevant signal assignments are given in the WT spectra (top). Assignments for pseudouridine H1 imino protons are given in blue. Relevant chemical shift differences between the WT and the pseudouridine containing variants are highlighted by red arrows. (C) Loop sequence of the WT NSR. Nucleotides that were replaced with pseudouridine are highlighted in bold italic. Nucleotides with relevant chemical shift differences in A and B are colored red.
Another marker for the folding state of the NSR is provided by its 1D-31P-NMR spectrum. The 31P-NMR spectra of the WT NSR (Fig. 2B, top) and the U14C NSR bound to ribostamycin (Supplemental Fig. S2B, top) feature a number of signals for nucleotides in the apical loop outside the chemical shift range for the bulk of the 31P-NMR signals (∼0 to −2 ppm) from nucleotides in A-form helical parts of the molecule. This is due to the presence of unusual backbone torsion angles and hydrogen bonding interactions in the apical loop. The 31P-NMR spectra of the pseudouridine replacement variants yielded similar unusual 31P chemical shifts directly demonstrating the presence of a backbone conformation close to the WT and the U14C NSR, respectively (Fig. 2B; Supplemental Fig. S2B).
Imino proton and 31P chemical shifts suggest the modulation of hydrogen bonding by pseudouridine replacements
The imino proton chemical shifts in nucleic acids can be related not only to base pairing or hydrogen bonding type (Fürtig et al. 2003) but also to differences in covalent bond length between the donor group heavy atom and the hydrogen as well as to the hydrogen bond length (Barfield et al. 2001). Theoretical studies for nucleic acids (Barfield et al. 2001) and experimental data for many other systems (Wagner et al. 1983; Jeffrey and Yeon 1986; Herschlag and Pinney 2018) suggest that “downfield” (to higher ppm) proton chemical shifts are generally correlated with shorter and stronger hydrogen bonds whereas “upfield” (to lower ppm) chemical shifts correspond to longer hydrogen bond lengths and thereby “weaker” less stable hydrogen bonds. Thus, given the very similar chemical nature of uridine and pseudouridine and the conserved overall structure of the RNA, the observed chemical shift differences for the H3 imino protons in the WT NSR and the pseudouridine replacement variants can be attributed mainly to differences in hydrogen bond and base pairing stability. However, other subtle structural differences for example, in stacking interactions and ion binding sites might also contribute to the chemical shift differences between the different RNAs.
For the U14ψ variant of the WT NSR, where the pseudouridine H3N3 imino group is directly involved in a hydrogen bond with the A17 phosphate group (HB1), we observe a ∼0.25 ppm upfield shift of the ψ14 H3 imino proton compared to the U14 H3 of the WT RNA (Fig. 2A), pointing to a weakening of the HB1 hydrogen bond. In addition, the U18 H3 proton shifts upfield by ∼0.2 ppm suggesting that also the hydrogen bonding interaction between the U18 imino proton and the U13 O2 in the U13:U18 base pair is destabilized (Fig. 2A). In contrast, the U13 imino proton shift is virtually identical between the WT and the U14ψ variant. Furthermore, for this variant we observe a significant downfield shift (∼0.7 ppm) of the A17 5′-phosphate 31P resonance (Fig. 2B). While no quantitative experimentally derived relationships between 31P chemical shifts in RNA and hydrogen bonding exist, we have previously shown by NMR and theoretical studies (Gottstein-Schmidtke et al. 2014; Krepl et al. 2018) that the replacement of the uridine imino group in HB1 with the imino group of a protonated C resulted in a significantly shorter and stronger hydrogen bond which is accompanied by a significant upfield chemical shift for the 31P signal of the hydrogen bond acceptor—the A17 phosphate group. Thus, the observed 31P chemical shift differences for A17 between the WT and the U14ψ variant is also in agreement with a weakening of the HB1 hydrogen bonding interaction in the U-turn. Taken together, the chemical shift data imply that the U14ψ replacement not only destabilizes the HB1 interaction of the U-turn but also the adjacent U13:U18 base pair.
For the U13ψ variants of the WT and the U14C NSR, we find that the chemical shifts of the U14/C14+ imino protons as well as the A17 phosphate 31P chemical shifts are very similar to the unmodified RNAs (Fig. 2; Supplemental Fig. S2). In contrast, the H3 imino proton signals of U18 and ψ13 are shifted downfield by ∼0.4 and ∼0.2 ppm, respectively, in both systems. Thus, the chemical shift changes for U13 and U18 suggest a higher stability of both hydrogen bonds in the ψ13:U18 base pair compared to the U13:U18 base pair in the unmodified RNAs.
The replacement of U18 with ψ in the WT NSR significantly impacts the chemical shifts of the H3 imino protons of U13, U14 and U18 as well as the 31P shift of the A17 phosphate group (Fig. 2). The imino proton of U14 shifts downfield by ∼0.25 ppm compared to the WT whereas the 31P signal of A17 shifts upfield by ∼0.9 ppm. These shift changes suggest a stabilization of the HB1 signature hydrogen bonding interaction of the U-turn in this variant. Furthermore, the ψ18 H3 imino proton is shifted downfield by ∼0.2 ppm compared to the U18 H3 in the WT indicating a more stable hydrogen bonding interaction between the ψ18 imino proton and the U13 O2 compared to the equivalent hydrogen bond in the U13:U18 base pair of the WT. In contrast, the imino proton of U13 is shifted upfield by ∼0.4 ppm, indicating a less stable hydrogen bond between the U13 imino proton and the ψ18 O2 in comparison to the equivalent U13 imino proton—U18 O4 hydrogen bond in the WT. The effects of the U18ψ replacement on the imino proton and 31P chemical shifts in the U14C NSR are very similar to those observed for the WT NSR (Supplemental Fig. S2).
The chemical shifts of the pseudouridine H1N1 imino group protons differ widely among the pseudouridine variants (Fig. 2; Supplemental Fig. S2). A visual inspection of static structural models based on the NMR structure of the NSR bound to ribostamycin assuming no structural changes reveals that these imino groups would be solvent exposed in both the U14ψ and the U13ψ variants, and yet their proton chemical shifts differ significantly with ∼9.7 and ∼10.6 ppm. The H1 imino proton of the pseudouridine in the U18ψ variants is buried in the interior of the RNA structure and located in a distance of ∼2.6 Å to the bridging oxygen (O5′) of the A17 phosphate group. Its chemical shift (∼9.4 ppm) is the most upfield H1 imino proton chemical shift of all variants. However, an interpretation of the H1N1 imino group chemical shifts in structural terms is not possible at this stage (see below).
We also tested the effects of a simultaneous replacement of U13 and U18 with ψ on the imino proton and 31P spectra of the WT NSR. Overall, the chemical shift differences to the WT RNA correspond closely to the sum of the effects of the individual replacements (Fig. 2, bottom) as expected when the structural and dynamical effects of the two pseudouridine replacements are additive.
Scalar couplings across hydrogen bonds suggest changes in hydrogen bond lengths for the signature HB1 interaction upon pseudouridine replacements
A class of NMR-parameters that are directly related to hydrogen bond lengths and angles are scalar couplings across hydrogen bonds (Grzesiek et al. 2004). Such scalar couplings are in principle present between the hydrogen atoms and NMR active nuclei of the hydrogen bond acceptor groups or between NMR active heavy nuclei of the donor and acceptor groups. Due to the limits of chemical RNA synthesis 15N, 13C-labeled pseudouridine or uridine could not be introduced with the required site specificity in the NSR. Thus, only the scalar coupling across the HB1 hydrogen bond between the imino proton of U14 and the 31P nucleus of the phosphate group of A17 (2hJHP) can be readily measured in this system. In previous work (Gottstein-Schmidtke et al. 2014; Krepl et al. 2018), we established that a shorter average hydrogen bond distance is related to a larger 2hJHP scalar coupling constant in this kind of hydrogen bond. Using previously established methods (Duchardt-Ferner and Wöhnert 2017), we therefore quantified the 2hJHP scalar coupling constants across the HB1 hydrogen bond for the pseudouridine replacement variants (Fig. 3). For the U14ψ variant of the WT NSR we found a 2hJHP scalar coupling constant of 2.60 Hz which is 0.22 Hz smaller than the one measured for the unmodified WT NSR in agreement with a longer average hydrogen bond length and a weaker HB1 interaction. For the HB1 interaction in U13ψ we measured a 2hJHP scalar coupling constant of 2.59 Hz which is again smaller than the one measured for the WT NSR also suggesting a weakened HB1 interaction in this system. For U18ψ the 2hJHP scalar coupling constant (3.03 Hz) across the HB1 hydrogen is significantly larger than in the WT NSR. This suggests a shorter average hydrogen bond length and a strengthened HB1 interaction in U18ψ in comparison to the WT in agreement with the chemical shift difference observed for the U14 imino proton in these two RNAs. The largest 2hJHP scalar coupling constant across the HB1 interaction (3.09 Hz) is measured for the U13ψ/U18ψ double replacement variant of the WT NSR.
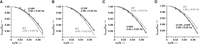
Determination of the 2hJHP scalar coupling constants across the HB1 hydrogen bond in quantitative H,P-experiments for the U14ψ variant (A), the U13ψ variant (B), the U18ψ variant (C) and the U13ψ_U18ψ variant (D) of the WT NSR. For comparison, the corresponding curve for the unmodified WT NSR is given in gray in each panel.
Water exchange rates and hydrogen bond dissociation free-energy differences reveal context-dependent stabilizing and destabilizing effects of pseudouridine replacements
Another NMR parameter directly related to the stability of intramolecular hydrogen bonding interactions is the exchange rate of the imino protons with the protons of the bulk water. This chemical exchange can only occur when the hydrogen bond involving the imino proton is broken and the imino proton becomes exposed to the solvent. Thus, this rate is a reporter of the opening frequency of intramolecular hydrogen bonding interactions and therefore also of hydrogen bonding strength. We measured the temperature dependence of these exchange rates in the different pseudouridine replacement variants. A steeper increase of the exchange rates with rising temperatures agrees with a stronger increase in the population of the open “broken” state of the respective hydrogen bonding interaction and therefore a weaker hydrogen bond. The temperature dependence of the exchange rate can also be directly used to calculate the free-energy difference (ΔG) between the hydrogen bonded “closed” state and an exchange-prone “open” state at a given temperature (Rinnenthal et al. 2010). The comparison between the ΔG values for a hydrogen bonding interaction in the unmodified NSR and the modified NSR variants then yields quantitative differences (ΔΔG) in the stabilities of these interactions.
For the U14ψ variant of the WT NSR we find a slightly increased exchange rate for the ψ14 imino proton in the HB1 hydrogen bond at higher temperature and significantly increased exchange rates for the imino protons of U13 and U18 (Supplemental Fig. S6) in comparison with the WT NSR in agreement with an overall destabilization of the apical loop region. At 25°C, this corresponds to a destabilization of the HB1 hydrogen bond by ∼0.05 kcal/mol, a destabilization of the U13 NH–U18 O4 hydrogen bond by ∼0.3 kcal/mol and a destabilization of the U18 NH–U13 O2 hydrogen bond by ∼1.0 kcal/mol (Fig. 4).
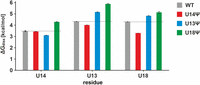
Dissociation free-energy ΔGdiss at T = 25°C for the signature HB1 interaction (left) and the hydrogen bonds involving the U/ψ13 H3 and U/ψ18 H3 protons (middle and right, respectively) in the WT NSR (gray) and the variants U14ψ (red), U13ψ (blue), and U18ψ (green). The dashed lines correspond to ΔGdiss for the interactions in the WT NSR.
For the U13ψ variant of the WT NSR, the temperature dependence of the exchange rate of the U14 imino proton is similar to the unmodified NSR while the absolute values of the exchange rate are slightly higher for the variant (Supplemental Fig. S7) in agreement with a weaker HB1 hydrogen bond in the U-turn. In contrast, the exchange rates of both the ψ13 and the U18 imino protons increase significantly slower with rising temperature than those of the equivalent protons in the WT NSR (Supplemental Fig. S7). At 25°C, the HB1 hydrogen bond is ∼0.4 kcal/mol less stable while the U13 NH–U18 O4 and the U18 NH–U13 O2 hydrogen bonds are stabilized by ∼0.8 kcal/mol and ∼0.5 kcal/mol, respectively, compared to the WT (Fig. 4).
Remarkably, for the U18ψ variant of the WT NSR (Supplemental Fig. S8), the exchange rates for all three relevant imino protons increase significantly slower with temperature than those for the WT variant. This translates into a stabilization of the HB1 hydrogen bond by ∼0.8 kcal/mol, the U13NH–U18 O4 hydrogen bond by ∼1.5 kcal/mol and the U18 NH–U13 O2 hydrogen bond by ∼0.8 kcal/mol at 25°C compared to the WT NSR (Fig. 4). Thus, in the U18ψ variant of the WT NSR the pseudouridine replacement not only leads to a local stabilization of the closing base pair but an overall stabilization of the entire apical loop.
The replacements of U13 and U18 with ψ in the context of the U14C NSR lead to similar effects on the imino proton exchange rates and comparable changes of the hydrogen bond dissociation free energies as those observed in the context of the WT NSR (Supplemental Fig. S9).
Overall, the measured imino proton and 31P chemical shifts, 2hJHP scalar couplings and the temperature dependence of the imino proton exchange rates (Supplemental Table 1) paint a qualitatively consistent picture on the diverse effects of the pseudouridine replacements at the three different positions. According to all measured parameters, the U14ψ replacement leads to a simultaneous destabilization of the U-turn and the U13:U18 closing base pair in comparison to the WT NSR. For the U13ψ variants, all parameters agree with a weakened HB1 signature interaction in the U-turn while the closing base pair is stabilized by the nucleotide modification. For the U18ψ variants, all parameters with the exception of the U13 imino proton chemical shift change agree with an overall stabilization of the entire apical loop in comparison with the WT NSR.
Pseudouridine replacements have no influence on potassium ion binding
The WT NSR contains a potassium ion binding site stabilizing the U-turn and the U:U closing base pair involving the C4 carbonyl groups of U13, U14, and U18 as well as the phosphate group of A17 (Krepl et al. 2018). This potassium binding site was mapped by NMR-based KCl-titrations of the WT NSR in BisTris buffer which also allowed the determination of the potassium ion affinity at this site. The WT NSR binds potassium ions at this site with a KD of ∼11 mM and at a second site at the G2:U26 wobble base pair at the end of the helix with a KD of ∼3 mM (Krepl et al. 2018). The addition of KCl to the U14ψ, U18ψ, and U13ψ variants in BisTris buffer led to chemical shift changes of the G2 and U26 imino proton signals and the signals for U13, U14, and U18 of the apical loop (Supplemental Figs. S10–S12) similar to the WT NSR. Imino proton spectra recorded at different temperatures in the presence and absence of potassium ions showed that the apical loops of the U14ψ, U18ψ, and U13ψ variants are stabilized by potassium ion binding similar to the WT NSR (Supplemental Figs. S10–S12). Plotting the chemical shift changes against the KCl concentration yielded dissociation constants. The U-turn of the U14ψ mutant binds K+ with a KD of ∼21 mM; the U18ψ and U13ψ mutants bind K+ with a KD of ∼14 mM and ∼9 mM, respectively. Thus, the affinity for potassium ion binding in the pseudouridine variants is similar to the wild-type RNA and not significantly influenced by the pseudouridine replacements.
Not all pseudouridine replacements create stable water binding sites
The presence of pseudouridine modifications has been associated with an increase in the stability of RNA helices due to the creation of an additional stable water binding site involving the H1N1 imino group and the phosphate group of the pseudouridine and the phosphate group of its preceding nucleotide, respectively, which form hydrogen bonds to the water molecule. The presence of a bound water molecule in the vicinity of the pseudouridine H1N1 imino group can be detected by CLEANEX-PM experiments, in which NOEs between RNA protons and water protons are measured (Newby and Greenbaum 2002a). A comparison of the CLEANEX-PM spectra of the WT NSR and its three single pseudouridine replacement variants showed that the strongest NOE by far between an imino group and water is present for the H1N1 imino group of the pseudouridine in the U13ψ variant. Thus, the presence of pseudouridine at position 13 apparently creates a stable water binding site in vicinity to the H1N1 imino group of ψ13 suggesting an explanation for the observed local stabilization of the U13:U18 base pair. In the U14ψ variant, the NOE between the H1N1 imino group of the pseudouridine and water is significantly smaller than the one observed for the U13ψ variant suggesting a less stable pseudouridine:water interaction. For the H1N1 imino group of the pseudouridine in the U18ψ variant an NOE with water is completely absent suggesting that this proton is effectively shielded from interactions with the solvent (Fig. 5). The presence of a strong hydrogen bond interaction between the H1N1 imino group of ψ13 in the U13ψ variant and the oxygen atom of a stably bound water molecule also offers a rationalization for the unusual downfield chemical shift of the ψ13 H1 imino proton (∼10.6 ppm) in comparison to the H1 imino proton chemical shifts in the other pseudouridine variants (<10.0 ppm) where such a strong interaction is not present. In line with such an interpretation are the H1 imino proton chemical shifts of pseudouridines in tRNAs and in A-form helical duplexes where structurally similar water binding sites are present that are also >10.2 ppm (Davis and Poulter 1991; Durant and Davis 1999; Yarian et al. 1999; Kierzek et al. 2014).
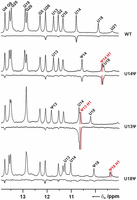
Interactions of pseudouridine H1N1 imino groups with bound water. Comparison of the 1D imino proton spectra (positive signals) and the corresponding CLEANEX-PM spectra (negative signals) for the unmodified WT NSR (top), the U14ψ variant (second row from top), the U13ψ variant (second row from bottom), and the U18ψ variant (bottom) of the WT NSR. Signal positions of the respective pseudouridine H1 imino protons are indicated in red lettering, other relevant signal assignments are given in black. Significant NOEs between H1 imino protons and water are highlighted with light red boxes.
MD simulations
To complement the NMR data and gain additional insights into the consequences of the individual pseudouridine replacements on the structure and dynamics of the NSR bound to ribostamycin at atomic resolution we carried out a series of MD simulations (Table 1). The MD simulations were accompanied by supplementary QM and QM/MM computations. In full agreement with the NMR results, the global fold of the NSR and its ability to bind ribostamycin are not affected in any of the pseudouridine variants and the pseudouridine bases always maintain their anti conformation. It should be noted that the force-field parameters used in our simulations favor the anti conformation of pseudouridine (Deb et al. 2014, 2016). However, the simulations revealed specific differences in the dynamics and the structural stability of the intramolecular interactions which define the U-turn motif and its closing U:U base pair between the different variants. These are discussed in detail below.
List of simulations
U14ψ
MD revealed that the U14ψ substitution increases the dynamics of the ribose-phosphate backbone for the entire U-turn loop and the loop-closing U13:U18 base pair compared to the WT (Fig. 6, left). Base dynamics was enhanced significantly for A17 and to a lesser degree for U13 (Fig. 6, right). However, no loss of intermolecular contacts with the ribostamycin was observed as the increased dynamics remained localized to the U-turn region, at least on our simulation timescale. The population of simulation frames in which the critical HB1 interaction of the U-turn was temporarily disrupted increased from ∼10% in the WT NSR to ∼35% in U14ψ (Table 2). For the WT it was suggested earlier that these reversible disruptions might represent genuine thermal fluctuations of the NSR on the microsecond timescale (Krepl et al. 2018) that are not directly captured by the NMR structural ensemble. This suggestion is now substantiated by the U14ψ variant since even the threefold increase in the population with a disrupted HB1 interaction does not translate into a violation of the ensemble-averaged NOE distances (Table 2; Duchardt-Ferner et al. 2016). Simulations of the U14ψ variant also showed altered H-bonding populations of the U13:U18 loop-closing base pair. Earlier it was shown for the WT NSR (Krepl et al. 2018) that this base pair can fluctuate between a major conformation with direct U13(H3)–U18(O4) and U18(H3)–U13(O2) hydrogen bonds and a minor conformation where the U13(H3)–U18(O4) interaction is direct and the U18(H3)–U13(O2) one is water mediated. For the U14ψ variant, the minor conformation was observed in 22% of simulation frames which is a significant increase compared to the 7% observed for the WT. The increased population of the minor conformation of the U13:U18 base pair for the U14ψ variant is in excellent agreement with the NMR chemical shift differences of the imino protons which indicated a destabilization of the U18(H3)–U13(O2) but not the U13(H3)–U18(O4) hydrogen bond compared to the WT. Thus, in agreement with the NMR measurements, the MD simulations suggest a destabilizing effect of the U14ψ replacement which spreads through the U-turn and its closing U:U base pair.

Average heavy atom per residue RMSF for the RNA backbone (left) and the RNA bases (right) in MD simulations of selected ψ replacement variants of the WT NSR. The inset on the right highlights the region corresponding to the U-turn loop. RNA backbone refers to the atoms P, OP1, OP2, O5′, C5′, C4′, C3′, and O3′ of each nucleotide. Average structures of each MD simulation ensemble were used as references.
Statistical analysis of the U-turn signature interactions and their overall stability in MD simulations
The structural basis for this effect revealed by the simulations is the ability of the ψ14 H1N1 imino group to form either water-mediated or direct hydrogen bonds (41% and 3% of the simulation time, respectively) with the phosphate groups of either ψ14 or U13 (Fig. 7A) that cannot be formed by the WT RNA. Due to overall structural constraints for the U-turn loop geometry, these hydrogen bonds involving ψ14 H1N1 directly compete with the HB1 interaction and are therefore responsible for the larger incidence of its temporary loss in U14ψ compared to the WT (Table 2). Even when the HB1 interaction was present, the ψ14 H1N1 imino group still often formed simultaneous water-bridged interactions with the phosphate groups of either ψ14 or U13 (Fig. 7A). This reduced the average N1–OP2 distance in ψ14 to ∼4.7 Å compared to the equivalent C5–OP2 distance of ∼5.3 Å for U14 in the WT NSR. It also slightly increased the average donor-acceptor distance for the HB1 interaction (Table 2) compared to the WT. Although all these differences are not large, their combination apparently translates into a notable destabilization of the highly optimized U-turn loop structure, as evidenced by the more frequent and longer disruptions of the HB1 in the U14ψ variant.
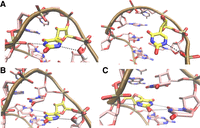
Structural consequences of pseudouridine modifications as seen in MD trajectories. (A) In addition to the signature HB1 interaction of the U-turn (left), ψ14 is able to reversibly form alternative interactions via its H1N1 imino group, which compete with, or can temporarily disrupt HB1 (right). (B) In addition to the native U13:U18 base pair, ψ13 formed a major water-bridge interaction via its H1N1 imino group. (C) The H1N1 imino group of ψ18 forms an additional H-bond with the A17 O5′ bridging oxygen atom, increasing stability of the U-turn motif and all its signature interactions.
QM calculations of small model systems representing the isolated HB1 interaction (Supplemental Fig. 13) reveal a comparable extension of the H3N3 covalent bond and a similar H-bond distance for both pseudouridine and uridine, even when an additional water molecule interacts with the H1N1 imino group (Table 3). The calculations thus do not suggest any direct electronic structure effect of the pseudouridine substitution on the HB1 interaction. In other words, we propose that the destabilization of the HB1 signature interaction by the U14ψ replacement is an indirect effect related to the competing hydrogen bonding interactions between the H1N1 imino and neighboring phosphate groups that are not compatible with the geometry of the U-turn. This results in increased backbone dynamics and enhanced conformational exchange for the loop-closing U13:U18 base pair, thereby rationalizing the experimental observations described above.
QM optimized small model systems representing the HB1 signature interaction in uridine- and pseudouridine-based NSR systems and reference QM optimizations of uridine and pseudouridine basesa
Surprisingly, the HB2 A16(N7)-U14(2′-OH) signature H-bond is significantly more populated in U14ψ than in the WT NSR simulations. In all previous simulations of NSR systems, the HB2 interaction was generally less populated than the HB1 interaction. Furthermore, there is a general trend that when the HB1 interaction is tighter, the HB2 population decreases (Krepl et al. 2018; Zhang et al. 2021). We suggest that there is a competition between the HB1 and the HB2 interaction reflecting genuine geometrical constraints inherent to the extraordinarily stiff U-turn motif. This leads to an anticorrelation of their strength. Unfortunately, we cannot address the U14ψ influence on HB2 experimentally by NMR-spectroscopy since this would require simultaneous site-specific isotope labeling with 15N–adenine nucleotides and pseudouridine during chemical RNA synthesis. We note that the force field might underestimate the population of the HB2 interaction as the U14 2′-OH group reversibly fluctuates toward spurious hydrogen bonds with U15 O5′ (Krepl et al. 2018; Zhang et al. 2021). The force field apparently over- and understabilizes the spurious U15 O5′ and native HB2 interaction, respectively (Krepl et al. 2018). However, the force field should be accurate enough to capture the above noted anticorrelation between the HB1 and the HB2 interaction. Finally, the anion-π interaction between A16 OP2 and the base of nucleotide 14 is unaffected by the U14ψ replacement (Table 2).
U13ψ
The dynamics of the U13ψ variant of the WT NSR during MD simulations was very similar to the unmodified RNA (Fig. 6). The geometry and stability of the U-turn signature interactions were also very similar (Table 2). The only notable difference was the dynamics of the U13:U18 closing base pair where the population of the major conformation with two direct H-bonds further increased compared to the wild-type (Table 2). Again, this excellently agrees with the NMR data, which indicate a stabilization of both hydrogen bonds in the U13:U18 base pair compared to the WT. The simulations also revealed a highly populated (76%) water-bridge between the ψ13 H1N1 imino group and either the ψ13 or C12 phosphate group or both (Fig. 7), in agreement with the CLEANEX-NMR experiments (Fig. 5). In contrast to what was observed for the U14ψ variant, the presence of the water-bridge involving the ψ13 H1N1 imino group does not change the N1–OP2 distance (4.7 Å) in ψ13 compared with the equivalent C5–OP2 distance of U13 in the WT and therefore does not alter the position or the conformation of this nucleotide.
U18ψ
The U18ψ replacement attenuates the dynamics of both the U-turn and the U13:U18 closing base pair compared to the WT NSR (Fig. 6). Visual inspection of the trajectories showed that the H1N1 imino group of ψ18 points towards the O5′ of the A17 phosphate group (Fig. 7C). The average H1–O5′ distance along the MD trajectory is 2.26 ± 0.26 Å with an interatomic N1–H1–O5′ angle of 141 ± 11°. This corresponds to an H-bond with a nonideal geometry. For comparison, the HB1 interaction has an average hydrogen–acceptor distance and H-bond angle of 1.95 ± 0.20 Å and 161 ± 10°, respectively. The relatively long interatomic distance and especially the deviation from linearity are very likely responsible for our inability to directly detect the H1–O5′ interaction by NMR in cross-hydrogen-bond long-range H,P correlation experiments (Duchardt-Ferner et al. 2011; Duchardt-Ferner and Wöhnert 2017) in line with expectations based on previous theoretical calculations (Del Bene et al. 2002). Notably, the formation of this additional hydrogen bonding interaction in U18ψ requires virtually no geometrical changes within the U-turn since the C5–H5 group of U18 was already suitably positioned in the WT system. Thus, it is seamlessly accommodated into the structural framework of the NSR U-turn without any competition with the other signature interactions. Despite its nonideal geometry, the presence of the weak additional hydrogen bond between the ψ18 H1N1 imino group and the O5′ atom of the A17 phosphate group has substantial consequences for the dynamics of the entire U-turn loop. On one hand, the conformational fluctuations were reduced (Fig. 6) and the conformational substates of the U-turn with a temporarily disrupted HB1 interaction were almost entirely eliminated from the simulation ensemble (1% in U18ψ compared to 11% in WT and 35% in U14ψ; see Table 2). On the other hand, compared to the WT NSR, the average donor-acceptor distance of the HB1 interaction is slightly shorter, the HB2 interaction is more populated, and the anion-π interaction more compact in U18ψ (Table 2). The major base pairing arrangement of the U13:ψ18 pair with two direct H-bonds becomes almost exclusively populated (Table 2). There was also a slight alteration of the base pair geometry in comparison with the WT with the interatomic distance of the ψ18(H3)–U13(O2) hydrogen bond decreased and U13(H3)–ψ18(O4) hydrogen bond increased by 0.05 and 0.03 Å, respectively. Although being a small change, it could help to rationalize the observed downfield and upfield NMR chemical shifts of the ψ18 and U13 imino groups, respectively, in the U18ψ variant.
We also note that the potassium ion binding site in the vicinity of the U14(O4) atom was slightly altered in the presence of ψ18 (Supplemental Fig. S14). Namely, the interaction between the potassium ion and the A17(OP2) and ψ18(O4) atoms was strengthened compared to the wild-type. This very minor restructuring of the ion binding site did not change the average residency time of the ion. However, it nicely demonstrates that the U18ψ replacement influences all signature interactions of the U-turn, including those facilitated by the solvent (Krepl et al. 2018). The other tested pseudouridine replacements had only a negligible effect on this potassium binding site.
Other simulated systems
Simulations of the NSR with simultaneous U13ψ and U18ψ replacements showed a behavior corresponding to a linear combination of the effects exhibited by both modifications individually, which agrees with the NMR data. Likewise, the results observed for pseudouridine replacements in the context of the U14C NSR follow the same trends as those in the WT NSR system (Table 2).
QM calculations and MM free-energy calculations suggest the H1N1 imino group hydration provides no intrinsic stabilization for the pseudouridine systems
In the earlier literature, the stabilizing effect of pseudouridine substitutions in various RNAs was primarily attributed to the water-bridge facilitated by the presence of the ψ(H1N1) hydrogen bonding donor group. Our data suggest that this observation is likely correct for the most explored classical A-form RNA helix where the interatomic distance between the U(C5) atom and the backbone phosphate oxygen in the unmodified RNA is ∼4.9 Å. A similar distance is observed also for U13 in the WT-NSR, allowing an overall stabilizing effect of the U13ψ replacement by enabling water-bridge formation without additional structural adaptations. However, the same is not universally true for all RNA structural motifs as shown by the U14ψ NSR variant where the water-bridge does not fit into the highly optimized and stiff structure of the U-turn loop, leading to its overall destabilization.
To further explore the intrinsic structural role of the H1N1 hydration in pseudouridine replacements, we performed high-level QM optimizations of the U and ψ models engaged in interaction with a phosphate group as seen for the HB1 interaction in the NSR (see Materials and Methods). These calculations revealed very similar geometries of the HB1 interaction with both bases as well as a negligible influence of the H1N1 donor hydration (Table 3). In other words, we detected no significant electronic structure effect on the H3N3 imino group—phosphate interaction attributable to the H1N1 donor hydration in pseudouridine.
We then performed MD thermodynamic integration calculations of the free energy of solvation of the N1-methyluracil and thymine models representing U and ψ, respectively (see Materials and Methods). The calculations predicted almost identical free energies of solvation (−14.0 ± 0.2 and −13.9 ± 0.1 kcal/mol for U and ψ, respectively); the former agreeing with the computational data reported earlier (Miller and Kollman 1996), despite the ψ possessing an additional H-bond donor group. We suggest that the enthalpy gain from the ψ(N1) hydration is cancelled by the increased order and therefore lower entropy of the solvent, leading to no difference in the hydration free energy.
DISCUSSION
Our combined NMR and MD results show that the replacement of uridine nucleotides with pseudouridine can have divergent effects that depend on the exact structural context of the replacement site. These effects range from a simultaneous destabilization of the U-turn motif and its adjacent U:U mismatch base pair in the U14ψ variant to the local stabilization of the U:U mismatch base pair accompanied by a destabilization of the U-turn HB1 signature interaction observed for the U13ψ variant to a more global stabilizing effect on the U-turn structure and the U:U base pair in the U18ψ variant. Both the stabilizing and the destabilizing effects can be explained by the presence of the additional hydrogen bond donor H1N1 imino group in pseudouridine. In the U14ψ variant, the additional imino group enables access to alternative conformational states of the U-turn not present in the unmodified RNA. These states are stabilized by hydrogen bonds involving the H1N1 imino group that compete with the canonical hydrogen bonding pattern of the U-turn motif, thereby increasing its dynamics. In the U13ψ variant, the U13:U18 base pair is locally stabilized by the presence of a stable hydration site coordinated by the ψ13 H1N1 imino group as well as the ψ13 and/or the C12 phosphate groups that is structurally prearranged already in the unmodified RNA but not occupied due to the absence of the H1N1 imino group in uridine. The geometry of this hydration site strongly resembles the one described previously in for example, tRNA and the U2 snRNA branch point helix (Arnez and Steitz 1994; Lin and Kielkopf 2008). In the U18ψ variant, the entire U-turn motif is globally stabilized by formation of an additional hydrogen bond, albeit with nonideal geometry, between the H1N1 imino group of the pseudouridine and the O5′ of the A17 phosphate group. This rigidifies the RNA backbone without causing structural perturbations in comparison to the structure of the unmodified RNA. While the presence of the H1N1 imino group in pseudouridine enables hydrogen bonding interactions not possible with uridine, our QM calculations on simple model systems show that the hydrogen bonding properties of the H3N3 imino group are very similar between pseudouridine and uridine, even when the H1N1 imino group of pseudouridine is involved in additional hydrogen bonding interactions. Thus, for example, the weakening of the HB1 signature interaction of the U-turn in the U14ψ variant is solely due to the stabilization of alternative structures as described above and not due to an intrinsic change of the H3N3 imino group hydrogen bonding properties induced by the presence of the H1N1 imino group and its additional interactions. Furthermore, the solvation free energies of pseudouridine and uridine are very similar according to our MD thermodynamic integration calculations, excluding general differences in hydration as a common source for the structural and dynamical effects of pseudouridine substitutions.
The destabilizing effect of the U14ψ substitution on the HB1 interaction in the U-turn might appear as a particularly surprising finding from our work since a structurally equivalent ψ (ψ55) involved in the HB1 interaction of a U-turn is found in the TψC-loop of virtually all tRNAs. Studies comparing fully modified tRNA and unmodified tRNA-transcripts usually find a stabilizing effect for tRNA modifications (Derrick and Horowitz 1993; Arnez and Steitz 1994; Serebrov et al. 1998; Vermeulen et al. 2005; Biedenbänder et al. 2022). However, the U-turns in the NSR and the TψC-loop of tRNAs differ significantly in their structural context. Namely, the U-turn in the NSR is closed by the U13:U18 base pair whereas the one in tRNA is closed by the rT54:A58 reversed Hoogsteen base pair. The larger size of the latter enforces a backbone geometry with an almost ideal geometry for a stable hydration site with N1–OP2 distances of ∼4.5 and 4.8 to the ψ55 and rT54 phosphate groups, respectively (PDB: 1ehz, Shi and Moore 2000). This is not the case in the NSR where the formation of a stable water-bridge between the H1N1 imino group and the phosphate groups of ψ14 and U13 is hindered by N1-OP2 distances that are larger than ideal, leading to backbone geometries that disrupt the U-turn. This suggests that pseudouridine replacements might only have a stabilizing effect due to H1N1 hydration when the unmodified RNA already has a structure compatible with hydration as for example, in A-form helices or as observed here for the C12–U13 segment in the NSR. This is supported by the observation that in the X-ray structure of an unmodified tRNA transcript (PDB: 3l0u, Byrne et al. 2010) the distances between the C5 atom of U55 and the OP2 atom of U55 and U54 are ∼4.9 Å and 5.2 Å, respectively, and therefore significantly shorter than the corresponding distances for U14 in the WT NSR (5.3 ± 0.3 and 7.3 ± 0.3 Å in the NMR structural ensemble). Furthermore, it is interesting to note that ψ55 is held in place by a tertiary interaction of its O4 atom with the H1N1 imino group of G18, which is likely to suppress structural excursions of ψ55 to alternative conformations similar to those observed for the U14ψ variant of the NSR. Finally, since there are often multiple modifications present in the T-arm and structurally neighboring regions of tRNAs, it is possible that the structural stabilization is a result of a cooperative effect of these modifications.
Thus, overall our results suggest that pseudouridine modifications of RNA might be stabilizing in structural contexts where either hydration sites involving the additional H1N1 imino groups or additional intra-RNA hydrogen bonds are already preorganized in the structure of the unmodified RNA. However, even in such situations it might be difficult to predict a priori if the stabilizing effect is a local one or also includes spatially close structural elements. On the other hand, by replacing stabilizing native interactions with alternative hydrogen bonding interactions, pseudouridine is also able to destabilize RNA structural motifs, thereby further complicating predictions of the effects of pseudouridine replacements.
MATERIALS AND METHODS
RNA sample preparation
Unmodified wild-type and C14+ NSR RNAs were synthesized via in vitro transcription using T7 polymerase, linearized plasmid DNA as template and 4 mM unlabeled commercially available nucleotide triphosphates (Merck). RNA transcripts were purified by preparative PAGE according to standard protocols and desalted via PD-10 columns (GE Healthcare). Chemically synthesized NSR variants with pseudouridine modifications were commercially obtained (Horizon Discovery Ltd.) and deprotected according to the instructions of the manufacturer.
Monomeric hairpin forms of all RNAs were obtained by heating to 95°C for 10 min and subsequent injection into 10 equivalents of ice cold water. RNA samples were concentrated and exchanged into NMR buffer (25 mM KPi and 50 mM KCl, pH 6.2), BisTris buffer (50 mM BisTris pH 6.3) for KCl-titrations or into ITC buffer (20 mM Na-cacodylate, 200 mM NaCl, 10 mM MgCl2, 1 mM spermidine, pH 6.8, Duchardt-Ferner et al. 2016) using Vivaspin concentrators (MW cutoff 3000 kDa, Sartorius).
Isothermal titration calorimetry (ITC)
ITC measurements were carried out on a MicroCal iTC200 instrument (Malvern Instruments) at 37°C using 10 µM RNA in the sample cell and 100 µM ribostamycin in the injector syringe as described previously (Duchardt-Ferner et al. 2016). An initial delay of 120 sec was followed by an initial injection of 0.2 µL and 19 injections of 2 µL with intervals of 180 sec and a stirring speed of 750 rpm. For all measurements, the reference power was set to 11 µcal*sec−1 and the high feedback mode was selected. Thermograms were processed and analyzed using Origin 7.0 software (OriginLab). Thermodynamic parameters were derived from a curve fit to the data using the one-site binding model. All measurements were repeated three times and the reported KD values are the average of these experiments.
NMR experiments
RNA concentrations in the NMR samples varied between 150 and 500 µM. All NMR measurements were carried out on 600 and 700 MHz Bruker Avance NMR-spectrometers equipped with 5-mm cryogenic triple resonance TCI-N and TCI-P or quadruple resonance QCI-P probes. 1D-31P spectra were collected at 25°C, all other NMR spectra were recorded at 10°C in 5% (v/v) D2O/95% H2O if not indicated otherwise and NMR data were processed using Topspin 4.0.6 (Bruker Biospin). For measurements of the ligand saturated RNA, samples were titrated with 1 to 1.2 equivalents of commercially available ribostamycin (Merck). Imino proton resonance assignments of the pseudouridine variants could be transferred from the wild-type and C14+ NSR in the majority of cases (Duchardt-Ferner et al. 2010; Gottstein-Schmidtke et al. 2014). The remaining imino proton resonances were assigned using the imino-imino cross-peaks in 2D-1H,1H-NOESY spectra and 2D-1H,13C-HSQC experiments recorded using standard pulse sequences (Fürtig et al. 2003). For detection of NH···O = P hydrogen bonds long-range 2D-1H,31P-HSQC experiments (Duchardt-Ferner et al. 2011; Duchardt-Ferner and Wöhnert 2017) were performed.
For quantification of the 2hJH,P cross-hydrogen bond scalar couplings constant time 1D-1H-experiments with and without 31P decoupling were recorded (Duchardt-Ferner et al. 2011) under identical buffer conditions at the same spectrometer for all RNAs to ensure comparability. For both the cross and
reference experiments seven different constant-time delays were used and all measurements were repeated twice. Signal intensity
ratios between the coupled cross (Icross) and the decoupled reference experiment (Iref) as a function of the constant-time delay (τm) were fitted to the equation
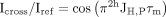 using Origin 2019 (OriginLab).
using Origin 2019 (OriginLab).
Solvent exchange rates for the imino protons were measured with inversion recovery experiments using a 180° RE-BURP soft pulse applied on the water frequency (Rinnenthal et al. 2010). A variable inversion recovery delay (τm) between 4 µsec and 4 sec and a recycle delay of 6 sec were used. Signal intensities were plotted against τm and exchange rates were determined as described (Rinnenthal et al. 2010) using Origin 2019 (OriginLab). Experiments were recorded within a temperature range between 5°C and 45°C and exchange rates were plotted as a function of temperature. ΔG at 25°C was calculated from the exchange rates at different temperatures as described previously (Rinnenthal et al. 2010).
CLEANEX-PM spectra were measured using a standard pulse sequence available in the Bruker sequence library as described (Hwang et al. 1997). The experiments were performed with a mixing time of 100 msec.
Molecular dynamics (MD) simulations
The high-resolution NMR-structure of the NSR in complex with ribostamycin (PDB: 2n0j; first frame) (Duchardt-Ferner et al. 2016) was used as the starting structure for all simulations. Based on this structure, we prepared systems with pseudouridine substitutions of nucleotides U13, U14, U18, and U13/U18, respectively, or systems where in addition to the pseudouridine substitutions of U13 or U18, the nucleotide U14 was replaced with a protonated cytosine (C14+). Based on the NMR results, the pseudouridine bases were initially positioned in the anti conformation with the χ dihedral angle at the same value as in the WT system. All simulation files were prepared in the tleap module of AMBER 18 (Case et al. 2018). The OL3 (Zgarbová et al. 2011) and GAFF (Wang et al. 2004) force fields were used to describe the RNA and ribostamycin, respectively. Partial charges and parameters for ribostamycin and the C14+ nucleotide were taken from an earlier simulation study of the NSR riboswitch (Krepl et al. 2018). We have used the Gaussian 09 (Frisch et al. 2009) to calculate the electrostatic potential of 5-methyl-uracil, using the HF/6-31G* level of theory and following the protocol which was previously used for calculations of the standard nucleotides (Cornell et al. 1995). The partial atomic charges for pseudouridine were subsequently derived by antechamber using the RESP procedure (Wang et al. 2000). The missing bond, angle and dihedral parameters for the ψ nucleotide were adapted from the OL3 RNA force field parameters for uridine (Zgarbová et al. 2011). We note that there are also alternative ψ dihedral parameters available that in our opinion correctly account for its experimentally known preference for the syn base orientation in isolated nucleosides and nucleotides (Deb et al. 2014, 2016). However, because our experiments unambiguously established the anti orientation of all the tested ψ replacements in the folded NSR, the standard OL3 parameters were used. The derived partial charges and adapted parameters for ψ are available in the Supplemental Information.
In all MD simulations, the NSR solute was surrounded with SPC/E water molecules (Berendsen et al. 1987) in an octahedral box, with a minimal distance of 13 Å between the solute and the box border. KCl (Joung and Cheatham 2008) ions were added to neutralize the system and to obtain a bulk ion concentration of ∼0.15 M. The simulations were performed in an NPT (constant pressure) ensemble, using the Monte Carlo Barostat and the Langevin thermostat for the regulation of pressure and temperature, respectively (Case et al. 2018). All other simulation settings were the same as in the previous simulation studies of the NSR (Krepl et al. 2018).
The models of the isolated U and ψ bases were constructed by removing the sugar-phosphate backbone atoms from the standard nucleotide, retaining only the base and the C1′ and H1′ atoms. Two additional hydrogen atoms were attached to the C1′ atom to form a methyl group cap for the N1 and the C5 atom in U or ψ, respectively, obtaining either N1-methyl uracil or thymine (5-methyl uracil). Partial atomic charges of the methyl group hydrogens were manually adjusted to obtain a zero net-charge of the system. The constructed models were surrounded in a water box and used to perform thermodynamic integration calculations (Case et al. 2018). We used five lambda windows, each simulated for 20 nsec at a temperature of 300 K, in which we gradually decoupled the nonbonded interactions between the base and the water. Soft-core potentials were used to describe the disappearing atoms (Steinbrecher et al. 2011). The free energy of solvation was derived by a five-point Gaussian quadrature (Case et al. 2018) and the error was estimated by the batch method where each lambda window was divided into clusters of one million lambda values and their averages computed. The standard deviation was then derived from these averages.
QM and QM/MM calculations
To complement the MD simulations, we performed QM calculations of small model systems representing the HB1 interaction of the NSR (Supplemental Fig. S13). The systems were constructed from carefully selected MD simulation snapshots of uridine and pseudouridine variants of the system. The main selection criteria for the snapshots were the geometry of the HB1 bond close to the MD simulation average and the presence of the essential K+ ion bound near U14(O4) or ψ14(O2). The QM optimizations were performed with PBEh-3c (Grimme et al. 2015), either in vacuum or using the COSMO (Klamt and Schüürmann 1993) solvation model. The hybrid density functional based PBEh-3c composite method is well tested for general chemistry (Brandenburg et al. 2014) and has also been successfully used for biomolecules (Pokorná et al. 2018; Mráziková et al. 2020). It combines a small basis set with empirical basis set correction to enable quality structure optimizations of large molecules with affordable computational costs. For this work in particular, it enables routine treatment of very large QM regions with an accuracy comparable to large basis set dispersion-corrected hybrid DFT methods. The calculations were performed using Turbomole together with the Xopt optimizer (Kruse 2020) using penalty-function restraints (Kruse and Šponer 2015) to maintain a conformation corresponding to the HB1 interaction. In addition to the small model QM calculations, we also performed QM/MM optimizations of the entire NSR, details of which are described in the Supplemental Information.
Analysis of the computational results
We used cpptraj (Roe and Cheatham 2013) to analyze all simulation trajectories and to evaluate the structural influence of the ψ substitutions on the NSR dynamics with atomistic resolution. Selected MD simulations were extended up to 10 µsec and the analysis above describes these long trajectories while the other shorter simulations were used to verify their reproducibility. For systems where the trajectories were not extended, we analyzed their combined simulation ensemble. VMD (Humphrey et al. 1996) and PyMOL (Schrödinger 2010) were used for structural visualization. H-bond interactions were analyzed by monitoring distances and angles between the relevant heavy atoms. An H-bond was considered to be present when the distance between the heavy-atoms was below 3.5 Å and the angle formed by the donor/hydrogen/acceptor atoms was larger than 120°. The anion-π interaction was evaluated by measuring the distance between the A16(OP2) atom and the geometrical center of the plane of the U14/ψ14/C14+ base. The experimental NOE distance violations in the simulations were evaluated by calculating the r−6 weighted average of the NOE distances (r) in the simulation ensemble and by comparing these values with the experimental upper-bound NOE distances. Average simulation values larger than the experimental upper bounds +0.3 Å were considered as NOE violations. We have used per-residue RMSF (root mean square fluctuation) values calculated against the average structure of each simulation ensemble to evaluate the dynamics of the RNA bases and the backbone, respectively. All simulation trajectories were visually inspected.
DATA DEPOSITION
The data related to the molecular dynamics simulations, including the trajectories and the input files necessary to reproduce the calculations, have been deposited at Zenodo under the accession code 10.5281/zenodo.7555856.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We are grateful to Christian Richter and Manfred Strupf for the excellent maintenance of the NMR facility and to Boris Fürtig for helpful discussions. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) Wo901/7-1 in the framework of the SPP 1784 “Chemical biology of native nucleic acid modifications” and the CRC 902 “Molecular mechanisms of RNA-based regulation” (project B17 to J.W.), the Czech Science Foundation (grant number 23-05639S to Z.Z., M.K., and J.S.), the European Commission H2020-MSCA-ITN project 765266 LightDyNAmics (to Z.Z.), and the State of Hesse—Center for Biological Magnetic Resonance (BMRZ) of the Goethe University Frankfurt.
Footnotes
-
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079506.122.
- Received November 3, 2022.
- Accepted February 10, 2023.
This article is distributed exclusively by the RNA Society for the first 12 months after the full-issue publication date (see http://rnajournal.cshlp.org/site/misc/terms.xhtml). After 12 months, it is available under a Creative Commons License (Attribution-NonCommercial 4.0 International), as described at http://creativecommons.org/licenses/by-nc/4.0/.








